
Abstract
Background
Sea-lavenders (Limonium Mill., Plumbaginaceae) are a cosmopolitan group of diploid and polyploid plants often adapted to extreme saline environments, with a mostly Tethyan distribution, occurring in the Mediterranean, Irano-Turanian, Euro-Siberian and in the New World. The halophylic Limonium vulgare polyploid complex in particular, presents a large distribution throughout extreme salt-marsh habitats and shows little morphological but high taximetric variation, frequently blurring species delimitation. In this work we pursue three main goals: assert whether SNP data from polyploid individuals has the resolution to distinguish the seven sampled species, to better understand how genetically structured Limonium vulgare is, and attempt to identify specific molecular mechanisms for the differentiation between L. maritimum and L. vulgare. For this purpose, 95 individuals were genotyped using Genotyping by Sequencing (GBS), which were assembled as two independent datasets using ipyrad. All analyses performed downstream of assembly were fully automated. Phylogenetic inference, PCA, and admixture plots were used to infer answers to the study’s main goals.
Results
Close to 10,000 SNPs were obtained for each dataset. Phylogenetic analyses reveal that polyploid data can be used to infer species relationships. Population structure analyses suggest a genetically structured L. vulgare. A set of 34 SNPs were found to be fully segregated between L. vulgare and L. maritimum, two of which are potentially linked to proteins that might be involved in the speciation process.
Conclusion
Despite polyploid data analyses shortcomings, GBS generated SNPs have the resolution to discern all seven included species. Limonium vulgare revealed pronounced genetic structure along a geographical north-south cline. L. maritimum always appears as a distinct genetic entity. Segregated SNPs between L. vulgare and L. maritimum indicate salinity response and morphological trait control genes as potentially interesting to follow up for studying these species’ divergence process.
Similar content being viewed by others


Background
Sea-lavenders (Limonium Mill., Plumbaginaceae) are cosmopolitan organisms, that survive in environments containing significant concentrations of soluble salt (halophytes) in which they grow, such as, rocky cliffs, and salt marshes that can be inundated with seawater during high tide. Many of these halophytes show a high level of endemicity since they grow in very specialized microhabitats that occupy small areas, usually exposed to intense anthropogenic disturbance [1,2,3,4].
This plant group has a mostly Tethyan distribution, occurring in the Mediterranean, Irano-Turanian and Euro-Siberian regions. The Limonium vulgare complex is represented by species such as Limonium vulgare sensu lato (west Atlantic, tetraploid), Limonium maritimum (west Atlantic, tetraploid), Limonium humile (west Atlantic, hexaploid [5]) or Limonium narbonense (west Mediterranean, tetraploid) [6, 7] but also includes New World native species, such as Limonium carolinianum (east Atlantic, tetraploid [8]), Limonium californicum (western Pacific, diploid [9]) and Limonium brasiliense (east Atlantic, unknown ploidy) [10]. Limonium from subfamily Limonioideae (= Staticoideae [10]) has a high enough incidence of polyploidy [11,12,13] and several taxonomical complex groups have been identified that include ca. 600 species [6, 7, 11, 12, 14,15,16]. Species from L. vulgare complex present diverse breeding systems such as selfing (L. californicum, L. carolinianum, L. humile), outcrossing (L. brasiliense, L. narbonense, L. vulgare) and probable apomixis (agamospermy, asexual reproduction through seeds; L. maritimum) [7, 9, 12, 17,18,19]. The complex’s nominal species, L. vulgare, is particularly interesting as a study subject, since due to its distribution, it is subject to a wide spectrum of environmental conditions and is sympatric to other closely related species such as L. maritimum, or L. humile [7, 12, 20, 21].
Phylogenetic relationships between Limonioideae subfamily members are intricate and complex, but thoroughly explored using plastidial and nuclear markers in [10, 22,23,24,25]. Relationships between L. vulgare, L. narbonense, L. humile, L. carolinianum, L. brasiliense and L. californicum are not yet entirely clear, with different markers proposing different optimal trees, but always segregating the mentioned species [10, 22,23,24,25]. Some studies, however, are unable to show a phylogenetic differentiation between L. maritimum and L. vulgare [25]. This not only triggers the question of how well differentiated these two species are on a genomic level, but also of what could have caused this segregation. Figure 1 conceptually summarizes the current known phylogenetic relationships and uncertainties among species within the L. vulgare complex.

Conceptual cladogram showing phylogenetic relationships found between species of the polyploid Limonium vulgare complex (Plumbaginaceae) based on previous genetic studies on these species (Malekmohammadi, Akhani & Borsch, 2017; Koutroumpa et al., 2018; Róis et al., 2018). The continuous black line represents relationships found in Malekmohammadi, Akhani & Borsch, (2017) and the red dashed line represents those reported in Koutroumpa et al., (2018). Relationships between L. vulgare and L. maritimum are based in Róis et al. (2018)
Relatively recent technological advances – “reduced representation libraries” (RRLs) such as “Genotyping by Sequencing” (GBS) [26] or “Restriction site-Associated DNA Sequencing” (RAD-Seq) [27] are able to prove solutions to both presented problems in a cost-effective way [28]. Resulting markers from these techniques – “Single Nucleotide Polymorphisms” (SNPs), can be used to both resolve difficult phylogenies [29], and provide insights regarding population genetics [30]. Use of these techniques, however, is not without its challenges, especially when applied to polyploid individuals [31, 32]. Although analyses pipelines for polyploid individuals do exist, each has limitations that prevent their generalized application [31]. Further downstream, polyploidy presents more data analyses barriers, for example regarding genotype assessment, association studies or even with FST based methods [31, 33]. Nonetheless, polyploid groups provide some of the most interesting organisms to study from an evolutionary point of view, especially regarding speciation dynamics [13, 34]. As such, this study’s first objective is to establish whether SNP data from polyploid individuals are able to differentiate the sampled species. For that, GBS was used to obtain SNPs from a set of 95 individuals of different Limonium species: L. vulgare, L. maritimum, L. narbonense, L. humile, L. carolinianum, L. brasiliense and L. californicum. Once this is demonstrated, three main follow-up questions ensue: a) Which phylogenetic hypothesis from Fig. 1 is supported by the GBS data, b) How genetically structured is L. vulgare across its distribution range, and c) Can SNPs that segregate the closely related species L. maritimum and L. vulgare be found in genes that help explain the species’ differentiation?
