
Abstract
Background
Pistil development is a complicated process in plants, and female sterile mutants are ideal material for screening and cloning pistil development-related genes. Using the female sterile mutant (fsm1), BraA04g009730.3C was previously predicted as a candidate mutant gene encoding the STERILE APETALA (SAP) transcriptional regulator. In the current study, a parallel female sterile mutant (fsm2) was derived from EMS mutagenesis of a Chinese cabbage DH line ‘FT’ seeds.
Results
Both fsm2 and fsm1 mutant phenotypes exhibited pistil abortion and smaller floral organs. Genetic analysis indicated that the phenotype of mutant fsm2 was also controlled by a single recessive nuclear gene. Allelism testing showed that the mutated fsm1 and fsm2 genes were allelic. A single-nucleotide mutation (G-to-A) in the first exon of BraA04g009730.3C caused a missense mutation from GAA (glutamic acid) to GGA (glycine) in mutant fsm2 plants. Both allelic mutations of BraA04g009730.3C in fsm1 and fsm2 conferred the similar pistil abortion phenotype, which verified the SAP function in pistil development. To probe the mechanism of SAP-induced pistil abortion, we compared the mutant fsm1 and wild-type ‘FT’ pistil transcriptomes. Among the 3855 differentially expressed genes obtained, 29 were related to ovule development and 16 were related to organ size.
Conclusion
Our study clarified the function of BraA04g009730.3C and revealed that it was responsible for ovule development and organ size. These results lay a foundation to elucidate the molecular mechanism of pistil development in Chinese cabbage.
Similar content being viewed by others


Background
Female sterility refers to the phenomenon in which pistil fertility is reduced or completely aborted due to the abnormal development of female organs in plants. The pistil structure is complex, and an abnormal female organ development may lead to female sterility in the sporophyte and gametophyte stages. According to the specific period of pistil abortion, female plant sterility can be divided into three types: (1) abnormal pistil, (2) abnormal ovule, and (3) abnormal egg cell [1]. Sterile female plants are highly useful in studying the developmental mechanism and genetic breeding of female organs in higher plants [2,3,4].
Among the floral organs, the pistil has the most complex structure, and its reproductive growth and development processes are regulated by a large number of transcription factors and functional genes [5,6,7]. Several genes regulating pistil development and physiological and biochemical changes during pistil abortion have been identified through mapping and cloning of female sterile mutant genes in Arabidopsis thaliana, rice, cotton, maize, and rapeseed, leading to a gradual understanding of the morphological model and genetic regulation of pistil development [8,9,10,11,12]. High-throughput transcriptome sequencing of female sterile mutants enables to explore the metabolic pathway changes that occur during pistil abortion at the transcriptional level; screening of key genes regulating pistil development is helpful in studying pistil and ovule development, as well as the genetic regulation mechanism [13,14,15,16,17].
Pistil development is a complex process controlled by multiple genes. Coen and Meyerowitz [18] proposed the ABC model of floral organ development, then Colombo et al. [19] and Theissen [20] extended the ABC model to the ABCD and ABCDE models. Among these, class D genes regulate ovule development, and most genes in the ABCDE model belong to the MADS-box gene family [21]. There are several collateral homologous genes in the MADS-box gene family that jointly regulate flower development [19, 22,23,24]. In A. thaliana, the ovule identity genes identified include AGAMOUS (AG), BELL1 (BEL1), SEEDSTICK (STK), SHATTERPROOF1/2 (SHP1/SHP2), CUP-SHAPED COTYLEDON3 (CUC3), and PRETTY FEW SEEDS2 (PFS2); these genes are closely related to ovule morphogenesis and encode proteins that contain the conserved MADS-box functional domains [25,26,27,28,29,30]. Genes associated with ovule primordia formation include AINTEGUMENTA (ANT), WUSCHEL (WUS), NOZZLE (NZZ), INNER NO OUTER (INO), BEL1, and PHABULOSA (PHB) [4, 31,32,33,34]. In addition, several genes related to integument development have been isolated and divided into two categories: one category controls the early development of integument and includes HUELLENLOS (HLL), ANT, NZZ, and INO [35,36,37,38]; the other controls the later development of integument and includes SHORT INTEGUMENT1 (SIN1), SUPERMAN (SUP), STRUBBELIG (SUB), ARABIDOPSIS CRINKLY4 (ACR4), ABERRANT TESTA SHAPE (ATS), KANADI1/2 (KAN1/2), UNICORN (UCN), and TSO1 [39,40,41,42,43].
In our previous study, we obtained a female sterile mutant fsm (namely fsm1 here) by isolated microspore culture combined with ethyl methanesulfonate (EMS) mutagenesis of the Chinese cabbage (Brassica rapa L. ssp. pekinensis) double haploid (DH) line ‘FT’ [44]. Pistil abortion in the fsm1 mutant was caused by abnormal ovules and BraA04g009730.3C (version 3.0) was presumed to be the candidate gene in the fsm1 mutant based on map-based cloning and whole-genome re-sequencing [12]. BraA04g009730.3C encodes STERILE APETALA (SAP), a transcriptional regulator that plays an important role in floral organ development. In A. thaliana, SAP not only regulates flower and ovule development [45], but also controls organ size by affecting cell proliferation [46].
