
Abstract
Background
Plant phylogeographic studies of species in subtropical China have mainly focused on rare and endangered species, whereas few studies have been conducted on taxa with relatively wide distribution, especially polyploid species. We investigated the cytotype and haplotype distribution pattern of the Actinidia chinensis complex, a widespread geographically woody liana with variable ploidy in subtropical China comprising two varieties, with three chloroplast fragments DNA (ndhF-rpl132, rps16-trnQ and trnE-trnT). Macroevolutionary, microevolutionary and niche modeling tools were also combined to disentangle the origin and the demographic history of the species or cytotypes.
Results
The ploidy levels of 3338 individuals from 128 populations sampled throughout the species distribution range were estimated with flow cytometry. The widespread cytotypes were diploids followed by tetraploids and hexaploids, whereas triploids and octoploids occurred in a few populations. Thirty-one chloroplast haplotypes were detected. The genetic diversity and genetic structure were found to be high between varieties (or ploidy races) chinensis and deliciosa. Our results revealed that these two varieties inhabit significantly different climatic niche spaces. Ecological niche models (ENMs) indicate that all varieties’ ranges contracted during the Last Inter Glacial (LIG), and expanded eastward or northward during the Last Glacial Maximum (LGM).
Conclusions
Pliocene and Plio-Pleistocene climatic fluctuations and vicariance appear to have played key roles in shaping current population structure and historical demography in the A. chinensis complex. The polyploidization process also appears to have played an important role in the historical demography of the complex through improving their adaptability to environmental changes.
Similar content being viewed by others
Background
In the last several decades, many phylogeographic studies have focused on the Qinghai-Tibetan Plateau and adjacent areas (the Himalayas and Hengduan Mountains) in China [1,2,3,4]. As an area harboring many ancient endemic genera and families, the subtropical region in China has recently attracted much attention due to its complex topography and fluctuated paleoclimate [5,6,7]. Nevertheless, most of these phylogeographic studies in these two hotspots of China have mainly focused on rare and endangered species [8,9,10,11], with few cases examined in the species with a relatively widespread range [12, 13], especially for polyploid species. Polyploidization is a common phenomenon in higher plants and plays an important role in plant diversification and adaptation to novel environments [14]. Although the wide recognition of the prevalence of polyploidy and accumulative cytological evidence for recent whole-genome duplication(s) in many plant groups [15, 16], studies on the phylogeography of polyploid species in these two hotspots in China [17,18,19] are few relative to those in other hotspots in the world (e.g. North America and Alps in Central Europe).
The Actinidia chinensis complex is a geographically widespread woody liana with variable ploidy in subtropical China. It is widely planted around the world as an important horticultural fruit tree crop. In the newly revised infrageneric system of Actinidia, the complex comprises three varieties, A. chinensis var. chinensis (hereafter: var. chinensis), A. chinensis var. deliciosa (hereafter: var. deliciosa), and A. chinensis var. setosa (hereafter: var. setosa) [20]. However, phylogenetic studies in Actinidia based on chloroplast DNA sequences indicate that var. setosa should not be included in A. chinensis complex but rather treated as a separate species (A. setosa) [21]. Previous studies have shown that the vast majority of var. chinensis and var. deliciosa can be distinguished by morphological characteristics such as the presence of overwintering buds or the hair types covering on flowering branchlets and fruits [22]. However, there are still morphological intermediates of these two varieties identified in their area of overlap in central China [23,24,25]. Actinidia chinensis var. chinensis grows mainly in eastern and central China, whereas var. deliciosa is more inland in central and western China [26]. They coexist in many localities of the overlap region but are slightly separated vertically with var. chinensis being found at lower elevations. A cytogeographic analysis has revealed that ploidy types of the A. chinensis complex vary from diploid to hexaploid and that vars. chinensis and deliciosa comprise different cytotypes (var. chinensis: 2 × and 4x, var. deliciosa: 6 × with few 4x) [25]. The existing population genetic studies of the A. chinensis complex have been based on few sample sets and do not cover the entire geographic range of this species [22, 24, 25, 27]. Thus, the genetic diversity and population divergence history of the A. chinensis complex and correlations based on ploidy level are still unclear.
The development of ecological niche models (ENMs) is a more popular tool to predict the changes in species distribution in response to past or future climatic events and which has been used widely in recent phylogeographic studies [28]. In combination with the methods of molecular phylogeography, ENMs can help to reveal the processes that have shaped current distribution patterns of populations and species [29, 30].
In this study, flow cytometric measurement (FCM) [31] was used to identify cytotypes of the A. chinensis complex, and cpDNA markers were used to investigate the genetic diversity and population structure of the A. chinensis complex. We used mismatch distribution analysis (MDA) together with ENMs to investigate the evolutionary history and demographic structure. The main objectives of our study were to (a) reveal the pattern of cytotype distribution; (b) evaluate the level of genetic diversity and the degree of genetic differentiation among populations for each variety; and (c) test the origin of polyploidy and explore the demographic history of the A. chinensis complex. Our aim was to explore patterns and mechanisms of population diversity and historical demography of a typical polyploidy plant in the subtropical forest that generally correspond to the Metasequoia Flora (Sino-Japanese Kingdom), one of the two major floristic regions in the East Asian Floras [7].
Results
Cytotype diversity and distribution
Ploidy was estimated for 3,338 individuals with fresh leaves harvested from hydroponic shoots as 248 individuals failed to obtain fresh leaves. Five ploidy types (di-, tri-, tetra-, hexa- and octoploid, Additional file 1: Figure S1) were identified and the frequency of these ploidy types were 51.56%, 0.48%, 22.65%, 25.22% and 0.09%, respectively (Additional file 2: Table S1). All the diploids belonged to var. chinensis, whereas all the hexaploids belonged to var. deliciosa. The tetraploids were shared between both var. chinensis and var. deliciosa. Sixteen triploids having similar morphological characteristics with var. chinensis were observed in the diploid, tetraploid, and mixed populations (2x, 4x), whereas rare octoploids were found in two hexaploid var. deliciosa populations and one mixed population (2x, 4x, and 6x), respectively.
Diploid var. chinensis and hexaploid var. deliciosa exhibited clear spatial segregation from east China to west China, and the main distribution of the two varieties overlaps in mountainous regions of midwestern China (Fig. 1). The tetraploid distribution area was mainly in the Wuyi and Mufu mountains in east China and Xuefeng mountain in the midwestern China. The subsequent phylogenetic analysis also showed that two parts of the tetraploids belonged to two different lineages (Fig. 3a) which have differentiated at 2.22 Ma.

Cytotype distribution of the A. chinensis complex. Each location is represented by a filled circle or pie chart, with ploidy levels indicated by color. Map generated in ESRI ArcGIS 10.3
Genetic diversity and population structure
To investigate the genetic diversity and population structure, three cpDNA fragments (ndhF-rpl132, rps16-trnQ, and trnE-trnT) across 608 sampled individuals (Additional file 2: Table S2) from 50 populations were successfully sequenced. The total length of the concatenated sequence was 1,854 bp. A total of 35 polymorphic sites and 31 haplotypes were identified (Additional file 2: Table S3). Twenty-eight haplotypes were present in var. chinensis and three haplotypes in var. deliciosa. Four haplotypes were shared vars. chinensis and deliciosa (Fig. 2, Additional file 2: Table S4). The total genetic diversity of var. chinensis (HT = 0.920, HS = 0.470) was higher than that of var. deliciosa (HT = 0.552, HS = 0.380). The coefficient of genetic differentiation NST was significantly larger than GST for var. chinensis, and vice versa for var. deliciosa (Table 1).