Answering these questions will contribute to better understanding the controversial phylogenetic relationships between the studied Limonium complex species, especially regarding the relationship between L. vulgare and L. maritimum, as well as providing a comprehensive overview of L. vulgare’s population genetic structure.
Results
The obtained raw data FASTQ files contained ~ 408 M reads in each pair file. After demultiplexing in 96 samples (95 plant samples plus a “blank” negative control), 9,308,503 pairs were lost due to barcode mismatch and 19,372 were assigned to the “blank” barcode, which left ~ 398 M read pairs distributed among all 95 samples and used in the “species” dataset. For the “lvu_lma” dataset ~ 263 M read pairs from 65 samples were used.
Each above-mentioned set of reads was independently assembled with ipyrad, resulting in 111,855 SNPs in 16,520 contigs for the “species” dataset, and 439,831 SNPs in 142,032 contigs for the “lvu_lma” dataset.
After filtering the resulting assemblies, 10,720, and 10,485 SNPs (in the same number of contigs) represented across 86, and 57 individuals remained in the “species”, and “lvu_lma” datasets respectively.
Phylogenetic analysis
Phylogenetic analyses were performed on 86 samples comprising a 1,950,198 base-pairs alignment, with 257,083 unique patterns (number of unique columns). The resulting tree can be seen in Fig. 2. These results indicate, first and foremost, that the analysed dataset is able to distinguish all species, with each forming its own monophyletic group, except the L. vulgare group, which contains L. maritimum, which consequently makes it a paraphyletic species. Furthermore, this distinction is always supported by very high bootstrap support values (always 100).

Maximum Likelihood tree of Limonium samples analysed for this study. New world species are represented in Yellow, L. narbonense in green, L. humile in teal, L. maritimum in magenta, and L. vulgare in blue. Circles in each node represent tree support in the form of bootstrap values, colour coded as per the top right corner information. Country of sample origin is stated as ISO 3166-1 alpha-2 codes in brackets after the species’ name
Two major, well-supported clades can be seen in this tree. The first one comprises all L. humile and New World species individuals. Each of these species forms its own, well differentiated clade, with L. humile as a basal group to the American species, which reveal L. carolinianum as basal to L. brasiliense and L. californicum clades. The second clade is sub divided in two groups. One comprising L. narbonense samples, and another comprising L. vulgare and L. maritimum individuals. The L. narbonense clade has a highly supported geographic structure, clearly separating samples from France and Spain. The L. vulgare group is also geographically structured, with well-supported sub-clades, but relationships between them are not as strongly supported as in previously observed groups. Phylogenetic reconstruction shows a series of ladder-like clades (with varying degrees of support) comprised of individuals from L. vulgare’s northern range: Sweden, United Kingdom, France, Belgium, Germany, and Northern Spain (Galicia). After this group, a single clade is formed comprised of samples from the species’ southern range (and L. maritimum) samples.
It is worth noting that in southern samples, the L. maritimum clade is grouped within a monophyletic clade of L. vulgare samples from the same geographic region, but at considerable distance from the latter. A similar pattern can also be observed regarding L. vulgare samples from Larache (Morocco), whose clade is also at a considerable distance from its sister taxa (Ayamonte).
Ordination and clustering analyses
PCA results of the “species” dataset (Fig. 3) reveal that 10,720 SNPs segregate each of the recognized species, similar to phylogenetic analysis results. This PCA further revealed L. vulgare and L. maritimum as the closest species, with L. narbonense as the closest species to the previous two. Further away from all other species it is possible to find L brasiliense and L. californicum. It is worth noting that Principal Component (PC) 1 and 2 explain a relatively high amount of variance: 15.29 and 8.60% respectively. Results of the clustering analyses performed by ALStructure can be seen in Fig. 4. The observed pattern is concordant with both PCA and phylogenetic analysis results, since each species can be identified by a different cluster (given a large enough K value). It is worth pointing out, however, that L. humile and L. carolinianum are always similarly classified, even though for higher values of K (K > 5), L. vulgare and L. narbonense are split in multiple intra-specific clusters.

PCA of the “species” dataset. Each species is represented in a different colour. The percentage of variance explained is 15.29 and 8.60% for Principal Component 1 and 2 respectively

Clustering analyses results of the “species” dataset based on ALStructure for values of K between 2 and 8. Each colour represents a different cluster
PCA results of the “lvu_lma” dataset (Fig. 5) reveal three groups – one with L. maritimum individuals, another one with southern L. vulgare samples, and one other with northern L. vulgare samples. Segregation between northern and southern individuals occurs along PC1, whereas segregation within groups occurs mostly along PC2 (9.02 and 6.02% variance explained respectively), suggesting that different groups of SNPs are responsible for the observed pattern. Within southern L. vulgare samples, it is further possible to identify two geographically distinct subgroups – one with samples from northern and centre Portugal, and another with southern Portugal, southern Spain and Moroccan samples. In the northern L. vulgare samples group, Galician samples appear relatively isolated. ALStructure results (Fig. 6) are generally compatible with PCA patterns, displaying the same clustering gradient between northern and southern L. vulgare individuals, and with L. maritimum samples isolated from L. vulgare in their own cluster from K ≥ 3. It is worth highlighting that the two southern samples’ clusters can be observed in both the PCA and ALStructure (for high enough values of K) plots, but the Galician samples’ isolation is not discriminated in ALStructure. Furthermore, ALStructure results reveal a cluster composed exclusively of samples from Larache for every K ≥ 4. This pattern is not as evident in the PCA, but is also present and well-supported in the phylogenetic tree (Fig. 2).

PCA of the “lvu_lma” dataset. The percentage of variance explained is 9.02 and 6.02% for Principal Component 1 and 2 respectively. The two capital letters before each location in the legend stand for the ISO 3166-1 alpha-2 codes from where L. vulgare samples were collected

Clustering analyses results of the “lvu_lma” dataset based on ALStructure for values of K between 2 and 8. Each colour represents a different cluster. The two capital letters before each location in the legend stand for the ISO 3166-1 alpha-2 codes from where L. vulgare samples were collected
L. vulgare and L. maritimum differentiation
Thirty-four SNPs were found to be fully segregated between L. vulgare and L. maritimum. Of these, three were matched to L. bicolor’s transcriptome sequences (Supplementary Table 3). In total, eight sequences were matched to UniParc peptides, but two of these hits were discarded since they did not match plant peptides, resulting in six hits that are potentially worth further studying. No relevant matches were found with NCBI’s “nt” database.