In this study, the DH line ‘FT’ was used as mutagenic material; germinating ‘FT’ seeds were treated with EMS solution to develop another parallel female sterile mutant (fsm2) whose phenotype was consistent with that of mutant fsm1. Allelism testing indicated that the mutant genes fsm1 and fsm2 were allelic. These two parallel mutants were used to verify the function of BraA04g009730.3C. To investigate the potential mechanism of ovule development, RNA-sequencing was used to compare the pistil transcriptome of mutant fsm1 and wild-type ‘FT’ plants. Genes related to ovule development and organ size were identified and screened, laying a foundation to further reveal the pistil abortion mechanism in Chinese cabbage.
Results
Comparison of morphological characteristics between the fsm1 and fsm2 mutants
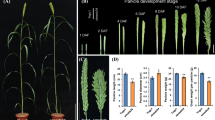
The fsm2 mutant phenotype was highly consistent with that of the fsm1 mutant. Compared with the wild-type ‘FT’ plants, the mutant fsm2 plants exhibited pistil abortion. As shown in Fig. 1b, the ovary was thin and short. In addition, the four-whorled floral organs of the mutant fsm2 plants were significantly smaller those of the wild-type plants (Fig. 1).

Morphological characterization of the wild-type ‘FT’ plants and female-sterile fsm1 and fsm2 mutants. a. Flowers of the wild-type ‘FT’ plants and fsm1 and fsm2 mutants. b. Pistils of the wild-type ‘FT’ plants and fsm1 and fsm2 mutants. c. Sepals of the wild-type ‘FT’ plants and fsm1 and fsm2 mutants. d. Petals of the wild-type ‘FT’ plants and fsm1 and fsm2 mutants. e. Stamens of the wild-type ‘FT’ plants and fsm1 and fsm2 mutants. Scale bar: 1 mm
As shown in Table 1, when a mutant fsm2 plant was used as the female parent, irrespective of whether its pollen or foreign (wild-type ‘FT’) pollen was employed as the male parent, no seed was harvested from the offspring. However, when a mutant fsm2 plant was used as the male parent, seeds could be collected from the offspring. Therefore, stamen fertility was normal but the pistil was abortive. Furthermore, female sterility of the fsm2 mutant was stable.
Genetic analysis of mutant fsm2
When a wild-type ‘FT’ plant was used as the female parent and the fsm2 mutant was used as the male parent for hybridization, the phenotype of all plants was consistent with that of the wild-type ‘FT’ in the F1 generation. In the F2 generation, the segregation ratio was 3:1, whereas the segregation ratio was approximately 1:1 in the BC1 generation (F1 × fsm2). These results suggest that the fsm2 mutant phenotype was controlled by a single recessive nuclear gene (Table 2).
Allelism testing
The reciprocal cross of both F1 (‘FT’ × fsm1) and F1 (‘FT’ × fsm2) exhibited character segregation. In the offspring, the segregation ratios of the wild-type-to-mutant plants were 137:32 (χ2 = 1.24 < χ20.05, 1 = 3.84) and 147:45 (χ2 = 0.09 < χ20.05, 1 = 3.84), respectively, consistent with the 3:1 segregation ratio. These results indicated that the mutant genes fsm1 and fsm2 were allelic and caused by mutations in the same gene.
BraA04g009730.3C clone in mutant fsm2
A BraA04g009730.3C clone in mutant fsm2 plants showed a single nucleotide mutation (G-to-A, A04: 7543809) in the first exon, causing an amino acid change from glutamic acid (G) to glycine (E), which differed from the mutation site of the fsm1 mutant (Fig. 2a, b). The three-dimensional structures of the proteins showed that the amino acid conformation between the wild-type ‘FT’ and mutant fsm2 were different at the mutant site (Fig. 2c). These results indicated that the two allelic mutations in BraA04g009730.3C conferred the similar pistil abortion phenotype and verified SAP function in pistil development.

Comparison of BraA04g009730.3C gene structure and sequence alignment between wild-type ‘FT’ and mutant fsm2 plants. a. Gene structure of BraA04g009730.3C and the single nucleotide polymorphism (SNP) site. b. Alignment of BraA04g009730.3C encoding sequences and amino acid sequences. The green underline and red frame show the sites where the SNP and the non-synonymous mutation occurred, respectively. c. Partial three-dimensional structure of BraA04g009730.3C in wild-type (left) and mutant fsm2 (right). Amino acid sequences include the SNPs. The red arrow represents different amino acid conformations at the mutation site
Illumina paired-end sequencing and global data analysis
The fsm1 and fsm2 plants are allelic mutants with different mutations in BraA04g009730.3C that encode a SAP transcriptional regulator. To examine the possible pathways through which SAP may regulate pistil development in the fsm plants, a comparative transcriptome analysis of ‘FT’ and fsm1 pistils was conducted. A total of 128,643,734 and 130,558,248 clean reads were obtained from the three biological replicates of the ‘FT’ and fsm1 plants, respectively. Of the total clean reads, the percentage of reads mapped to the reference genome ranged from 90.66 to 91.76% in the six libraries and 97.48 to 97.88% of the mapped reads were matched to unique genomic locations (Table 3) that could be used for the differential analysis of gene expression between the ‘FT’ and fsm1 plants.