Geographical distribution of cpDNA haplotypes. For the A. chinensis complex, geographical locations of the 50 populations and distributions of 31 chloroplast haplotypes of the A. chinensis complex examined in this study. The populations of var. chinensis and var. deliciosa are indicated different color. Map generated in ESRI ArcGIS 10.3
No spatial population structure was detected by the SAMOVA analysis of cpDNA haplotypes from 50 populations; the FCT values did not have significant peaks as the value of K increased from 2–20 (Additional file 1: Figure S2). Therefore, the populations were divided into two groups by taxonomy for the AMOVA analysis. The AMOVA analysis (Additional file 2: Table S5) revealed that 25.06% of the molecular variation was distributed between varieties (FCT = 0.251, P < 0.01). The inter-variant population fixation indices (FST) were 0.654 and 0.352 (P < 0.01) for var. chinensis and var. deliciosa, respectively. The isolation-by-distance (IBD) tests based on the pairwise FST/(1- FST) genetic distances against the geographic distances detected no significant correlation for the cpDNA (R2 = 0.1234, P = 0.988) among the 50 populations of the A. chinensis complex.
Phylogenetic relationships and divergence dating
Phylogenetic analysis of the concatenated cpDNA haplotype data sets recovered three haplotype lineages (lineages 1, 2, and 3; Fig. 3a), a result consistent with the phylogenetic network based on the 31 haplotypes (Fig. 3b). Phylogenetic relationships of the haplotype lineages broadly correspond to distinct geographical regions (Figs. 2 and 3). The haplotypes in lineage 1 were mainly found in mountainous regions of midwestern China where the two varieties overlap. In lineage 1, the haplotype H16 was shared by var. chinensis and var. deliciosa, but other haplotypes only belong to the diploid population of var. chinensis. The haplotypes in lineage 3, which belong to var. chinensis (2x) were found in eastern China, such as the Wuyi mountains, Dabie mountains, and Luoxiao mountains. Finally, almost all polyploid haplotypes were found in lineage 2. The dominant haplotype H10 and H11 occurred in 154 and 111 individuals, respectively. Although these two haplotypes were shared by var. chinensis and var. deliciosa, they still mainly belong to var. deliciosa. Nearly half of the haplotypes (16/31) were restricted to a single population. For example, in lineage 3, H3, H6, and H8 were found exclusively in Dabie mountain and Huang mountain, whereas H28, H30, and H31 appeared only in the Zhejiang-Fujian Hilly Region.

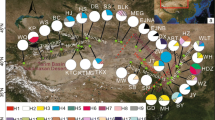
BEAST-derived chronograms and haplotypes network of the A. chinensis complex. a BEAST-derived chronograms of the A. chinensis complex based on cp DNA sequences. Blue bars indicate 95% HPD clade credibility intervals for nodes of particular interest with ages (in Ma). The cytotypes were also labeled for each haplotype. b The network of 31 chloroplast haplotypes. Each circle corresponds to a unique haplotype, with circle size reflecting its frequency. Small black circles indicate missing haplotypes
The time-calibrated evolutionary tree (Fig. 3a) showed that the divergence time between the A. chinensis complex and the outgroup (A. eriantha) dates back to an estimated 11 million years ago (Ma) during the late Miocene. For the 31 cpDNA haplotypes of the A. chinensis complex, the mean crown-group age of the A. chinensis complex was ca. 4.43 Ma (95% HPD 2.81–6.39 Ma, node A), thereby indicating the recent diversification of the cpDNA haplotypes of the A. chinensis complex during the Pliocene. Two polyploid haplotype lineages diverged at ca. 2.22 Ma (95% HPD 1.14–3.57 Ma, node C).
Demographic history and ancestral geographical area
All populations were divided into four groups E2x, E4x, M2x, and M4 × 6x which represent the eastern diploid populations, eastern tetraploid populations, midwestern diploid populations, and midwestern tetraploid and hexaploid populations, respectively. The mismatch distributions of group E4x and M4 × 6x were unimodal (Additional file 1: Figure S3), and the Fs test and Tajima’s D test of group E4x and M4 × 6x were also negative, although only the tests for group E4x were significant (P < 0.05, Table 2), closely fitted to the expected distribution under the sudden expansion model. For the other two groups (group M2x and E2x), there were no explicit signals of population expansion or equilibrium, as evidenced from the neutral tests and mismatch analysis (Table 2; Additional file 1: Figure S3), although the variance of the sum of square deviations and Harpending’s raggedness index tests (SSD and HRI, P > 0.05; Table 2) failed to reject the spatial expansion model. The tau values (τ) presented a rough estimate when population expansion began. The approximate expansion times (year) for groups E4x and M4 × 6x were measured as 0.084 (0.033–0.136) Ma BP, 0.137 (0.081–0.162) Ma BP, respectively.
The BBM (Bayesian Binary MCMC) analysis of ancestral distribution areas (Fig. 4) revealed two vicariance events and twelve dispersal events. As based on the topology of the intraspecific chronogram, a likely ancient vicariance event (node I, EA, and CE) of two sister lineages was recovered. A subsequent vicariance event (node II) between populations of tetraploid var. chinensis in eastern China (EA) and tetraploid/hexaploid var. deliciosa in central-western (CE and WE) China was also recovered. Recent colonization events from the eastern region (EA) to the central region (CE) were inferred, as based on the genetic admixture (H1, H2, and H7) in the adjacent mountains (e.g. Qinling and Dabie Mountains) or between the distant mountains (e.g. Qinling and Wuyi Mountains), with multiple expansions at different times (Fig. 4). For the diploid cytotypes, the mountains of eastern China (EA) were inferred as the most likely ancestral area. The ancient haplotypes (H12, Fig. 3b) identified in haplotype network analysis were also distributed in populations ZJTS and FJGT of eastern China. However, for the polyploid varieties, the mountains of central China (CE), e.g., Xuefeng mountain were deduced as the most likely ancestral area, which was also supported by the results of ENM analysis. No extinction events were discovered in any lineage by BBM analysis.

Ancestral areas, reconstructed with the Bayesian Binary MCMC method
Ecological niche modeling and niche comparison
The values of Area Under the Curve (AUC) between 0.7 and 0.9 indicate good discrimination. The AUC values of ENM for each variety and climate scenario were high (0.978/0.982), indicating that all models performed well in predicting the suitable habitat for each variety. The current distributional predictions were good representations of the actual distribution of both varieties. The main geographic distributional areas for both varieties were predicted mainly in the mountains of the subtropical region in China. The elevations predicted for var. deliciosa was often higher than that of var. chinensis. The geographic areas of the two varieties overlapped in the mountainous regions of midwestern China. The predicted geographic range for var. deliciosa during the LIG (LIG, Last Inter Glacial period) was more southward than that of the present (Fig. 5a, e). The variety var. chinensis also had a more restricted distribution during the LIG than that in the present, and the range was more broken than currently (Fig. 5b, f). The predicted species distribution during the LGM for vars. chinensis and deliciosa were both significantly larger than the predicted distributions under the LIG or current conditions (Fig. 5), suggesting that the distribution ranges of the two varieties had expanded during the LGM.

Potential distributions of var. chinensis and var. deliciosa predicted with MaxEnt. shown at 2.5 arc minute resolution and projected from six bioclimatic variables representing the current (a, b), LGM (c, d), and LIG (e, f) climatic conditions, respectively. Warmer colors denote areas with a higher probability of presence
Observed measures of niche-overlap and identity (D = 0.433 and I = 0.708, see Fig. 6a) were lower than the null distributions for var. chinensis vs. var. deliciosa, suggesting high niche differentiation between these two varieties. In addition, more detailed discrepancies can be found in the niches of three different cytotypes (Additional file 1: Figure S4). Thus, each variety or cytotype occupied a different niche with distinct environments, which was also supported by the results of PCA analysis as based on the data from elevation and 19 bioclimatic variables (Fig. 6b).