Locus 984 was matched to uncharacterized proteins, but in the vicinity to reverse transcriptase proteins, which are associated with retrotransposons in plants [35].
Locus 3813 was matched to “Tetratricopeptide-like helical domain superfamily” proteins, which is a structural motif present in a wide range of proteins. In this case, it was found associated with “Pentatricopeptide repeat”, which is involved in multiple aspects of RNA metabolism [36].
Locus 4214 (GBRK01021417.1) was matched to a “TCP transcription factor family”, which is known to be involved in the evolution of key morphological traits, including plant growth and development [37], including flower development [38, 39].
Locus 12,565 was matched to an “Endonuclease/exonuclease/phosphatase superfamily” protein, which is known to be involved in intracellular signalling [40].
Locus 46,709 (GBRK01023424.1) was matched to an “MFS transporter superfamily” protein. These are known to play important roles as transport proteins, essential for the movement of a wide range of substrates across biomembranes [41], and are known to be linked to salinity tolerance in plants [42].
Locus 95,053 (GBRK01036747.1) was matched to a “Zinc finger, PHD-finger” protein. These proteins modify chromatin as well as mediate molecular interactions in gene transcription, which can work as epigenome readers that can influence gene expression [43].
Discussion
Species of L. sect. Limonium subsect. Genuinae (Boissier 1848) are among the most taxonomically difficult groups in Limonium due to the occurrence of intermediate characters [7, 10, 21]. Previous phylogenetic works are not in accordance with this group’s relationships [10, 22,23,24,25] (Fig. 1). The present work attempts to bring new insights into the group’s relationships resorting to high throughput sequencing technology. It is worth highlighting that L. vulgare, L. maritimum, L. narbonense and L. carolinianum are tetraploid, whereas L. californicum is diploid and L. humile is hexaploid [5, 8, 20, 21]. This presented additional data analyses challenges, since most methods (both before and after SNP calling) are designed for diploid, or sometimes haploid species [44].
The used GBS dataset was “assembled” into two separate SNP datasets, each of which optimized for reaching this work’s proposed objectives. In both cases, conservative approaches to SNP filtering were used, which caused a near 10-fold decrease in number of “raw” to filtered SNPs on each dataset. Of these, the most impactful is the linkage disequilibrium mitigation strategy, which discards all but one SNP from each contig to avoid bias [45]. A close second is the decision to remove all loci with a minimum allele frequency below 2 alleles [46]. The approach to handling missing data, which is limited to a maximum of 20% per locus, a strategy devised from [47] further reduced the number of resulting SNPs. Despite these stringent filtering strategies, the number of obtained SNPs in each dataset is similar to those found in other comparable studies [48, 49]. It is worth noting, however, that using less conservative missing data thresholds did have noticeable impact. Leaving individuals with high amounts of missing data would cause them to be grouped in unexpected positions, while leaving SNPs with much missing data would substantially decrease the PCA’s amount explained variance.
Does the GBS data from polyploid individuals have the resolution to distinguish the sampled species?
To provide an answer for the first question proposed in this work, the “species” dataset was used. All performed analyses, (Phylogenetics, PCA and clustering analyses) seem to indicate that indeed the obtained GBS data has the resolution to distinguish sampled individuals at species level. Results seem to corroborate what most of the literature already sates – L. vulgare and L. maritimum are genetically very close, L maritimum seems to have originated from L. vulgare [7, 12, 25], and L. narbonense is sister taxa to L. vulgare [10, 22,23,24,25]. On the other hand, data suggests that L. californicum and L. brasiliense are closer to each other than to L. carolinianum, which is compatible with results from [24], but not from [10], where this relationship is missing. This group is in turn closer to L. humile than to any other included species. Although this grouping is very strongly supported, it is important to highlight that a previous study [24] positioned L. humile closer to L. narbonense than to L. carolinianum. Limonium humile and L. carolinianum are both Atlantic species [9, 10, 17], and share traits not common to L. narbonense (which has a southwestern Mediterranean distribution [6, 7, 16, 50]) such as the selfing breeding system [9, 17, 21, 51]. However, L. humile is hexaploid, while the other two species are tetraploid, and this difference is known to bias clustering patterns [44], which might explain the observed arrangement. As such, this particular relationship should be interpreted with caution, and confirmed with more studies, specifically resorting to long read whole genome sequencing approaches.
How genetically structured is L. vulgare across its distribution range?
Analyses of the “lvu_lma” dataset provide some insights regarding L. vulgare’s population structure. The most evident segregation in this dataset is between samples from the northern (Sweden, United Kingdom, Germany, Belgium, France and northern Spain) and southern (Portugal, southern Spain and Morocco) distribution ranges. The gradient like clustering pattern is especially evident Fig. 6’s K = 2 plot, which suggests some degree of isolation by distance. Intermediate K values reveal a fragmented southern group, with a differentiated Larache (Morocco) cluster and even a third cluster comprised of Odemira, Tavira and Ayamonte individuals (southern Iberian Peninsula). Only for very high values of K (7 and 8) can evidence of fragmentation in the northern group be found, with samples from Nieupoort (Belgium) and Brem-Sur-Mer (France) each forming independent clusters. These results suggest that L. vulgare is likely genetically structured throughout its distribution range. This is contrasting with morphometric [21] and taximetric [7] studies of L. vulgare complex, where no correlation is found between taximetry and geographic distribution.
Future studies can explore these insights to understand the reasons for this structure, using local adaptation detection and genomic offsets [52]. This will require a specifically designed sampling strategy, comprised of both more locations and more samples per location than those found in this study.
Differentiation between L. maritimum and L. vulgare
Previous works have identified L. maritimum and L. vulgare as distinct, albeit close species [7, 25]. Both phylogenetic and population genetics results in this work seem to support this hypothesis, but do not provide context on the genetic basis for this differentiation. Fully segregated SNPs between these species, however, do provide some clues in this regard. Although only a very small percentage of the analysed SNPs were completely segregated between both species (34 of 10,485), it was possible to match some of their flanking regions to known proteins.