In addition, 24,494 (FT1), 24,668 (FT2), 24,863 (FT3), 24,918 (M1), 24,860 (M2), and 25,418 (M3) genes expressed were generated from these six libraries, respectively (Additional file 1: Table S1).
Differentially expressed genes (DEGs) between the ‘FT’ and fsm1 plants
A comparison of ‘FT’ vs. fsm1 plants revealed 3855 DEGs, of which 2356 were upregulated and 1499 were downregulated (Additional file 2: Table S2). The number of upregulated DEGs was significantly higher than that of the downregulated DEGs in the fsm1 mutant. Of the DEGs, 106 were specifically expressed, with 21 and 85 specifically expressed in the ‘FT’ and fsm1 plants, respectively (Additional file 3: Table S3).
Functional enrichment analysis of DEGs
The GO functional enrichment analysis was performed to identify the biological functions of DEGs. We identified 1304 enriched GO terms. Of these, 768, 126, and 410 GO terms were in the “biological process,” “cellular component,” and “molecular function” categories, respectively (Additional file 4: Fig. S1). Two GO terms related to flower development were identified (“flower development” (GO: 0009908; four DEGs) and “regulation of flower development” (GO: 0009909; one DEG)). In addition, numerous GO terms associated with plant hormone metabolism were also identified, including “response to hormone” (GO: 0009725; 21 DEGs), “hormone-mediated signaling pathway” (GO: 0009755; one DEG), and “response to auxin” (GO: 0009733; 16 DEGs). The significantly enriched GO terms are shown in Additional file 5: Table S4.
The KEGG pathway analysis was performed to confirm the genes involved in metabolic or signal transduction pathways. We identified 117 enriched KEGG pathways. The 20 most significantly enriched KEGG pathways are shown in Fig. 3 and Additional file 6: Table S5. Of these, the “plant hormone signal transduction” pathway (KO04075) was significantly enriched, with 67 DEGs grouped into the auxin (IAA), cytokinin (CK), gibberellin (GA), abscisic acid (ABA), ethylene (ETH), brassinosteroid (BR), jasmonic acid (JA), and salicylic acid (SA) signal transduction pathways. Previous studies have shown that the plant hormones IAA, CK, ETH, GA, JA, and BR can influence the pistil development [11, 47, 48]. Of these, 31 DEGs were enriched in the IAA signal transduction pathway, followed by BR (7 DEGs), JA (5 DEGs), ETH (2 DEGs), CK (1 DEG), and GA (1 DEG) (Additional file 7: Table S6). The DEGs involved in the IAA signal transduction process included auxin1 (AUX1), auxin/indole-3-acetic acid (AUX/IAA), auxin response factors (ARF), GH3, and small auxin up RNA (SAUR); the majority of these genes were downregulated in the fsm1 mutant (Fig. 4).

Twenty most significantly enriched KEGG metabolic pathways

Expression of differentially expressed genes involved in the auxin signal transduction pathway. Note: The bar on the right represents the relative expression values. Gene expression is measured by counting the log2-based fragments per kilobase of transcript per million read values. Green arrows indicate genes that were downregulated and yellow arrows indicate genes that were both upregulated and downregulated
Analysis of DEGs related to ovule development and organ size
Among the DEGs, 29 were related to ovule development. Of these, the AGAMOUS-like (AGL) genes (BraA09g048000.3C, BraA03g051930.3C, BraA03g058700.3C, BraA09g006770.3C, and BraA06g039170.3C) were involved in ovule morphogenesis; genes encoding ANT (BraA03g061040.3C and BraA05g011060.3C), CUC3 (BraA07g039980.3C), and WUS (BraA03g038230.3C, BraA05g011060.3C, BraA05g029540.3C, and BraA10g022260.3C) were involved in ovule primordia formation; and genes encoding ANT, BEL1 (BraA03g000480.3C, BraA04g013970.3C, BraA03g058930.3C, BraA04g025560.3C, BraA03g018820.3C, BraA10g033530.3C, BraA07g027760.3C, BraA05g009430.3C, BraA04g017450.3C, and BraA07g038990.3C), TSO1 (BraA09g005800.3C, BraA01g021830.3C, and BraA05g024430.3C), SUB (BraA06g007870.3C and BraA06g001180.3C), SUP (BraA05g002900.3C), and KAN1 (BraA02g031490.3C) were involved in integument development. These genes may be associated with pistil abortion in the fsm1 mutant.