Niche comparison of var. chinensis and var. deliciosa. a Niche-identity tests by comparing the niches of var. chinensis and var. deliciosa. b Principal component analysis (PCA) with environmental variables from 240 occurrence data points
Discussion
High ploidy diversity in A. chinensis complex
To our knowledge, this study represents the most comprehensive investigation of the ploidy variation of A. chinensis complex by examining 128 populations and multiple individuals per population (28 on average) across their entire geographic distribution. Var. deliciosa predominantly comprises tetraploids and hexaploids, whereas var. chinensis consists of diploids and tetraploids, which is largely consistent with the results of Li et al. [25] where only the populations from Hunan and Guizhou provinces, China, were studied. For the A. chinensis complex, hexaploids were centered in the western portion of their ranges and diploids and tetraploids in the eastern portions. For var. deliciosa, the most frequent and widespread cytotype is hexaploid, followed by tetraploid. For var. chinensis, diploid is geographically most widespread followed by tetraploid. This is the first study to detect octoploids in var. deliciosa. In addition, natural triploid var. chinensis is described for the first time. The low frequency of triploids within diploid-tetraploid populations suggest the probability of interaction between diploids and tetraploids in the contact zone and highlights the potential for contemporary gene flow from diploid to polyploid populations.
In total, eight different mixed-ploidy populations were observed, including two majority cytotypes (2x + 4x, 4x + 6x,) and six minority cytotype (2x + 3x, 2x + 3x + 4x, 2x + 4x + 6x, 2x + 3x + 4x + 6x, 2x + 4x + 6x + 8x, 6x + 8x) (Fig. 1). The coexistence of cytotypes at the infraspecific or even intrapopulation levels has been documented for many plant species [32]. The maintenance of multiple cytotypes within the same sites may be explained by the divergence in pollinator spectra and/or visit frequency, different flowering time, and other factors associated with reproductive isolation [25]. For example, our field investigation shows diploids often flower slightly earlier than tetraploids and hexaploids, which suggests a reproductive barrier between cytotypes in the overlapping niches. The overall cytotype distribution pattern may arise from divergent adaptive abilities to the local environment. Generally, polyploid species have been suggested to be more adaptive to new ecological conditions than the ancestral diploid [14, 33, 34]. The overall geographical pattern of cytotype segregation in the species complex revealed in the present study implies kiwifruit species enhanced their adaptability to harsh climate conditions (e.g. colder climate) by reaching a higher ploidy level [25].
Multiple origins of polyploids
Despite autopolyploids being challenging to identify, several lines of evidence, such as isozymes and DNA sequences derived from the polygalacturonase gene, suggest that tetraploid var. chinensis is autoploid and hexaploid var. deliciosa is alloploid [35, 36]. The polyploids of the A. chinensis complex may experience multiple origins regardless of whether they are autopolyploids or allohexaploids. In this study, 31 haplotypes were detected in A. chinensis complex, of which 19 were found in diploids only, six were found in polyploids (tetraploid or hexaploid) only, and six were shared by different ploidy. Moreover, none of these polyploidy haplotypes were clustered together in the phylogenetic analysis (e.g. H16 and H17 in Fig. 3a). If we assume that diploids produced the tetraploids with identical cpDNA haplotypes (e.g. H16, H10, H7, and H26 in Fig. 3a), each haplotype has only evolved once (without parallel evolution). In that case, we can conclude that the tetraploids in the A. chinensis complex had experienced at least four independent origins in the evolutionary history of this species.
Previous studies have proposed that another hypothesis of sharing multiple haplotypes between diploids and tetraploids resulted from the hybridization among the different ploidy race [37, 38], where plastids and nuclear genes can be transferred to tetraploids by unreduced gametes or triploid bridges [39,40,41,42]. In the present study, a few triploid individuals were found in the mixed 2x-4 × populations. This finding implies that the hybridization may exist in the A. chinensis complex among the different ploidy races. Our data are still insufficient for distinguishing between independent vs. hybrid origins. Futher study is needed to test this hypothesis with a more in‐depth analysis and an increased marker density. However, multiple independent origins of both autopolyploids and allopolyploids through new polyploidization events, rather than hybridization have been widely reported in previous studies [17, 43,44,45,46,47]. For example, multiple independent origins were observed in the Allium przewalskianum based on the same methods [17]. In the present study, whatever the type of origin, our data reveal the multiple origins of polyploids in the A. chinensis complex.
Genetic divergence between var. chinensis and var. deliciosa
In this study, 31 haplotypes were obtained by sequencing the cpDNA of 50 populations of A. chinensis complex, and the genetic diversity (HT) at the species level was 0.884. Among these haplotypes, the chloroplast genetic diversity (HT) of var. chinensis was 0.920, which was significantly higher than that the relict plants in this region, such as Cercidiphyllum japonicum (0.757) [48], Davidia involucrata (0.882) [49], and Eupleea pleiosperma (0.893) [50]. However, the genetic diversity (HT) of var. deliciosa was 0.552, which is significantly lower than that of other species distributed in the same region. Although the evolutionary rate of mutation of the chloroplast genome is generally slow and the level of intraspecific genetic diversity low [51], there are still many genetic variants accumulated. Var. chinensis originated about 4.43 million years ago in China, whereas var. deliciosa originated about 2.22 million years (Fig. 3). Moreover, var. chinensis has a wide distribution with high environmental heterogeneity, which results in an increased probability of isolation, drift, and mutation [50]. Although var. deliciosa is also widely distributed, its suitable habitats are relatively less than those for var. chinensis (see below). In addition, previous studies have also found that widespread species tend to have a higher level of genetic diversity than local species [48]. The same distinction also exists in their genetic structure: var. chinensis has a clear genetic structure (GST / NST = 0.490/0.591, P < 0.01), while var. deliciosa does not have such genetic structure (GST / NST = 0.312 /0.252, P > 0.01). Although the above results indicate a significant genetic divergence between the two varieties. The finding of the lineage 1 haplotypes in the sympatric region suggested potential hybridization. In particular, the haplotype H16 was shared by the two varieties, indicating that genetic mixture occurred between these two varieties. Hybridization and introgression have been considered to be common in the evolution of Actinidia [52, 53].
Pliocene intraspecific divergence and population dynamic history
Global climate cooling is hypothesized to be a key trigger for climate change in East Asia, which enhances the monsoon climate in East Asia simultaneously [54, 55]. The rapid rise in elevation on the eastern edge of the Qinghai-Tibet Plateau in the mid-Pliocene (c. 3.6 Ma) led to dramatic changes in the landscape of southwestern China [55, 56]. The estimated divergence time between the two major lineages of the A. chinensis complex was 4.43 Ma (95% CI: 2.81–6.39; see node A in Fig. 3), which coincides with the accelerated rate of cooling of the early Pliocene of the Tertiary period [10]. Therefore, we suggest that climate and geological changes since the Pliocene may have played important roles in habitat fragmentation and the geographical barrier to gene flow, which in turn led to early genetic differentiation of the A. chinensis complex. Such isolation and differentiation were also used to explain similar patterns of east–west differentiation in other plants from subtropical China such as Primula obconicain [57], Cyclocarya paliurus [58], and Quercus acutissima [59].
The BBM result of cpDNA and the distribution of an ancient haplotype (H12) revealed that the most ancient area of var. chinensis and eastern refugium is located in Fujian hilly region (Figs. 2 and 4). The moist and warmer climate conditions in Fujian hilly region enhance the survival of warm-temperate deciduous forest species since the LGM [1]. Restricted to Fujian hilly region in the LIG period, var. chinensis then rapidly migrated eastward, westward, and northward from this refugial area, reaching the vast regions since the LGM. In addition, seed dispersal mechanism (animal dispersal), polyploidization, and interspecific/intraspecific hybridization may also have contributed to its rapid spread and expansion [52].