Only six of the 34 segregated SNPs were mapped to known protein regions. Of these, four were matched to “house-keeping” genes (Loci 984, 3813, 12,565 and 95,053), which provide no indication regarding the differentiation’s context. However, two loci (Loci 4214, and 46,709) were matched to proteins with functions which may help to pinpoint what could have caused these two species to diverge. Although locus 46,709, which can be linked to salinity response, is a very good candidate for follow-up studying in these halophyte species, as a differential tolerance to salinity could help explain the observed divergence, it is locus 4214, linked to TCP transcription factors, that provides the most important clue to explain the mentioned divergence. The current speciation hypothesis, proposed in [7] is based on the fact that L. vulgare presents heterostylic flowers and dimorphic populations with self-sterile pollen-stigma combinations (A or B), associated with sexual reproduction (outcrossing) [7, 17, 21, 53,54,55], whereas L. maritimum specimens show exclusively homostylic flowers with a unique self-incompatible pollen-stigma morph (B) and very low fertility [7], probably associated to apomixis (agamospermy, asexual reproduction through seeds). TCP transcription factors are involved in various flower development aspects, including shape and symmetry [38, 56]. These genes’ activities are also essential for leaf development [57] and ovule development [58]. Other key biological process is in hormone regulation of auxin homeostasis (auxin perception and response) in the gynoecium by binding the IAA3 promoter in Arabidopsis [59]. Further, changes in TCP expression result in male and female sterility [56]. In a recent study on floral sRNAs originated from apomictic and sexual genotypes of Paspalum notatum transcription factors from the TCP subfamily is affected in apomictic ovules [60].
The segregation in locus 4214 between these species, which is linked to flower shape morphology supports this hypothesis, by providing a genetic basis to the morphological observations. Although this link does not confirm causality, further studying these species’ TCP transcription factor proteins and reproductive systems, through wet-lab validation methods may yield further insights into this subject which is a strong candidate for follow-up studies.
Conclusions
In this work, three main biological questions were addressed, which provide new insights on part of the Limonium complex evolutionary history, as well as intra and interspecific relationships.
Results demonstrate that genome wide SNP data such as the one used here can match those of sequence data regarding species discrimination within the complex. Although this has been widely demonstrated before [61], in this particular case, polyploidy could have biased the results, which meant that validation was in order. At population level, polyploidy also played a role, limiting the type of analyses that can be accurately performed, such as F statistics-based methods [44]. As such, alternative methods, like the model-free approach from ALStructure, were found to answer the proposed questions with a polyploid dataset.
This study is, to date, using the largest data matrix to infer phylogenetic relationships in the L. vulgare complex. As such, it is able to provide detailed interspecific relationships with unprecedented support levels, while also confirming L. maritimum as a genetically distinct entity from L. vulgare, making the latter a paraphyletic species.
At an intra-specific level, this study has two main highlights. L. vulgare is revealed to be a genetically structured species, with clear northern and southern clusters. Furthermore, samples from Larache (Morocco), reveal a differentiation pattern from other individuals, somewhat similar to L. maritimum’s divergence pattern.
Both inter and intra-specific revealed results are worth further exploring in follow-up studies with more fine-grained approaches, such as WGS, and/or with more comprehensive sampling strategies.
Regarding L. vulgare and L. maritimum divergence, this study’s findings provide novel evidence supporting the hypothesis proposed in [7] based on morphological traits. These results highlight the potential role of TCP transcription factors in these species’ divergence, and suggest a starting place to further study these two close species’ divergence.
Methods
Biological samples
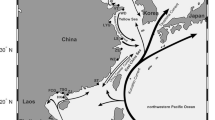
Leaf material was obtained from 95 individuals: 33 plants were sampled in natural populations (Supplementary Table 1), and 62 were originated from seeds planted and germinated at “Instituto Superior de Agronomia” (ISA). Of these seeds, 25 were field collected (Supplementary Table 1), and 37 were requested from seed banks (Supplementary Table 2). A total of 30 sites were sampled, as shown in Fig. 7.

A map of the sites sampled for this work. In European samples the first part of the label represents the species: “Lvu” - L. vulgare; “Lma” - L. maritimum; and “Lna” - L. narbonense. The second part represents the country of origin according to ISO 3166. The third part is the name of the location where samples were collected
Formal sample identification was undertaken by Ana D. Caperta following the treatments of [6, 7, 62,63,64]. Voucher specimens were deposited in the Herbarium João de Carvalho e Vasconcellos (LISI), Instituto Superior de Agronomia, Portugal (Supplementary Table 1).
Plants of ex situ collections were established from seeds provided by seed banks at the Botanic Garden Meise (Belgium), Freie Universität Berlin (Germany), Institut de Recherche pour la Conservation des Zones Humides Méditerranéennes (France), Millenium Seed Bank (UK), Museum National d’ Histoire Naturelle (France) Universidad Nacional del Sur (Argentina), and University of Sussex (UK). For establishing experimental collections, seeds were germinated under controlled conditions, in a growth chamber (Phytotron, Rumed) with a photoperiod of 18 h / 6 h of light and dark, respectively, and a temperature of 25 °C until germination [65]. Seedlings were transferred to jiffy pots and maintained in similar growth conditions for 2 months. After this period, they were transplanted into plastic pots with substrate and grown under greenhouse conditions for 1 year as described in [65].
Library preparation and sequencing
Genomic DNA was extracted from liquid nitrogen grounded leaves of all samples collected for this work using the kit “innuPREP Plant DNA Kit” (Analytik Jena AG) with the SDS based Lysis Solution OPT protocol, which produced the best results for Limonium species. DNA was quantified in Qubit 2.0 (Invitrogen) using Qubit dsDNA HS Assay kit, quality checked using a Nanodrop 1000 (Thermo Fisher Scientific, Wilmington, DE, United States), and integrity verified using agarose gel electrophoresis (1%). Isolated DNA samples with a minimum concentration of 30 ng/μl were sent to Elshire GBS services (The Elshire Ltd., Manawatu, New Zealand) to perform library preparation and sequencing. Library preparation for deep sequencing was carried out using the TruSeq Nano DNA Library Preparation Kit (350 bp insert size). The genotyping-by-sequencing data was generated following the method described in [26] and included the following changes: 100 ng of genomic DNA were used, 3.6 ng of total adapters were used, genomic DNA was restricted with ApeKI enzyme and the library was amplified with 18 PCR cycles. Sequencing of Limonium cDNA libraries was carried out using the Illumina HiSeq Ten platform with 2 × 150 bp paired end reads.