Compared with the wild-type ‘FT’ plants, the fsm1 mutant showed pistil abortion, and the four-whorled floral organs were significantly small. We identified a number of genes related to organ size regulation including the following: ANT, AUXIN-REGULATED GENE INVOLVED IN ORGAN SIZE (ARGOS; BraA09g049790.3C), TEOSINTE BRANCHED1/CYCLOIDEA/PCF (TCP; BraA02g042770.3C, BraA05g005380.3C, BraA05g032060.3C, BraA01g036950.3C, BraA07g034590.3C, BraA08g023460.3C, BraA07g030260.3C, BraA03g036760.3C, and BraA05g041050.3C) and AINTEGUMENTA-LIKE (AIL; BraA06g029000.3C, BraA03g004320.3C, BraA10g027380.3C, and BraA02g012300.3C). Most of these genes were downregulated in the fsm1 mutant. This downregulation may play a role in regulating floral organ size in the fsm1 mutants.
Analysis of gene expression patterns by quantitative reverse-transcription PCR (RT-qPCR)
To further confirm DEG expression patterns, 24 DEGs related to ovule development were selected for the RT-qPCR analysis including genes encoding AGL (BraA09g048000.3C, BraA03g051930.3C, BraA03g058700.3C, BraA09g006770.3C, and BraA06g039170.3C), WUS (BraA03g038230.3C, BraA05g011060.3C, BraA05g029540.3C, and BraA10g022260.3C), ANT (BraA03g061040.3C and BraA05g011060.3C), BEL1 (BraA03g058930.3C, BraA10g033530.3C, BraA07g027760.3C, BraA04g017450.3C, and BraA07g038990.3C), KAN1 (BraA02g031490.3C), TSO1 (BraA09g005800.3C, BraA01g021830.3C, and BraA05g024430.3C), CUC3 (BraA07g039980.3C), SUB (BraA06g007870.3C and BraA06g001180.3C), and SUP (BraA05g002900.3C). As shown in Fig. 5, the gene expression patterns showed a tendency similar to those detected by RNA-Seq, indicating the reliability of our transcriptome analysis.

RT-qPCR analysis of gene expression patterns. * indicates a significant difference at the 0.05 level determined by t-test
Discussion
In our previous study, we developed a female sterile mutant (fsm1) using isolated microspore culture combined with EMS mutagenesis in DH line ‘FT’ Chinese cabbage. BraA04g009730.3C was predicted as the candidate gene in mutant fsm1 that encodes a SAP transcriptional regulator; the coding sequence of BraA04g009730.3C is 1374 bp long [12]. In the present study, ‘FT’ seeds were also employed as the mutagenic material and germinating ‘FT’ seeds were treated with EMS solution to develop another parallel female sterile mutant (fsm2) that displayed a phenotype consistent with the fsm1 mutant. Allelism testing and the gene cloning analysis indicated that fsm1 and fsm2 were allelic and caused by mutations in the same gene, which verified the SAP function in pistil development. To probe the pistil abortion mechanism caused by SAP, comparative transcriptome sequencing of the pistil of the fsm1 mutant and wild-type ‘FT’ plants was conducted and the genes related to ovule development and organ size were identified. These results lay a foundation to reveal the mechanism of pistil abortion caused by the SAP gene in Chinese cabbage.
In plants, gene overexpression, RNA interference, gene knockout, site-directed mutagenesis, gene trapping, and biochip technology are commonly used methods for studying gene function. However, all these methods depend on the complete transgenic technology system, and they are time consuming. In contrast, allelism detection of mutant genes in different mutants with similar phenotype is a believable approach to verify whether they are controlled by the allelic genes, and can be applied to clarify gene function. Zhang et al. [49] identified two round-leaf mutants, rl-1 and rl-2, with a similar smooth leaf margin from a cucumber EMS mutagenic population. The map-based cloning strategy combined with a modified MutMap method suggested that CsPID (encoding a serine/threonine protein kinase) was the most likely candidate for rl-1. An allelism test of a cross between the rl-1 F1 (rl-1 × wild-type CCMC) and rl-2 F1 (rl-2 × wild-type CCMC) plants showed that the segregation ratio of normal leaf shape-to-round leaf shape was approximately 3:1, suggesting that rl-1 and rl-2 were allelic mutants with mutations in the same gene. These results suggest that CsPID is the gene responsible for the round leaf phenotype. Similar results have been reported in maize [11] and rice [50]. In the present study, the separation ratio of reciprocal crosses between the F1 (‘FT’ × fsm1) and F1 (‘FT’ × fsm2) plants was 3:1, suggesting that the fsm1 and fsm2 mutants had allelic mutations in the same gene. Based on our previous study [12], an SNP (C-to-A) occurred in the first exon (A04: 7544007) of BraA04g009730.3C, resulting in a premature stop codon in mutant fsm1; another SNP (G-to-A) was located in the first exon (A04: 7543809) of BraA04g009730.3C, causing a non-synonymous mutation in mutant fsm2. These results indicated that the allelic mutations in BraA04g009730.3C encoding an SAP transcriptional factor were responsible for pistil development in fsm1 and fsm2 mutants.