The ENMs analysis based on paleoclimate shows that the distribution of the A. chinensis complex has experienced Last Interglacial contraction and Last Glacial Maximum expansion, which is mutually supported by the results of population dynamic analysis. This dynamic pattern is the same as that of Pinus kwangtungensis [60] and Emmenopterys henryi [9], which belong to the region of subtropical China. The A. chinensis complex is a montane deciduous broad-leaved plant distributed throughout the subtropical zone of China with an elevation of 200–2500 m [26]. Previous studies have that changes in temperature can cause montane species to migrate up and down mountainous slopes [61, 62]. In the LIG period, the temperature was 2 °C higher than that of the present [63], in which case montane species will migrate to higher elevation, and the distribution of species will be more fragmented or even disappear because of theabsence of appropriate habitat. However, in the LGM period of lower temperature, these species will migrate to lower elevations in more suitable habitats with population expansion, and groups on different mountain may even reunite. Moreover, polyploidization would promote a broader niche, which has been verified in previous studies [64,65,66,67]. In this study, the ecological niche differentiation between var. chinensis and var. deliciosa has been confirmed. Our field survey data directly show that the elevational distribution range of var. deliciosa (including tetraploid of var. chinensis) is higher than that of var. chinensis (mainly diploid). Therefore, based on the MDA analysis of demographics, two groups (group E4x and M4 × 6x, Additional file 1: Figure S3) including polyploid individuals show a clear history of population expansion.
Polyploids often differ in physiological and life-history characteristics that may confer adaptive advantages compared with their diploid ancestors. Previous studies have also revealed a causal relationship between polyploidy and population expansion or ecological radiation in plants [68,69,70]. For example, Ramsey [71] compared the adaptability to the environment for Achillea borealis with different ploidy and found that the adaptability to the environment of hexaploid A. borealis was stronger than that of tetraploid and more than 70% of the fitness advantage was produced in the polyploidization process. In the present study, the polyploid populations (group E4x and group M4 × 6x) showed a historical population expansion (Additional file 1: Figure S3), but the population expansion was not observed in diploid populations (group E2x and M2x). The results of niche overlap and identity tests between three main ploidy (2x, 4x, and 6x) indicate that the environmental niche of hexaploid var. deliciosa was distinguishable from both diploid and tetraploid A. chinensis (Additional file 1: Figure S4). In particular, the hexaploid var. deliciosa colonized westward to the areas with cooler and slightly drier environments compared with their diploid parents (Fig. 2). Although the ecological niche of tetraploid overlaps with that of diploid, the tetraploid usually occupied higher elevations in the sympatric region. This ecological radiation pattern is revealed in other plants such as Aster amellus [72], Larrea tridentata [73], or Senecio carniolicus [74] where higher ploidy cytotypes could experience niche expansion whereas low levels were unable to occupy their full potential niche [32]. Thus, polyploidization appears to have played an important role in the historical demography of the A. chinensis complex through adaptation to environmental changes.
Conclusions
We sampled populations of the A. chinensis complex across its geographic distribution in subtropical China to study its cytotype distribution and phylogeography. The most frequent and widespread cytotypes were diploids followed by tetraploids and hexaploids, whereas triploids and octoploids were only observed in a few populations. The ENMs and BBM results suggested that Pliocene and Plio-Pleistocene climatic fluctuations and vicariance played key roles in shaping the current population structure and historical demography of the A. chinensis complex. This is consistent with the hypothesis that tracking suitable habitat by expansion or contraction would be the main pathway responding to climate changes for the species with a conservative ecological niche. Moreover, the polyploidization of the A. chinensis complex also appears to have played an important role in the historical demography of adaptation to environmental changes.
Methods
Plant sampling
From 2015–2018, 3586 samples were collected from 128 populations representing the species complex A. chinensis Planch. including the vars. chinensis and deliciosa. Another variety, A. chinensis var. setosa, narrowly distributed in the Taiwan mountains was not included in the present study because its taxonomic status is controversial. The taxonomy of the varieties is adopted from Li et al. [20, 75], which is based mainly on morphological characteristics such as the hair types covering flowering branchlets and fruits (var. chinensis: finely tomentose; var. deliciosa: brown strigose hairs), the presence of over-wintering buds (var. chinensis: exposed; var. deliciosa: buried in the bark) and the color of the flesh of the mature fruit (var. chinensis: mostly yellow or yellow-green; var. deliciosa: mostly green). Field collection followed the ethics and legality of the local government and was permitted by the government. The formal identification of the plant material was undertaken by the National Actinidia Germplasm Repository (NAGR) of China, and voucher specimens were deposited at NAGR. Each population consisted of either var. chinensis or var. deliciosa, and there were some populations containing both varieties as based on morphology in the sympatric regions. For each population, 6 to 72 individuals were sampled (Additional file 2: Table S1). To avoid collecting closely related individuals, sampled individuals were separated by ca. 50–100 m. For the subsequent cpDNA analysis, the fresh leaves were dried in silica gel in the field and then stored at -80 °C. One-year-old shoots were also collected in the field, providing fresh leaves for the ploidy level identification in the laboratory. These shoots were stored at 4 °C in a refrigerator for at least 30 days to overcome dormancy and then placed in fresh water at room temperature to stimulate bud breakage for the collection of fresh leaf tissue. The latitude, longitude, and elevation of each sampled individual were recorded with a global positioning system.
Ploidy analysis
The ploidy level of individuals were determined by estimating its relative DNA content with FCM. The detailed FCM method in this study was performed as in Li et al. [25]. Briefly, fresh leaves of the sample were chopped with leaves of the internal standard in 0.5 mL of ice-cold Otto I buffer (solution A of the High-Resolution Kit, Partec, Germany) and then were filtered through a 30 µm nylon sieve, followed by the addition of 1 mL Otto II buffer (4 µg/mL 4′,6-diamidino-2-phenylindole [DAPI]; solution B of the kit) held at 4 °C. Five minutes later, these samples were used to estimate the ploidy levels on a CyFlow Ploidy Analyser (Partec, Germany) automatically. Relative fluorescence intensity of a diploid var. chinensis cultivar, ‘Hongyang’ (2n = 58), whose chromosome number had been determined previously from chromosome counts and the estimated genome size of 750 Mb [76], was used as an internal standard for each measurement [25].
DNA isolation, amplification, and sequencing
For chloroplast DNA analysis, 50 out of 128 populations were selected to cover the entire geographic distribution of the A. chinensis complex. A modified CTAB method [77] was used to extract total genomic DNA from 608 individuals (Additional file 2: Table S2) which were selected in 50 population as evenly as possible by the predominant ploidy level (rare ploidy levels were excluded), then 1% agarose gels and NanoDrop 8000 (Thermo Fisher Scientific, Waltham, MA, USA) were employed to examine the quality and concentration of genomic DNA, respectively.
To identify chloroplast DNA (cpDNA) regions with sufficient variation, 12 individuals of var. chinensis and 12 var. deliciosa were randomly selected to undertake the preliminary screening of primer pairs for eight intron regions: ndhF-rpl132, rps16-trnQ, trnE-trnT, psbA-trnH, rpl16, trnL-trnF, trnL-trnT, and trnD-trnT (Additional file 2: Table S6). Finally, three consistently amplified and variable non-coding intergenic spacer (IGS) regions ndhF-rpl132, rps16-trnQ, and trnE-trnT [21], were selected for further analysis. A total of 608 individuals from all 50 populations of the A. chinensis complex were sequenced. Amplification was carried out in a volume of 20 μL reaction solution containing 10 μL 2 × Taq PCR MasterMix ( Bioteke), 0.5 μL each primer (0.2 μM), 1 μL template DNA (ca. 50–100 ng) and 8 μL ddH2O. The thermo-cycling conditions for PCR are given as follows: 94 °C, 4 min; 35 × (94 °C, 30 s; 56 °C, 60 s; 72 °C, 60 s); and 72 °C, 10 min. PCR products were sequenced in an ABI 377XL DNA sequencer (Applied Biosystems). All haplotype sequences identified in this study were submitted to GenBank (MT812986-MT813020). For subsequent analysis, A. eriantha and A. polygama were used as the outgroup and these chloroplast sequences were downloaded from GenBank (KY100979.1 and KX345297.1).