Raw data handling
Raw, paired end data was demultiplexed using axe-demux v0.3.3-2 [66]. The resulting demultiplexed “fastq” files were submitted to NCBI’s Sequence Read Archive (SRA) as “BioProject” PRJNA592300. These were then analysed using ipyrad v0.9.71 [67], using a “conda” [68] environment containing muscle v3.8.1551 [69] and vsearch v2.17.0 [70]. Read “assembly” was performed on two distinct sets of individuals. One comprised of all sampled sites and species (L. brasiliense, L. californicum, L. carolinianum, L. humile, L. maritimum, L. narbonense and L. vulgare) from here on designated as “species”, and a second one, comprising all L. vulgare and L. maritimum individuals, called “lvu_lma”. In both cases, the same assembly parameters were used, including GBS as datatype, a clustering threshold of 0.85, and a mindepth of 8. Other parameters were left at their defaults. In the “species” dataset, each species had to be represented by at least one individual for a SNP to be called. This restriction was not enforced on the “lvu_lma” dataset. Furthermore, in order to ensure only nuclear DNA was included in the analyses, a mitochondrial genome of Beta vulgaris (NCBI Accession BA000009.3) and a plastidial genome of Limonium sinense (NCBI Accession MN599096.1) were used to “bait” and discard mtDNA and cpDNA sequence reads. Full assemblies’ parameters can be found in a gitlab repository [71].
Data analyses
Analyses downstream of the assembly were automated using “GNU Make” (https://www.gnu.org/software/make) to centralizing all details of each analysis step. To facilitate reproduction, a “ready to use” docker image [72] with all the software, configuration files, parameters, and the Makefile, is also provided. Furthermore, a gitlab repository [71] was created to host all files (and change history) necessary for analyses reproduction, such as the Makefile, static analysis parameter files, and a Dockerfile, (containing instructions on how to build the static analysis environment). Availability of the analyses’ environment (and creation process) should ensure that this work remains reproducible for the foreseeable future. This automation is an improvement upon a previous, similar scheme described in [73].
Raw data filtering
Assembled data was filtered using VCFtools v0.1.14 [74]. Filtering parameters include, in this order: maximum percentage of missing data an individual can have in order not to be discarded (40 and 10% for the “species” “lvu_lma” datasets respectively); minimum number of individuals each SNP has to be represented in (80%); minimum allele frequency (MAF) of each SNP was set to 0.01 (any position where the rarer allele frequency is below 1% was discarded) – following the recommendations present in [46] to minimize population structure analyses bias.
In order to minimize the effects of linkage disequilibrium, the python script vcf_parser.py [75] as of commit “478a8e3” was used to keep only the SNP closest to each locus’s centre.
Phylip formatted files for phylogenetic analyses were filtered to include the same individuals that passed the above-mentioned filtering criteria.
PGDSpider v2.1.1.5 [76] was used to perform most file format conversions. Those not supported by PGDSpider were performed using scripts available in https://github.com/CoBiG2/RAD_Tools/ (technical details on these conversions can be found in the Makefile).
Phylogenetic analyses
Phylogenetic analysis was conducted using RAxML v8.2.12 [77], PTHREADS version, using the model GTRCAT, and an automatic number of rapid bootstrap replicates. Seed value for both parsimony and rapid bootstrap inference was 112,358. It is important to note that there is no outgroup in this analysis, and as such, the tree rooting is artificial and arbitrary (chosen based on branch size, optimized for visualization).
Ordination and clustering analyses
Two methods were used for grouping samples in order to understand the general pattern of individual or population structure, namely, Principal Components Analysis (PCA) and ALStructure (as of commit “e355411”) [78] – a likelihood-free, PCA based method to infer admixture plots. This method was chosen over the more popular likelihood based methods, such as STRUCTURE [79] due to the nature of polyploid data, which without phase information may bias likelihood model based methods.
The PCA was performed with the script snp_pca_static.R [75] as of commit “a69e9bd”.
To interpret admixture analyses results, an estimate of the optimal value of “K”, which represents how many demes the data can be clustered into is usually calculated. However, since ALStructure is PCA based, it can only produce such estimate in the absence of missing data. Since both used datasets have at least a fraction of missing data, an estimate of “K” was not possible to obtain. ALStructure was wrapped under Structure_threader v1.3.9 [80] and was run for values of “K” between 1 and 8.
L. vulgare and L. maritimum differentiation
In order to further understand the genetic basis behind the L. vulgare and L. maritimum differentiation, SNPs fully segregated between these two species (using the “lvu_lma” dataset) were identified using the script segregating_loci_finder.py [75] as of commit “146b483”. Each segregated SNP’s flanking region was then obtained using the script loci_consensus.py [75] as of commit “d277ed3”. Since the obtained sequences are relatively small (between 41 and 261 bp), these sequences were queried against the Limonium bicolor transcriptome (GenBank: GBRK00000000.1) [81], using NCBI BLAST+ v2.9.0 [82] and for any relevant hits (e-value ≤ 1e− 8) the GBS locus was replaced with the transcriptome matched sequence. Resulting sequences where then queried against the UniParc database (release 2022_01) using DIAMOND v2.0.15 [83] and against NCBI’s “nt” database (as of 10-05-2022) using NCBI BLAST+. Only relevant hits (e-value ≤ 1e− 8) with plant sequences were considered valid matches.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI SRA repository, under “BioProject” PRJNA592300. All analyses details and scripts are contained in a gitlab repository (dynamic) and in a zenodo repository(static, with DOI). A docker image containing the previous repository and all the required software, including usage instructions is available in docker hub. Filtered data can be found in this second zenodo repository.
Change history
04 February 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12870-023-04095-0
References
Ennos RA, French GC, Hollingsworth PM. Conserving taxonomic complexity. Trends Ecol Evol. 2005;20(4):164–8.
Laguna E, Deltoro V, Ferrer-Gallego PP, Navarro A, Ferrando Pardo I, Escribá M, et al. Relevant Plant Recovery Programmes. Conservation Management of Plant Micro-Reserves and Ecological Restoration; 2013. p. 127–40.
Laguna E, Fos S, Ferrando-Pardo I, Ferrer-Gallego PP. Endangered halophytes and their conservation. In: Grigore MN, editor. Handbook of halophytes: from molecules to ecosystems towards biosaline agriculture. Cham: Springer International Publishing; 2021. p. 661–723. https://doi.org/10.1007/978-3-030-57635-6_21. Cited 2022 Jul 14.