The SAP transcription factor was initially identified in A. thaliana, where SAP is essential for flower development. The sap mutant exhibited serious abnormalities in inflorescence, flower, and ovule development [45]. SAP encodes an F-box protein, which is a component of the SKP1/Cullin/ F-box (SCF) E3 ubiquitin ligase complex [46]. SAP affects organ size by regulating cell proliferation in A. thaliana [51]. In the genus Capsella, decreased SAP activity can result in small petals by shortening the cell proliferation period and reducing the number of petal cells [52]. Yang et al. [53] found a cucumber littleleaf (ll) mutant that exhibited smaller organ sizes and more lateral branches. Identification of the major-effect quantitative trait loci showed that LL in cucumber is an ortholog of Arabidopsis SAP and that they play similar roles in organ size control. In the present study, the fsm1 and fsm2 mutants presented an identical phenotype, exhibiting pistil abortion and small floral organs compared with the wild-type ‘FT’ plants, demonstrating that the SAP gene was involved in the mutant phenotype of Chinese cabbage.
Our previous study showed that an SNP was identified in BraA04g009730.3C, resulting in a premature stop codon in mutant fsm1, and BraA04g009730.3C expression had no obvious difference between the ‘FT’ and fsm1 mutant plants [12]. In this study, another SNP was located in BraA04g009730.3C, causing a non-synonymous mutation in mutant fsm2. We further analyzed the protein three-dimensional structures, and the results showed that the amino acid conformation were different at the mutant site between the wild-type ‘FT’ and mutant fsm2. The abnormal function of SAP in the fsm mutants was mainly manifested in amino acid coding. Given this, we speculated that the single nucleotide variation had no influence on BraA04g009730.3C expression, however, the variant protein may affect the expression of its downstream genes, eventually leading to phenotypic variation. We also demonstrated that mutant fsm1 pistil abortion was caused by abnormal ovule development [12, 44]. To further investigate the potential mechanism of ovule development induced by the SAP gene, we used RNA-Seq to compare the pistil transcriptome of the mutant fsm1 and wild-type ‘FT’ plants. There are two main regulatory pathways of pistil development in plants. In the first pathway, WUS-AG-related genes jointly regulate pistil development and in the second pathway, the KNOXI genes regulate pistil development. Of these, the first pathway is the main regulatory pathway [54, 55], suggesting that the AG genes play essential roles in the pistil development process. Studies have shown that SAP negatively regulates AG expression and that they jointly determine floral organ differentiation [45]. AG belongs to the MADS-box gene family and plays an important role in ovule development; its activity also contributes to ovule morphogenesis [21, 29, 56]. As important regulatory genes in ovule development, AG homologs also play essential roles in pistil formation [57]. For example, AGL11 plays an important regulatory role in ovule development [58], AGL8 and AGL19 play vital roles in regulating floral transition and female sterility [16, 59], and AGL62 can stimulate nucellus degeneration [60]. The homeobox gene WUS plays an essential role in regulating ovule development; it is mainly expressed in the nucellus of ovule primordia and is required for integument initiation [33]. ANT is a member of the AP2/ERF transcription factor family, which is involved in ovule development and ovule primordia formation [61, 62]. ANT mutation results in failed ovule integument formation, which then results in abnormal ovule development and female sterility [63, 64]. BEL1 is a homeodomain transcription factor that can control ovule patterning, particularly in determining integument identity and development; ovules develop a single integument-like structure in bel1 mutants [25, 65]. CUC3 is a putative NAC-domain transcription factor member of the CUC gene family [27]. CUC1, CUC2, and CUC3 are expressed during ovule primordia development. Of these, CUC1 and CUC2 play redundant roles in promoting ovule initiation in young gynecium with a fewer ovules [66]. CUC2 and CUC3 are also redundantly required for proper ovule development [30]. SUP, SUB, KAN1/2, and TSO1 regulate late integument development and play a role in the genetic control of integument morphogenesis [40, 41, 43]. Of these, KAN1 belongs to the KANADI family and is one of the most important genes in outer integument regulation [67]. In the current study, 29 DEGs related to ovule development were identified. Of these, the AGL genes regulate ovule morphogenesis; ANT, CUC3, and WUS regulate ovule primordia formation; and ANT, BEL1, TSO1, SUB, SUP, and KAN1 regulate integument development. These genes presented different expression patterns in the ‘FT’ and fsm mutant plants; we speculated that the interaction and regulation of these genes may influence ovule development in the fsm mutants (Fig. 6).

Possible SAP-mediated regulatory pathways involved in fsm mutants pistil development. The size of the circle represents the number of genes. Red and green indicate upregulated and downregulated genes, respectively. Yellow indicates genes that were both upregulated and downregulated
Compared with the wild-type ‘FT’ plants, the fsm1 mutant plants exhibited pistil abortion, and the floral organs were small. Plant organ size is determined by two developmental processes: cell proliferation and cell expansion [68]. In A. thaliana, the F-box protein SAP is a positive regulator of organ growth that regulates organ size by promoting cell proliferation [46, 51]. ANT not only regulates the formation of integument, but also regulates the size of plant organs [69]. ANT and ARGOS regulate organ size by influencing cell proliferation [61, 70, 71]. Loss-of-function ARGOS or ANT mutants exhibit smaller leaves and floral organs [53, 72]. ANT and AIL6 are important regulators of floral development as they can regulate floral meristem and organ growth [73, 74]. ANT, AIL5, and AIL7 play a redundant role in inflorescence meristem and flower development [75]. The TCP proteins are plant-specific transcription factors that can control leaf and flower size and shape [76]. In the current study, we identified 16 DEGs involved in the regulation of organ size, and they mainly included ANT, ARGOS, TCP, and AIL. The majority of these genes were downregulated in the fsm1 mutant. The interaction of these organ size-related DEGs might result in the smaller floral organs observed in the fsm mutant plants (Fig. 6).