Analyses of genetic diversity and population structure
Three cpDNA fragments were aligned separately and trimmed in MEGA 6 [78] and combined into a single dataset with the online tool FaBox [79] for the phylogeographic analyses. During the analyses, indels (gaps) were treated as a single mutation event and coded as substitutions A or T (a third G or C were used when three kinds of mutations coexisted). The number of cpDNA haplotypes, haplotype diversities (Hd), and nucleotide diversities (π) for each variety were calculated with DnaSP 5.10 [80]. Haplotype distribution maps were constructed with ArcGIS 10.3. Total genetic diversity (HT) and within-population diversity (HS) were calculated with PermutCpSSR 2.0 [81].
The phylogeographic structure was inferred by comparing population differentiation for phylogenetically ordered (NST) and unordered (GST) haplotype with PermutCpSSR 2.0. A test of significance comprising 1000 permutations was used to determine if NST > GST. A significantly higher NST than GST usually indicates the presence of phylogeographical structure. The geographically and genetically distinguishable groups and the potential barriers between groups were analyzed with SAMOVA 2.0 [82]. Various SAMOVA were run, increasing the number of K groups until the percentage of explained variance among groups reached a limit (K = 2–20). In addition, a molecular variance (AMOVA) analysis was performed using Arlequin v3.5 [83] with significance tested under 1023 permutations to test genetic differentiation among populations and between varieties. Finally, estimates of pairwise FST/(1 − FST) were regressed against the pairwise natural logarithm of the geographic distance by using a Mantel test with 999 random permutations in GENALEX 6.5 to assess IBD patterns among the populations [84, 85]. Measures of pairwise FST distances were calculated with Arlequin, and the pairwise natural logarithms of geographic distance were calculated with GENALEX 6.5.
Haplotype relationships and divergence time estimation
To determine phylogenetic relationships among haplotypes, median-joining networks were constructed with NETWORK v5.0 [86]. Actinidia eriantha and A. polygama as the outgroup were included in haplotype network construction as well as molecular dating analysis. We used Bayesian phylogenetic inference on all haplotypes (sequence without recoding) with Markov chain Monte Carlo (MCMC) methods. A strict clock model in BEAST2 v2.5.0 was performed to estimate divergence time among lineages [87]. A GRT model was selected from the result of Akaike Information Criterion (AIC) with jModelTest v 2.1.10 test [88]. As a tree prior, the Yule model was specified. Two calibration points from previous studies of Actinidia [53] were used to constrain the nodes with a normal distribution prior. One point is the estimated split between A. polygama and the other two species (19 Myr, 95%CI: 14–24), and another is the estimated split between A. eriantha and A. chinensis (11 Myr, 95%CI: 4.7–17.3). The Monte Carlo Markov chain runs were performed every 1 × 109 generations, with sampling every 1 × 105 generations, following a burn-in of the initial 10% cycles. The convergence and effective sample size (ESS) > 200 for all parameters were assessed with Tracer version 1.7.1. After discarding the first 25% trees as burn-in, the rest of the trees were summarized in a maximum clade credibility tree with treeAnnotator version 2.6.0. Finally, the maximum clade credibility tree was visualized in FigTree version 1.4.3 (available at http://tree.bio.ed.ac.uk/software/figtree/).
Demographic history analyses and ancestral geographical area reconstruction
The mismatch distribution analysis was conducted with Arlequin 3.5 [83] to test whether each group has undergone recent demographic or spatial population expansion events. Multimodal mismatch distributions of pairwise differences between individuals are expected for populations at demographic equilibrium with a relatively stable size over time, whereas unimodal distributions are expected for the population that has experienced recent demographic expansions [89]. The goodness-of-fit of observed mismatch distributions to the theoretical distributions under a model of sudden expansion was tested with the raggedness index [90]. The significance of the raggedness index was obtained by examining the null distribution of 1000 coalescent simulations of these statistics. Small values of the raggedness index suggest sudden expansion, whereas high values of the raggedness index suggest stationary or genetic bottleneck. Second, for expanding lineage, the expansion parameter (τ) and its 95% confidence interval were converted into generation time (T) since expansion following the equation: T = τ/2μ [91], where μ is the neutral mutation rate of the entire cpDNA sequences per generation. The value for μ was calculated as μ = uk, where u is substitution rate in substitutions/site/generation (s/s/g) (here, 1.0 × 10–8; refer to Gaut [92]), k is the average sequence length of the cpDNA region under this study (here, 1,854 bp), the expansion time was calculated by assuming a generation time of 7 years for A. chinensis under natural conditions [53]. Finally, we also calculated Tajima’s D [93] and FS statistics of Fu [94] using Arlequin 3.5 [83], which is based on 1000 random permutations. The mismatch distribution of the observed number of nucleotide differences between pairs of DNA sequences was computed with DnaSP 5.10 [80].
Ancestral range reconstruction was conducted to estimate possible historical patterns of geographical distribution for A. chinensis complex using the Bayesian binary MCMC method (BBM) implemented in RASP v.4.0 [95]. For BBM analysis, 10,001 BEAST-generated trees and consensus tree without the outgroup were used as topology input, excluding the first 1000 as burning. In the ancestral area reconstruction, the geographical areas of the A. chinensis complex were defined four biogeographical regions: EA, East; SO, South; CE, Central; and WE, West (Fig. 4a) [52]. Ten BBM chains were run for 100,000 generations with a sampling frequency of 100. F81 was used as a state frequencies model in accordance to the AIC.
Ecological niche models
To study the niche differentiation and distributional changes for each variety, the Maxent Version 3.4.0 program [96, 97] was used to undertake to test of the ecological niche models and projected their potential distributions during three periods: the present, the Last Glacial Maximum (LGM, 21kya), and the Last Interglacial (LIG,120–140 kya). The geographic distribution data were obtained from our field collection and the Chinese Virtual Herbarium (CVH, available at http://www.cvh.ac.cn/). For all CVH records, the taxonomic identity was verified by specimen images and records of specimens taken from cultivated plants removed to ensure that geolocations were consistent with the known distribution. After removing duplicates, 150 sites for var. chinensis and 90 sites for var. deliciosa were identified. The current and past environmental datasets of 19 bioclimate variables with spatial resolutions of 2.5 arc minute were downloaded from the WorldClim database [98]. To avoid over-fitting [99], strongly correlated bioclimatic parameters, according to Pearson’s coefficient (|r|> 0.8) with the Perl program ENMTools v.1.3 [100], were excluded. Finally, six bioclimatic variables (Additional file 2: Table S7) were retained for ecological niche model construction with Maxent. The model quality was assessed by cross-validation comprising 100 replicates with 25% of the data for model testing, and the maximun number of background points was 10,000. The accuracy of each cross-validation test was evaluated with the area under the ROC curve (AUC) [101].
Ecology divergence analysis
To measure niche divergence, the environmental niches of the two varieties were compared using niche overlap and identity tests, in which the difference between the actual niches was contrasted with null models generated from randomly reshuffled occurrence points [102]. Niche identity was calculated with Schoener’s D similarity index [103] and Warren’s I [102] implemented in ENMTools version 1.3 [100, 102]. Both Schoener’s D and Warren’s I ranged from 0 (no niche overlap) to 1 (identical niches). One hundred pseudoreplicates of shuffling were conducted to generate null models for these statistics, and tested for significance; histograms were drawn using R 3.6.0 (http://www.rproject.org/). In addition, the same tests and analyses were also carried out for the three main different level ploidy (2x, 4x, and 6x) populations.