Caperta AD, Espírito-Santo MD, Silva V, Ferreira A, Paes AP, Róis AS, et al. Habitat specificity of a threatened and endemic, cliff-dwelling halophyte. AoB Plants. 2014;6:plu032.
Rice A, Glick L, Abadi S, Einhorn M, Kopelman NM, Salman-Minkov A, et al. The chromosome counts database (CCDB) – a community resource of plant chromosome numbers. New Phytol. 2015;206(1):19–26.
Erben M. Limonium. In: Iberica F, editor. CSIC. Madrid: Real Jardín Botánico; 1993. p. 2–143.
Cortinhas A, Erben M, Paula Paes A, Espírito Santo D, Guara-Requena M, Caperta AD. Taxonomic complexity in the halophyte Limonium vulgare and related taxa (Plumbaginaceae): insights from analysis of morphological, reproductive and karyological data. Ann Bot. 2015;115(3):369–83.
Gervais C, Parent M, Trahan R, Plante S. IOPB chromosome data 12. Newslett Int Organ Pl Biosyst. 1997;28:16–8.
Baker HG. Dimorphism and monomorphism in the PlumbaginaceaeIII. Correlation of geographical distribution patterns with dimorphism and monomorphism in Limonium. Ann Bot. 1953;17(4):615–28.
Malekmohammadi M, Akhani H, Borsch T. Phylogenetic relationships of Limonium (Plumbaginaceae) inferred from multiple chloroplast and nuclear loci. TAXON. 2017;66(5):1128–46.
Erben M. Die gattung limonium im südwestmediterranen raum. Mitteilungen Bot Staatssamml Münch. 1978;14:361–631.
Caperta AD, Conceição SIR, Róis AS, Loureiro J, Castro S. Cytogenetic features of sexual and asexual Limonium taxa (Plumbaginaceae). TAXON. 2018;67(6):1143–52.
Marques I, Loureiro J, Draper D, Castro M, Castro S. How much do we know about the frequency of hybridisation and polyploidy in the Mediterranean region? Plant Biol. 2018;20(S1):21–37.
Erben M. Karyotype differentiation and its consequences in Mediterranean « Limonium ». Webbia. 1979;34(1):409–17.
Castro M, Rosselló JA. Karyology of Limonium (Plumbaginaceae) species from the Balearic Islands and the western Iberian Peninsula. Bot J Linn Soc. 2007;155(2):257–72.
Brullo S, Erben M. The genus Limonium (Plumbaginaceae) in Greece. Phytotaxa. 2016;240(1):1.
Baker HG. The evolution, functioning and breakdown of heteromorphic incompatibility systems. I. the Plumbaginaceae. Evol Int J Org Evol. 1966;20(3):349–68.
Baltzer J, Hewlin HL, Reekie E, Taylor PD, Boates J. The impact of flower harvesting on seedling recruitment in sea lavender (Limonium carolinianum, Plumbaginaceae). undefined. 2002. Available from: https://www.semanticscholar.org/paper/The-impact-of-flower-harvesting-on-seedling-in-sea-Baltzer-Hewlin/d4ca3d236d574c8ee8c170735ce79686d02a951e. Cited 2021 Sep 13.
Georgakopoulou A, Manousou S, Artelari R, Georgiou O. Breeding systems and cytology in Greek populations of five Limonium species (Plumbaginaceae)*. Willdenowia. 2006;36(2):741–50.
Dawson HJ. Chromosome numbers in two Limonium species. Watsonia U K. 1990. Available from: http://agris.fao.org/agris-search/search.do?recordID=GB9101144. Cited 2019 Feb 20.
Dawson HJ, Ingrouille MJ. A biometric survey of Limonium vulgare Miller and L. humile Miller in the British Isles. Watsonia. 1995;20:239–54.
Lledó MD, Crespo MB, Fay MF, Chase MW. Molecular phylogenetics of Limonium and related genera (Plumbaginaceae): biogeographical and systematic implications. Am J Bot. 2005;92(7):1189–98.
Akhani H, Malekmohammadi M, Mahdavi P, Gharibiyan A, Chase MW. Phylogenetics of the Irano-Turanian taxa of Limonium (Plumbaginaceae) based on ITS nrDNA sequences and leaf anatomy provides evidence for species delimitation and relationships of lineages. Bot J Linn Soc. 2013;171(3):519–50.
Koutroumpa K, Theodoridis S, Warren BH, Jiménez A, Celep F, Doğan M, et al. An expanded molecular phylogeny of Plumbaginaceae, with emphasis on Limonium (sea lavenders): taxonomic implications and biogeographic considerations. Ecol Evol. 2018;8(24):12397–424.
Róis AS, Castro S, Loureiro J, Sádio F, Rhazi L, Guara-Requena M, et al. Genome sizes and phylogenetic relationships suggest recent divergence of closely related species of the Limonium vulgare complex (Plumbaginaceae). Plant Syst Evol. 2018;304(8):955–67.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3(10):e3376.
Ree RH, Hipp AL. Inferring phylogenetic history from restriction site associated DNA (RADseq). In: Next-generation sequencing in plant systematics. Germany: Koeltz Scientific Books; 2015. p. 181–204. (Regnum vegetabile; vol. 158). Available from: www.iapt-taxon.org. Cited 2019 Feb 19.
Leaché AD, Oaks JR. The utility of single nucleotide polymorphism (SNP) data in Phylogenetics. Annu Rev Ecol Evol Syst. 2017;48(1):69–84.
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R. A practical guide to environmental association analysis in landscape genomics. Mol Ecol. 2015;24(17):4348–70.
Clark LV, Lipka AE, Sacks EJ. polyRAD: genotype calling with uncertainty from sequencing data in Polyploids and diploids. G3 Genes Genomes Genet. 2019;9(3):663–73.
Melo ATO, Hale I. Expanded functionality, increased accuracy, and enhanced speed in the de novo genotyping-by-sequencing pipeline GBS-SNP-CROP. Bioinformatics. 2019;35(10):1783–5.
Bourke PM, Voorrips RE, Visser RGF, Maliepaard C. Tools for genetic studies in experimental populations of Polyploids. Front Plant Sci. 2018;9 Available from: https://www.frontiersin.org/articles/10.3389/fpls.2018.00513/full. cited 2020 Feb 26.
Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, Rieseberg LH. The frequency of polyploid speciation in vascular plants. Proc Natl Acad Sci. 2009;106(33):13875–9.