Hormones play an important role in the regulation of plant growth and development. Multiple mutant genes involved in hormone signaling pathways including the IAA, CK, ETH, GA, JA, and BR pathways have been previously determined in female sterile mutants, resulting in morphological abnormalities in the pistil [11, 34, 48, 77,78,79,80]. The transcriptome analysis of female sterile materials showed that DEGs are significantly enriched in the hormone signal transduction pathways in rice, pomegranate, and Pinus tabuliformis, indicating that changes in the hormone metabolic pathway have an important effect on ovule development [15,16,17]. Auxin plays an important role in ovule development and promotes carpel initiation and gynecium growth [81]. Furthermore, ANT has been proposed to act downstream of auxin in flower growth and patterning [73]. The auxin-response genes mainly include ARF, GH3, SAUR, and AUX/IAA. ARF6 and ARF8 are involved in ovule adaxial/abaxial polarity formation and play a vital role in ovule and anther development [17, 82]. The downregulation of ARF6 and ARF8 can result in female sterility [77]. CK positively regulates ovule formation and pistil development [83]. Furthermore, the BEL1 transcription factors play an important role in cytokinin signaling pathways for correct ovule patterning [34]. Indeed, the CK levels have been positively correlated with ovule numbers. Reduced CK content results in a corresponding significant reduction in both ovule number and pistil size, thus resulting in female sterility [84,85,86]. ETH contributes to pistil development [78, 87] and ethylene-responsive factor (ERF) ethylene-response signal genes belong to the AP2 gene family, which positively regulate ETH. The ERF protein can also inhibit AG gene expression, thus affecting ovule development [21, 88, 89]. BR plays a role in the development of the ovule outer integument and gynecial medial domain [79]. GA plays a major role in the control of ovule integument development and ovule initiation [90] and negatively modulates the number of ovules in plants [48]. JA plays an important role in determining the fate of pistils as high JA levels promote pistil abortion [11]. The functional enrichment analysis in the present study showed that the GO terms related to hormones were enriched and that the plant hormone signal transduction pathways were also significantly enriched. The DEGs identified in our study were involved in IAA, CK, ETH, GA, JA, and BR signaling. Of these pathways, the IAA signaling pathway had the highest number of DEGs. Further study of these genes involved in hormone signal transduction may elucidate the pistil abortion mechanism; different hormones may have synergistic or antagonistic effects in regulating pistil development in Chinese cabbage.
Conclusions
The ovules play a major role in sexual reproduction, and they are the female reproductive organs in Chinese cabbage. Our study clarified the function of BraA04g009730.3C and revealed that it was responsible for ovule development and organ size. Comparative transcriptome analyses were performed to explore the regulatory effect of SAP in ovule development, and several DEGs related to ovule development were identified. Our study provides valuable information for future studies on pistil development and lays a solid foundation to elucidate the molecular mechanism of pistil development in Chinese cabbage.
Methods
Plant materials and mutagenic treatment
The wild-type ‘FT’ was a DH line derived from Chinese cabbage variety ‘Fukuda 50’, which was screened by Shenyang greenstar Chinese cabbage research institute (Shenyang, China) [91]. Germinated ‘FT’ seeds were immersed in 0.8% EMS solution for 12 h, and then thoroughly washed in running water for 12 h. After vernalization treatment at 2 °C for 15 d, the seeds were sown in a greenhouse in Shenyang Agricultural University. All live plants (M0 generation) were self pollinated. The mutant materials were screened and identified in the M1 generation to obtain the parallel female sterile mutant fsm2.
Observation of morphological characteristics
In the full-bloom stage, floral organ characteristics were observed and compared between the fsm1 and fsm2 mutants and wild-type ‘FT’ plants. According to our previous method [44], three each of ‘FT’ and mutant fsm2 plants were selected and artificial self-pollination of the fsm2 mutant and a reciprocal cross between ‘FT’ and the fsm2 mutant was performed. Seed-setting rates of each plant type were recorded and analyzed.
Genetic analysis
Mutant fsm2 and wild-type ‘FT’ plants were employed as parents to obtain the F1, F2, and BC1 populations. The phenotype of each plant in each generation was recorded to investigate the genetic characteristics of mutant fsm2 plants.