To better understand and visualize the similarities and differences between the environments in which var. chinensis and var. deliciosa occur, principal components analyses (PCA) were conducted with the elevation variable and 19 other bioclimatic variables from WorldClim [98, 104]. The same 240 occurrence data points used for ENMs were extracted values in ArcGIS 10.3. Before the PCAs analysis, the matrices of environmental values were standardized simultaneously. The distributions of species in environmental space were visualized in R 3.6.0.
Availability of data and materials
All sequences of A. chinensis complex generated during the current study are available in the NCBI GenBank database under accession numbers MT812986-MT813020.
Abbreviations
- ENMs:
-
Ecological niche models
- LIG:
-
Last Inter Glacial
- LGM:
-
Last Glacial Maximum
- FCM:
-
Flow cytometric measurement
- MDA:
-
Mismatch distribution analysis
- SSD:
-
Sum of square deviations
- HRI:
-
Harpending’s raggedness index
- IBD:
-
Isolation-by-distance
- RASP:
-
Reconstruct Ancestral State in Phylogenies
- BBM:
-
Bayesian Binary MCMC
- AUC:
-
Area Under the Curve
- LDD:
-
Long-distance dispersal
- NAGR:
-
The National Actinidia Germplasm Repository
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- CVH:
-
Chinese Virtual Herbarium
- MCMC:
-
Markov chain Monte Carlo
- AIC:
-
Akaike Information Criterion
- PCA:
-
Principal components analyses
References
Qiu YX, Fu CX, Comes HP. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol. 2011;59(1):225–44.
Liu J, Möller M, Provan J, Gao LM, Poudel RC, Li DZ. Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytol. 2013;199(4):1093–108.
Khan G, Zhang F, Gao Q, Fu P, Zhang Y, Chen S. Spiroides shrubs on Qinghai-Tibetan Plateau: multilocus phylogeography and palaeodistributional reconstruction of Spiraea alpina and S. Mongolica (Rosaceae). Mol Phylogenet Evol. 2018;123:137–48.
Fu PC, Sun SS, Khan G, Dong XX, Tan JZ, Favre A, Zhang FQ, Chen SL. Population subdivision and hybridization in a species complex of Gentiana in the Qinghai-Tibetan Plateau. Ann Bot. 2020;125(4):677–90.
Yu XQ, Gao LM, Soltis DE, Soltis PS, Yang JB, Fang L, Yang SX, Li DZ. Insights into the historical assembly of East Asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 2017;215(3):1235–48.
Fan D, Sun Z, Li B, Kou Y, Hodel RGJ, Jin Z, Zhang Z. Dispersal corridors for plant species in the Poyang Lake Basin of southeast China identified by integration of phylogeographic and geospatial data. Ecol Evol. 2017;7(14):5140–8.
Chen YS, Deng T, Zhou Z, Sun H. Is the East Asian flora ancient or not? Nat Sci Rev. 2018;5(6):920–32.
Zhang J, Li Z, Fritsch PW, Tian H, Yang A, Yao X. Phylogeography and genetic structure of a Tertiary relict tree species, Tapiscia sinensis (Tapisciaceae): implications for conservation. Ann Bot. 2015;116(5):727–37.
Zhang YH, Wang IJ, Comes HP, Peng H, Qiu YX. Contributions of historical and contemporary geographic and environmental factors to phylogeographic structure in a Tertiary relict species, Emmenopterys henryi (Rubiaceae). Sci Rep. 2016;6:24041.
Riddle BR. Comparative phylogeography clarifies the complexity and problems of continental distribution that drove A. R. Wallace to favor islands. Proc Natl Acad Sci U S A. 2016;113(29):7970–7.
Kou Y, Zhang L, Fan D, Cheng S, Li D, Hodel RGJ, Zhang Z. Evolutionary history of a relict conifer, Pseudotaxus chienii (Taxaceae), in south-east China during the late Neogene: old lineage, young populations. Ann Bot. 2020;125(1):105–17.
Gong W, Liu W, Gu L, Kaneko S, Koch MA, Zhang D. From glacial refugia to wide distribution range: demographic expansion of Loropetalum chinense (Hamamelidaceae) in Chinese subtropical evergreen broadleaved forest. Org Divers Evol. 2016;16(1):23–38.
Gao YD, Gao XF, Harris A. Species boundaries and parapatric speciation in the complex of alpine shrubs, Rosa sericea (Rosaceae), based on population genetics and ecological tolerances. Front Plant Sci. 2019;10:321.
Soltis PS, Marchant DB, Van de Peer Y, Soltis DE. Polyploidy and genome evolution in plants. Curr Opin Genet Dev. 2015;35:119–25.
Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, Rieseberg LH. The frequency of polyploid speciation in vascular plants. Proc Natl Acad Sci U S A. 2009;106(33):13875–9.
Husband BC, Baldwin SJ, Suda J. The incidence of polyploidy in natural plant populations: major patterns and evolutionary processes. In: Plant genome diversity volume 2. Vienna: Springer; 2013. p. 255–76.
Wu LL, Cui XK, Milne RI, Sun YS, Liu JQ. Multiple autopolyploidizations and range expansion of Allium przewalskianum Regel. (Alliaceae) in the Qinghai-Tibetan Plateau. Mol Ecol. 2010;19(8):1691–704.
He J, Wang S, Li J, Fan Z, Liu X, Wang Y. Genetic differentiation and spatiotemporal history of diploidy and tetraploidy of Clintonia udensis. Ecol Evol. 2017;7(23):10243–51.
Zhang X, Su H, Yang J, Feng L, Li Z, Zhao G. Population genetic structure, migration, and polyploidy origin of a medicinal species Gynostemma pentaphyllum (Cucurbitaceae). Ecol Evol. 2019;9(19):11145–70.
Li JQ, Soejarto D, Li XW. A revision of the Genus Actinidia from China. In: VI International symposium on kiwifruit, volume 753. 2007. 41–4.
Tang P, Xu Q, Shen RL, Yao XH. Phylogenetic relationship in Actinidia (Actinidiaceae) based on four noncoding chloroplast DNA sequences. Plant Syst Evol. 2019;305:787–96.
Liu Y, Li D, Yan L, Huang H. The microgeographical patterns of morphological and molecular variation of a mixed ploidy population in the species complex Actinidia chinensis. PLoS One. 2015;10(2):e0117596.
Tian Z. Genetic diversity, gene introgression and homoplasy in sympatric populations of the genus Actinidia as revealed by chloroplast microsatellite markers. Biodivers Sci. 2007;15(1):1.
Liu Y, Liu Y, Huang H. Genetic variation and natural hybridization among sympatric Actinidia species and the implications for introgression breeding of kiwifruit. Tree Genet Genomes. 2010;6(5):801–13.
Li D, Liu Y, Zhong C, Huang H. Morphological and cytotype variation of wild kiwifruit (Actinidia chinensis complex) along an altitudinal and longitudinal gradient in central-west China. Bot J Linnean Soc. 2010;164(1):72–83.
Ferguson AR, Huang H. Genetic resources of kiwifruit: domestication and breeding. Hortic Rev. 2007;33:1–121.
Wang YC, Liao L, Li ZZ. Genetic differentiation of Actinidia chinensis and analysis of gene flow barriers in the Qinling Mountains, the species’ northern distribution boundary. Genet Resour Crop Evol. 2017;65(3):881–95.
Wang YH, Jiang WM, Comes HP, Hu FS, Qiu YX, Fu CX. Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): insights into Plio-Pleistocene range dynamics of evergreen forest in subtropical China. New Phytol. 2015;206(2):852–67.