Dixit A, Ma KH, Yu JW, Cho EG, Park YJ. Reverse transcriptase domain sequences from Mungbean (Vigna radiata) LTR retrotransposons: sequence characterization and phylogenetic analysis. Plant Cell Rep. 2006;25(2):100–11.
Delannoy E, Stanley WA, Bond CS, Small ID. Pentatricopeptide repeat (PPR) proteins as sequence-specificity factors in post-transcriptional processes in organelles. Biochem Soc Trans. 2007;35(6):1643–7.
Cubas P, Lauter N, Doebley J, Coen E. The TCP domain: a motif found in proteins regulating plant growth and development. Plant J. 1999;18(2):215–22.
Manassero NGU, Viola IL, Welchen E, Gonzalez DH. TCP transcription factors: architectures of plant form. Biomol Concepts. 2013;4(2):111–27.
Li D, Zhang H, Mou M, Chen Y, Xiang S, Chen L, et al. Arabidopsis class II TCP transcription factors integrate with the FT-FD module to control flowering. Plant Physiol. 2019;181(1):97–111.
Dlakić M. Functionally unrelated signalling proteins contain a fold similar to Mg2+−dependent endonucleases. Trends Biochem Sci. 2000;25(6):272–3.
Quistgaard EM, Löw C, Guettou F, Nordlund P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol. 2016;17(2):123–32.
Acharya BR, Sandhu D, Dueñas C, Ferreira JFS, Grover KK. Deciphering molecular mechanisms involved in salinity tolerance in guar (Cyamopsis tetragonoloba (L.) Taub.) using transcriptome analyses. Plants. 2022;11(3):291.
Sanchez R, Zhou MM. The PHD Finger: a Versatile epigenome reader. Trends Biochem Sci. 2011;36(7):364–72.
Meirmans PG, Liu S, van Tienderen PH. The analysis of Polyploid genetic data. J Hered. 2018;109(3):283–96.
Shi M, Umbach DM, Wise AS, Weinberg CR. Simulating autosomal genotypes with realistic linkage disequilibrium and a spiked-in genetic effect. BMC Bioinformatics. 2018;2(19):2.
Linck E, Battey CJ. Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Mol Ecol Resour. 2019;19(3):639–47.
Huang H, Knowles LL. Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst Biol. 2016;65(3):357–65.
Mastretta-Yanes A, Arrigo N, Alvarez N, Jorgensen TH, Piñero D, Emerson BC. RAD sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol Ecol Resour. 2014:28-41.
White TA, Perkins SE, Heckel G, Searle JB. Adaptive evolution during an ongoing range expansion: the invasive bank vole ( Myodes glareolus ) in Ireland. Mol Ecol. 2013;22(11):2971–85.
Pandža M, Franjić J, Škvorc Ž. The salt marsh vegetation on the east Adriatic coast. Biologia (Bratisl). 2007;62(1):24–31.
Hamilton MB, Rand DM. Relatedness measured by oligonucleotide probe DNA fingerprints and an estimate of the mating system of sea lavender (Limonium carolinianum). Theor Appl Genet. 1996;93(1):249–56.
Rellstab C. Genomics helps to predict maladaptation to climate change. Nat Clim Chang. 2021;11(2):85–6.
Baker HG. Dimorphism and monomorphism in the PlumbaginaceaeI. A survey of the family. Ann Bot. 1948;12(3):207–19.
Costa J, Castro S, Loureiro J, Barrett SCH. Experimental insights on the function of ancillary pollen and stigma polymorphisms in plants with heteromorphic incompatibility. Evol Int J Org Evol. 2017;71(1):121–34.
Costa J, Torices R, Barrett SCH. Evolutionary history of the buildup and breakdown of the heterostylous syndrome in Plumbaginaceae. New Phytol. 2019;224(3):1278–89.
Nicolas M, Cubas P. TCP factors: new kids on the signaling block. Curr Opin Plant Biol. 2016;33:33–41.
Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, et al. Control of leaf morphogenesis by microRNAs. Nature. 2003;425(6955):257–63.
Wei B, Zhang J, Pang C, Yu H, Guo D, Jiang H, et al. The molecular mechanism of SPOROCYTELESS/NOZZLE in controlling Arabidopsis ovule development. Cell Res. 2015;25(1):121–34.
Lucero LE, Uberti-Manassero NG, Arce AL, Colombatti F, Alemano SG, Gonzalez DH. TCP15 modulates cytokinin and auxin responses during gynoecium development in Arabidopsis. Plant J. 2015;84(2):267–82.
Ortiz JPA, Leblanc O, Rohr C, Grisolia M, Siena LA, Podio M, et al. Small RNA-seq reveals novel regulatory components for apomixis in Paspalum notatum. BMC Genomics. 2019;20(1):487.
Tripp EA, Tsai YE, Zhuang Y, Dexter KG. RADseq dataset with 90% missing data fully resolves recent radiation of Petalidium (Acanthaceae) in the ultra-arid deserts of Namibia. Ecol Evol. 2017;7(19):7920–36.
Brummitt RK. A synonymized checklist of the vascular Flora of the United States. Canada and Greenland Kew Bull. 1996;51(3):615–9.
Stace C. New Flora of the British Isles: Cambridge University Press; 2010. p. 1267.
Brazilian Flora 2020. 2019. Available from: http://reflora.jbrj.gov.br/reflora/listaBrasil/ConsultaPublicaUC/ConsultaPublicaUC.do;jsessionid=7778ACC7C33B00726065211196FC9C88. Cited 2019 Feb 15.
Róis AS, Teixeira G, Sharbel TF, Fuchs J, Martins S, Espírito-Santo D, et al. Male fertility versus sterility, cytotype, and DNA quantitative variation in seed production in diploid and tetraploid sea lavenders (Limonium sp., Plumbaginaceae) reveal diversity in reproduction modes. Sex Plant Reprod. 2012;25(4):305–18.
Murray KD, Borevitz JO. Axe: rapid, competitive sequence read demultiplexing using a trie. Bioinformatics. 2018;34(22):3924–5.
Eaton DAR, Overcast I. Ipyrad: interactive assembly and analysis of RADseq datasets. Bioinformatics. 2020;36(8):2592–4.
Anaconda Software Distribution. Anaconda documentation: Anaconda Inc; 2020. Available from: https://docs.anaconda.com/
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Pina-Martins F. Francisco Pina-Martins / Limonium_GBS_data_analyses GitLab. GitLab. 2022. Available from: https://gitlab.com/StuntsPT/Limonium_GBS_data_analyses. Cited 2022 Sep 14.