Allelism test between the fsm1 and fsm2 mutants
Both F1 (‘FT’ × fsm1) and F1 (‘FT’ × fsm2) populations exhibited the normal phenotype. The F1 (‘FT’ × fsm1) and F1 (‘FT’ × fsm2) populations were used as parent plants. Reciprocal crosses were made to obtain phenotypic segregation ratios of the populations. The population segregation ratios were analyzed using the Chi-square (χ2) test at the 0.05 level.
Gene cloning and sequencing
The coding sequence of BraA04g009730.3C was amplified in mutant fsm2 plants using the primer sequences shown in Additional file 8: Table S7. Gene cloning was performed according to the method of Huang et al. [92] and samples were sequenced by GENEWIZ (Suzhou, China) using the Sanger method. Sequences were aligned and analyzed using DNAMAN software. In addition, the online software SWISS-MODEL (https://swissmodel.expasy.org/) was used to analyze the three-dimensional protein structures of wild-type ‘FT’ and mutant fsm2.
RNA extraction, cDNA library construction, and Illumina sequencing
In the full-bloom stage, five wild-type ‘FT’ and five fsm1 mutants were selected. Pistils within the mature flower buds of ‘FT’ and mutant fsm1 plants were randomly selected and mixed; the mixed samples were used as one biological replicate. Three independent biological replicates of both ‘FT’ and fsm1 mutant were used.
The total RNA of the six samples was extracted using the TRIzol kit (Invitrogen, USA) following the manufacturer’s instructions. The quality and purity of the total RNA were checked using a NanoDropND-1000 spectrophotometer (NanoDrop, USA), and integrity was detected using the Agilent 2100 Bioanalyzer (Agilent, USA).
The six samples were designated as FT1, FT2, and FT3 (three biological replicates of ‘FT’) and M1, M2, and M3 (three biological replicates of mutant fsm1). Equal amounts of total RNA from the six samples were pooled for RNA-Seq library construction. The six cDNA libraries were sequenced using the Illumina novaseq-PE150 sequencing platform at Novogene (Beijing, China).
RNA-Seq analysis, differential gene expression, and functional enrichment analysis
Clean reads were mapped to the Brassica reference genome (http://brassicadb.org/brad/datasets/pub/Genomes/Brassica_rapa/V3.0/) using HISAT2 software.
Fragments per kilo bases per million fragment (FPKM) values and DESeq software [93] were used to analyze differential gene expression. DEG screening criteria were defined as having a |log2(fold change)| > 1 and P-value < 0.05. Significantly enriched GO terms and KEGG pathways of DEGs were analyzed using the topGO and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, respectively.
RT-qPCR analysis
Twenty-four genes related to ovule development were selected for the RT-qPCR analysis and the gene-specific primers were designed using Primer Premier 5.0 software. The primer sequences are listed in Additional file 9: Table S8. The cDNAs of ‘FT’ and mutant fsm1 pistils (collected as described in RNA-Seq) were employed as templates for RT-qPCR using UltraSYBR Mixture reagent (CWBIO, China) and the QuantStudio 6 Flex Real-Time PCR System (ABI, USA). The reaction system and program were used according to the manufacturer’s instructions. The 2-ΔΔCt method was used to calculate the relative gene expression levels [94]. Actin and 18S rRNA were used as the internal controls [95]. All reactions were performed with three technical and biological replicates, and the data were analyzed using OriginPro8.0. Significant difference at the 0.05 level was determined using the t-test with SPSS 16.0 software.
Availability of data and materials
The data charts supporting the results and conclusions are included in the article and additional files. Transcriptome sequencing data have been deposited in the NCBI Gene Expression Omnibus (GEO) Database under accession number GSE147438 (https://www.ncbi.nlm.nih.gov/geo).
Abbreviations
- EMS:
-
Ethyl methanesulfonate
- DH:
-
Double haploid
- IAA:
-
Auxin
- CK:
-
Cytokinin
- GA:
-
Gibberellin
- ABA:
-
Abscisic acid
- ETH:
-
Ethylene
- BR:
-
Brassinosteroid
- JA:
-
Jasmonic acid
- SA:
-
Salicylic acid
- SNP:
-
Single nucleotide polymorphism
References
Li SC, Yang L, Deng QM, Wang SQ, Wu FQ, Li P. Phenotypic characterization of a female sterile mutant in rice. J Integr Plant Biol. 2006;48:307–14.
Bencivenga S, Colombo L, Masiero S. Cross talk between the sporophyte and the megagametophyte during ovule development. Sex Plant Reprod. 2011;24:113–21.
Tedder A, Helling M, Pannell JR, Shimizu-Inatsugi R, Kawagoe T, van Campen J, Sese J, Shimizu KK. Female sterility associated with increased clonal propagation suggests a unique combination of androdioecy and asexual reproduction in populations of Cardamine amara (Brassicaceae). Ann Bot. 2015;15:763–76.
Wei B, Zhang J, Pang C, Yu H, Guo D, Jiang H, Ding M, Chen Z, Tao Q, Gu H, Qu LJ, Qin G. The molecular mechanism of SPOROCYTELESS/NOZZLE in controlling Arabidopsis ovule development. Cell Res. 2015;25:121–34.