Scoble J, Lowe AJ. A case for incorporating phylogeography and landscape genetics into species distribution modelling approaches to improve climate adaptation and conservation planning. Divers Distrib. 2010;16(3):343–53.
Mellick R, Lowe A, Allen C, Hill RS, Rossetto M. Palaeodistribution modelling and genetic evidence highlight differential post-glacial range shifts of a rain forest conifer distributed across a latitudinal gradient. J Biogeogr. 2012;39(12):2292–302.
Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science. 1983;220(4601):1049–51.
Kolář F, Čertner M, Suda J, Schönswetter P, Husband BC. Mixed-ploidy ploidy speciesspecies: progress progress and opportunities opportunities in polyploid polyploid researchresearch. Trends Plant Sci. 2017;22(12):1041–55.
Levin DA. Polyploidy and novelty in flowering plants. Am Nat. 1983;122(1):1–25.
Marchant DB, Soltis DE, Soltis PS. Patterns of abiotic niche shifts in allopolyploids relative to their progenitors. New Phytol. 2016;212(3):708–18.
Atkinson RG, Cipriani G, Whittaker DJ, Gardner RC. The allopolyploid origin of kiwifruit, Actinidia deliciosa (Actinidiaceae). Plant Syst Evol. 1997;205(1–2):111–24.
Huang H, Dane F, Wang Z, Jiang Z, Huang R, Wang S. Isozyme inheritance and variation in Actindia. Heredity. 1997;78(3):328–36.
Kolář F, Štech M, Trávníček P, Rauchová J, Urfus T, Vít P, Kubešová M, Suda J. Towards resolving the Knautia arvensis agg. (Dipsacaceae) puzzle: primary and secondary contact zones and ploidy segregation at landscape and microgeographic scales. Ann Bot. 2009;103(6):963–74.
Sabara HA, Kron P, Husband BC. Cytotype coexistence leads to triploid hybrid production in a diploid-tetraploid contact zone of Chamerion angustifolium (Onagraceae). Am J Bot. 2013;100(5):962–70.
Mráz P, Španiel S, Keller A, Bowmann G, Farkas A, Šingliarová B, Rohr RP, Broennimann O, Müller-Schärer H. Anthropogenic disturbance as a driver of microspatial and microhabitat segregation of cytotypes of Centaurea stoebe and cytotype interactions in secondary contact zones. Ann Bot. 2012;110(3):615–27.
Zozomová-Lihová J, Malánová-Krásná I, Vít P, Urfus T, Senko D, Svitok M, Kempa M, Marhold K. Cytotype distribution patterns, ecological differentiation, and genetic structure in a diploid-tetraploid contact zone of Cardamine amara. Am J Bot. 2015;102(8):1380–95.
Hülber K, Sonnleitner M, Suda J, Krejčíková J, Schönswetter P, Schneeweiss GM, Winkler M. Ecological differentiation, lack of hybrids involving diploids, and asymmetric gene flow between polyploids in narrow contact zones of Senecio carniolicus (syn. Jacobaea carniolica, Asteraceae). Ecol Evol. 2015;5(6):1224–34.
Winkler M, Escobar García P, Gattringer A, Sonnleitner M, Hülber K, Schönswetter P, Schneeweiss GM. A novel method to infer the origin of polyploids from Amplified Fragment Length Polymorphism data reveals that the alpine polyploid complex of Senecio carniolicus (Asteraceae) evolved mainly via autopolyploidy. Mol Ecol Resour. 2017;17(5):877–92.
Beck JB, Allison JR, Pryer KM, Windham MD. Identifying multiple origins of polyploid taxa: a multilocus study of the hybrid cloak fern (Astrolepis integerrima; Pteridaceae). Amer J Bot. 2012;99(11):1857–65.
Mavrodiev EV, Chester M, Suarez-Santiago VN, Visger CJ, Rodriguez R, Susanna A, Baldini RM, Soltis PS, Soltis DE. Multiple origins and chromosomal novelty in the allotetraploid Tragopogon castellanus (Asteraceae). New Phytol. 2015;206(3):1172–83.
Servick S, Visger CJ, Gitzendanner MA, Soltis PS, Soltis DE. Population genetic variation, geographic structure, and multiple origins of autopolyploidy in Galax urceolata. Amer J Bot. 2015;102(6):973–82.
Vallejo-Marín M, Buggs RJA, Cooley AM, Puzey JR. Speciation by genome duplication: repeated origins and genomic composition of the recently formed allopolyploid species Mimulus peregrinus. Evolution. 2015;69(6):1487–500.
Vallejo-Marín M, Quenu M, Ritchie S, Meeus S. Partial interfertility between independently originated populations of the neo-allopolyploid Mimulus peregrinus. Plant Syst Evol. 2017;303(8):1081–92.
Qi XS, Chen C, Comes HP, Sakaguchi S, Liu YH, Tanaka N, Sakio H, Qiu YX. Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). New Phytol. 2012;196(2):617–30.
Chen JM, Zhao SY, Liao YY, Gichira AW, Gituru RW, Wang QF. Chloroplast DNA phylogeographic analysis reveals significant spatial genetic structure of the relictual tree Davidia involucrata (Davidiaceae). Conserv Genet. 2015;16(3):583–93.
Cao YN, Comes HP, Sakaguchi S, Chen LY, Qiu YX. Evolution of East Asia’s Arcto-Tertiary relict Euptelea (Eupteleaceae) shaped by Late Neogene vicariance and Quaternary climate change. BMC Evol Biol. 2016;16(1):1–7.
Hewitt GM. Speciation, hybrid zones and phylogeography or seeing genes in space and time. Mol Ecol. 2001;10(3):537–49.
Huang H, Liu Y. Natural hybridization, introgression breeding, and cultivar improvement in the genus Actinidia. Tree Genet Genomes. 2014;10(5):1113–22.
Liu Y, Li D, Zhang Q, Song C, Zhong C, Zhang X, Wang Y, Yao X, Wang Z, Zeng S, et al. Rapid radiations of both kiwifruit hybrid lineages and their parents shed light on a two-layer mode of species diversification. New Phytol. 2017;215(2):877–90.
Heermance RV, Chen J, Burbank DW, Wang C. Chronology and tectonic controls of Late Tertiary deposition in the southwestern Tian Shan foreland, NW China. Basin Res. 2007;19(4):599–632.
Ge J, Dai Y, Zhang Z, Zhao D, Li Q, Zhang Y, Yi L, Wu H, Oldfield F, Guo Z. Major changes in East Asian climate in the mid-Pliocene: Triggered by the uplift of the Tibetan Plateau or global cooling? J Asian Earth Sci. 2013;69:48–59.
Shi Y, Li J, Li B. Uplift of the Qinghai-Xizang (Tibetan) plateau and east Asia environmental change during late Cenozoic. Acta Geogr Sin Chin Ed. 1999;54:20–8.
Yan HF, Zhang CY, Wang FY, Hu CM, Ge XJ, Hao G. Population expanding with the phalanx model and lineages split by environmental heterogeneity: a case study of Primula obconica in subtropical China. PLoS One. 2012;7(9):e41315.
Kou Y, Cheng S, Tian S, Li B, Fan D, Chen Y, Soltis DE, Soltis PS, Zhang Z. The antiquity of Cyclocarya paliurus (Juglandaceae) provides new insights into the evolution of relict plants in subtropical China since the late Early Miocene. J Biogeogr. 2016;43(2):351–60.
Zhang XW, Li Y, Zhang Q, Fang YM. Ancient east-west divergence, recent admixture, and multiple marginal refugia shape genetic structure of a widespread oak species (Quercus acutissima) in China. Tree Genet Genomes. 2018;14(6):1–5.