Pina-Martins F. limonium_data_analyses Docker image. limonium_data_analyses. 2022. Available from: https://hub.docker.com/repository/docker/stunts/limonium_data_analyses. Cited 2022 Sep 14.
Pina-Martins F, Baptista J, Pappas G, Paulo OS. New insights into adaptation and population structure of cork oak using genotyping by sequencing. Glob Chang Biol. 2019;25(1):337–50.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Pina-Martins F, Silva DN, Balata D, Laurentino TG. RAD Tools. Computational Biology & Population Genomics Group; 2022. Available from: https://github.com/CoBiG2/RAD_Tools. Cited 2022 Sep 15.
Lischer HEL, Excoffier L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 2012;28(2):298–9.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Cabreros I, Storey JD. A likelihood-free estimator of population structure bridging admixture models and principal components analysis. Genetics. 2019;genetics:302159.2019.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Pina-Martins F, Silva DN, Fino J, Paulo OS. Structure_threader: an improved method for automation and parallelization of programs structure, fastStructure and MavericK on multicore CPU systems. Mol Ecol Resour. 2017;17(6):e268–74.
Yuan F, Lyu MJA, Leng BY, Zheng GY, Feng ZT, Li PH, et al. Comparative transcriptome analysis of developmental stages of the Limonium bicolor leaf generates insights into salt gland differentiation. Plant Cell Environ. 2015;38(8):1637–57.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18(4):366–8.
Acknowledgements
The authors gratefully acknowledge Ana Paula Paes (LEAF/ISA) for support in plant maintenance, Dalila Espírito Santo (LEAF/ISA) and Wanda Viegas (LEAF/ISA), and Tim Flowers (University of Sussex, UK) for their encouragement in Limonium studies. We acknowledge Laila Rhazi (University of Rabat, Morocco) and Mohammed El Madihi (University of Rabat, Morocco), Miguel Guara Requena (University of Valencia, Spain), and Mats Havström (Gothenburg Botanical Garden, Sweden) for help in field work. We would like to thank Ana Andrada and Maria Elena Gil (Universidad Nacional del Sur, Bahía Blanca, Argentina), Birgit Nordt (Dahlem Seed Bank, Freie Universität Berlin, Germany), Patrick Grillas and Hugo Fontes (Institut de Recherche pour la Conservation des Zones Humides Méditerranéennes, Tour du Valat, France), Janet Terry (Millenium Seed Bank, Kew, UK), Tim Flowers (University of Sussex, UK), Thierry Vanderborgtht (Seed Bank, Botanic Garden Meise, Belgium), and Yves Pauthier (Seed Bank, Museum National d’ Histoire Naturelle, Paris, France, for providing seeds that were used to established plant ex situ collections at ISA.
Funding
This work was supported by Portuguese national funds through Fundação para a Ciência e a Tecnologia (www.fct.pt/) projects PTDC/AGRPRO/4285/2014, LEAF UI/AGR/04129/2013 and UIDB/00329/2020 (study design, library preparation and sequencing), grants PTDC/AGRPRO/4285/BD/2014 (data analysis and manuscript writing), attributted to FPM, and PTDC/AGRPRO/4285/BM/2014 and PhD Scholarship ref. UI/BD/153730/2022 (wet-lab work) attributed to SIRC. UIDB/00239/2020 (CEF), under the Scientific Employment Stimulus—Individual Call (CEEC Individual)—2021.01107.CEECIND/CP1689/CT0001 (IM). The Linnean Society of London through the Percy Sladen Memorial Fund provided a grant to ADC for fieldwork in the project ‘Taxonomic reassessment of Limonium spp. (Plumbaginaceae) from Morocco Atlantic coast’. Computational resources were partially provided by INCD funded by FCT and FEDER under the project 01/SAICT/2016 n° 022153.
Author information
Authors and Affiliations
Contributions
ADC and OP conceived and coordinated the study. ADC did identification of Limonium specimens, and ADC and SIRC established and maintained the Limonium vulgare and related species ex situ collections. VN, SIRC and IM undertook Library preparation and sequencing. FPM processed the raw data and carried out genomic data analyses. ADC, FPM and OP drafted the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Collection of plant material complies with relevant institutional, national, and international guidelines and legislation. Limonium vulgare is not a protected species in Portugal nor in Spain. For L. vulgare plants collected in Morocco, the Haut Commissariat aux Eaux et Foréts et a la Lute Contre la Desértification (Morocco), provided scientific permission (No. 04/2015), and Länsstyrelsen Västra Götaland Lä (Sweden), provided permission 522-24302-2017, as well as Centre per a l’Investigació I Experimentació Forestal de la Conselleria d’Agricultura, Medi Ambient, Canvi Climatic i Desenvolupament Rural de la Generalitat Valenciana (Spain) for collecting L. narbonense samples.
All seeds used for establishing plant ex-situ collections institutions at ISA were obtained from seed banks in Universidad Nacional del Sur, Bahía Blanca, Argentina (Ana Andrada and Maria Elena Gil), Dahlem Seed Bank, Freie Universität Berlin, Germany (Birgit Nordt), Institut de Recherche pour la Conservation des Zones Humides Méditerranéennes, Tour du Valat, France (Patrick Grillas), Millenium Seed Bank, Kew, UK) (Janet Terry), Seed Bank, Botanic Garden Meise, Belgium (Thierry Vanderborgtht), Seed Bank, Museum National d’ Histoire Naturelle, Paris, France (Yves Pauthier).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: authors would like to update the Funding section by adding 'UIDB/00239/2020 (CEF), under the Scientific Employment Stimulus—Individual Call (CEEC Individual)—2021.01107.CEECIND/CP1689/CT0001 (IM)'.
Supplementary Information
Additional file 1: Supplementary Table 1.
List of specimens.
Additional file 2: Supplementary Table 2.
Seed provenance.
Additional file 3: Supplementary Table 3.
List of GBS loci hits to public databases.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pina-Martins, F., Caperta, A.D., Conceição, S.I.R. et al. A first look at sea-lavenders genomics – can genome wide SNP information tip the scales of controversy in the Limonium vulgare species complex?. BMC Plant Biol 23, 34 (2023). https://doi.org/10.1186/s12870-022-03974-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03974-2