Tian S, López-Pujol J, Wang HW, Ge S, Zhang ZY. Molecular evidence for glacial expansion and interglacial retreat during Quaternary climatic changes in a montane temperate pine (Pinus kwangtungensis Chun ex Tsiang) in southern China. Plant Syst Evol. 2010;284(3–4):219–29.
Davis MB, Shaw RG. Range shifts and adaptive responses to Quaternary climate change. Science. 2001;292(5517):673–9.
Colwell RK, Brehm G, Cardelús CL, Gilman AC, Longino JT. Global warming, elevational range shifts, and lowland biotic attrition in the wet tropics. Science. 2008;322(5899):258–61.
Otto-Bliesner BL, Marshall SJ, Overpeck JT, Miller GH, Hu A. Simulating Arctic climate warmth and icefield retreat in the last interglaciation. Science. 2006;311(5768):1751–3.
Visger CJ, Germain-Aubrey CC, Patel M, Sessa EB, Soltis PS, Soltis DE. Niche divergence between diploid and autotetraploid Tolmiea. Am J Bot. 2016;103(8):1396–406.
Muñoz-Pajares AJ, Perfectti F, Loureiro J, Abdelaziz M, Biella P, Castro M, Castro S, Gómez JM. Niche differences may explain the geographic distribution of cytotypes in Erysimum mediohispanicum. Plant Biol. 2018;20:139–47.
López-Jurado J, Mateos-Naranjo E, Balao F. Niche divergence and limits to expansion in the high polyploid Dianthus broteri complex. New Phytol. 2019;222(2):1076–87.
Molina-Henao YF, Hopkins R. Autopolyploid lineage shows climatic niche expansion but not divergence in Arabidopsis arenosa. Am J Bot. 2019;106(1):61–70.
Leitch A, Leitch I. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320(5875):481–3.
Van de Peer Y, Mizrachi E, Marchal K. The evolutionary significance of polyploidy. Nat Rev Genet. 2017;18(7):411.
Han TS, Zheng QJ, Onstein RE, Rojas-Andrés BM, Hauenschild F, Muellner-Riehl AN, Xing YW. Polyploidy promotes species diversification of Allium through ecological shifts. New Phytol. 2020;225(1):571–83.
Ramsey J. Polyploidy and ecological adaptation in wild yarrow. Proc Natl Acad Sci U S A. 2011;108(17):7096–101.
Münzbergová Z, Šurinová M, Castro S. Absence of gene flow between diploids and hexaploids of Aster amellus at multiple spatial scales. Heredity. 2013;110(2):123–30.
Laport RG, Minckley RL, Ramsey J. Ecological distributions, phenological isolation, and genetic structure in sympatric and parapatric populations of the Larrea tridentata polyploid complex. Am J Bot. 2016;103(7):1358–74.
Sonnleitner M, Hülber K, Flatscher R, García PE, Winkler M, Suda J, Schönswetter P, Schneeweiss GM. Ecological differentiation of diploid and polyploid cytotypes of Senecio carniolicus sensu lato (Asteraceae) is stronger in areas of sympatry. Ann Bot. 2016;117(2):269–76.
Li XW, Li JQ, Djendoel SD. Advances in the study of the systematics of Actinidia Lindley. Front Biol China. 2009;4(1):55-61.
Huang S, Ding J, Deng D, Tang W, Sun H, Liu D, Zhang L, Niu X, Zhang X, Meng M, et al. Draft genome of the kiwifruit Actinidia chinensis. Nat Commun. 2013;4:2640.
Doyle J, Doyle J. Isolation of DNA from small amounts of plant tissues. BRL focus. 1990;12(1):V15.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Villesen P. FaBox: an online toolbox for fasta sequences. Mol Ecol Notes. 2007;7(6):965–8.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Pons O, Petit R. Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144(3):1237–45.
Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11(12):2571–81.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7.
Rousset F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997;145(4):1219–28.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics. 2012;28(19):2537–9.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48.
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, Heled J, Jones G, Kühnert D, De Maio N. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2019;15(4):e1006650.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
Harpending HC. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol. 1994;66(4):591–600.
Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9(3):552–69.
Rogers AR. Genetic evidence for a Pleistocene population explosion. Evolution. 1995;49(4):608–15.
Gaut BS. Molecular clocks and nucleotide substitution rates in higher plants. In: Evolutionary biology. Boston: Springer; 1998. p. 93–120.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95.
Fu Y-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25.
Yu Y, Harris AJ, Blair C, He X. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol Phylogenet Evol. 2015;87:46–9.
Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol Model. 2006;190(3–4):231–59.
Phillips SJ, Dudík M. Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography. 2008;31(2):161–75.
Fick SE, Hijmans RJ. WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int J Climatol. 2017;37(12):4302–15.
Peterson AT, Nakazawa Y. Environmental data sets matter in ecological niche modelling: an example with Solenopsis invicta and Solenopsis richteri. Glob Ecol Biogeogr. 2008;17(1):135–44.
Warren DL, Glor RE, Turelli M. ENMTools: a toolbox for comparative studies of environmental niche models. Ecography. 2010;33(3):607–11.
Fawcett T. An introduction to ROC analysis. Pattern Recogn Lett. 2006;27(8):861–74.
Warren DL, Glor RE, Turelli M. Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution. 2008;62(11):2868–83.
Schoener TW. The Anolis lizards of Bimini: resource partitioning in a complex fauna. Ecology. 1968;49(4):704–26.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 2005;25(15):1965–78.
Acknowledgements
We appreciate the linguistic assistance provided by Peter W. Fritsch during the preparation of this manuscript. We are grateful to the editors and two reviewers for their valuable comments to improve this manuscript.
Funding
The work was supported by grants from the National Key Research and Development Program of China (2019YFD1000200) and the National Natural Science Foundation of China (31370251 and 31770374). These funding bodies did not have a role in the design of the study, the collection, analysis, and interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
XHY and ZZL designed the study; ZZL, CLY, and ZW collected the field samples; CLY and ZW carried out the experiments and analyzed the data; ZW, CHZ, DWL, ZZL, and XHY drafted the manuscript and carried out the critical revision of the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Schematic diagram of flow cytometric histogram of DAPI-stained nuclei of six type of ploidy levels (2x, 3x, 4x, 6x, and 8x) of the A. chinensis complex analyzed simultaneously with the internal standard Actinidia chinensis cv. ‘Hongyang’. Figure S2. Results of spatial analysis of molecular variance analysis (SAMOVA, K = 2-20) on the A. chinensis complex populations in subtropical China. K refers to the number of predefined groups used in the analyses. Figure S3. Observed and expected mismatch distributions for different groups of the A. chinensis complex. (a) group E2x; (b) group E4x; (c) group M2x; (d) group M4x6x. The solid line indicates observed distribution; The dashed line represents theoretically expected distribution under a population expansion model. Figure S4. The results of niche-identity tests by comparing the niches of three main ploidy level populations. (a) 2x vs. 4x (b) 2x vs. 6x (c) 4x vs. 6x.
Additional file 2: Table S1.
Characteristics of sampled populations and ploidy levels of the A. chinensis complex. Table S2. List of ploidy level and haplotype for 608 individuals in cpDNA analysis. Table S3. Description of haplotypes in the A. chinensis complex from three combined chloroplast DNA fragments. Table S4. Genetic characteristics of cpDNA for 31 var. chinensis populations and 25 var. deliciosa populations. Table S5. Results of AMOVA testing of the A. chinensis complex. Table S6. List of primers for the eight cpDNA loci. Table S7 The six environmental variables used for ecological niche modeling.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Z., Zhong, C., Li, D. et al. Cytotype distribution and chloroplast phylogeography of the Actinidia chinensis complex. BMC Plant Biol 21, 325 (2021). https://doi.org/10.1186/s12870-021-03099-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03099-y