Baker SC, Robinson-Beers K, Villanueva JM, Gaiser JC, Gasser CS. Interactions among genes regulating ovule development in Arabidopsis thaliana. Genetics. 1997;145:1109–24.
Zhou HC, Jin L, Li J, Wang XJ. Altered callose deposition during embryo sac formation of multi-pistil mutant (mp1) in Medicago sativa. Genet Mol Res. 2016;15:15027698.
Singh SK, Kumar V, Srinivasan R, Ahuja PS, Bhat SR, Bhat SR, Sreenivasulu Y. The TRAF Mediated Gametogenesis Progression (TRAMGaP) gene is required for megaspore mother cell specification and gametophyte development. Plant Physiol. 2017;175:1220–37.
Lee JJ, Hassan OSS, Gao W, Kohel RJ, Wei NE, Kohel RJ. Developmental and gene expression analyses of a cotton naked seed mutant. Planta. 2006;223:418–32.
Nonomura K, Morohoshi A, Nakano M, Eiguchi M, Miyao A, Hirochika H, Kurata N. A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell. 2007;19:2583–94.
Teng CC, Du DZ, Xiao L, Yu QL, Shang GX, Zhao ZG. Mapping and identifying a candidate gene (Bnmfs) for female-male sterility through whole-genome resequencing and RNA-Seq in rapeseed (Brassica napus L.). Front Plant Sci. 2017;8:2086.
Zhao Y, Zhang YZ, Wang LJ, Wang XR, Xu W, Gao XY, Liu BS. Mapping and functional analysis of a maize silkless mutant sk-A7110. Front Plant Sci. 2018;9:1227.
Liu WJ, Huang SN, Liu ZY, Lou TX, Tan C, Wang YH, Feng H. A missense mutation of STERILE APETALA leads to female sterility in Chinese cabbage (Brassica campestris ssp. pekinensis). Plant Reprod. 2019;32:217–28.
Kubo T, Fujita M, Takahashi H, Nakazono M, Tsutsumi N, Kurata N. Transcriptome analysis of developing ovules in rice isolated by laser microdissection. Plant Cell Physiol. 2013;54:750–65.
Fu WQ, Zhao ZG, Ge XH, Ding L, Li ZY. Anatomy and transcript profiling of gynoecium development in female sterile Brassica napus mediated by one alien chromosome from Orychophragmus violaceus. BMC Genomics. 2014;15:61.
Yang LY, Wu Y, Yu ML, Mao BG, Zhao BR, Wang JB. Genome-wide transcriptome analysis of female-sterile rice ovule shed light on its abortive mechanism. Planta. 2016;244:1011–28.
Chen LN, Zhang J, Li HX, Niu J, Xue H, Liu BB, Wang Q, Luo X, Zhang FH, Zhao DG, Cao SY. Transcriptomic analysis reveals candidate genes for female sterility in pomegranate flowers. Front Plant Sci. 2017;8:1430.
Yao Y, Han R, Gong ZX, Zheng CX, Zhao YY. RNA-seq analysis reveals gene expression profiling of female fertile and sterile ovules of Pinus Tabulaeformis Carr during free nuclear mitosis of the female gametophyte. Int J Mol Sci. 2018;19:2246.
Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353:31.
Colombo L, Franken J, Koetje E, van Went J, Dons HJM, Angenent GC, van Tunen AJ. The petunia MADS box gene FBP11 determines ovule identity. Plant Cell. 1995;7:1859–68.
Theissen G. Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol. 2001;4:75–85.
Pinyopich A, Ditta GS, Savidge B, Liljegren SJ, Baumann E, Wisman E, Yanofsky MF. Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature. 2003;424:85–8.
Theißen G, Saedler H. Plant biology: floral quartets. Nature. 2001;409:469–71.
Tsai WC, Chen HH. The orchid MADS-box genes controlling floral morphogenesis. Sci World J. 2006;6:1933–44.
Galimba KD, Di Stilio VS. Sub-functionalization to ovule development following duplication of a floral organ identity gene. Dev Biol. 2015;405:158–72.
Reiser L, Modrusan Z, Margossian L, Samach A, Ohad N, Haughn GW, Fischer RL. The BEL1 gene encodes a homeodomain protein involved in pattern formation in the Arabidopsis ovule primordium. Cell. 1995;83:735–42.
Favaro R, Battaglia R, Kooiker M, Borghi L, Ditta G, Yanofsky MF, Kater MM, Colombo L. MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell. 2003;15:2603–11.
Vroemen CW, Mordhorst AP, Albrecht C, Kwaaitaal MACJ, de Vries SC. The CUP-SHAPED COTYLEDON3 gene is required for boundary and shoot meristem formation in Arabidopsis. Plant Cell. 2003;15:1563–77.
Park SO, Zheng Z, Oppenheimer DG, Hauser BA. The PRETTY FEW SEEDS2 gene encodes an Arabidopsis homeodomain protein that regulates ovule development. Development. 2005;132:841–9.
Ó’Maoiléidigh DS, Wuest SE, Rae L, Raganelli A, Ryan PT, Kwasniewska K, Das P, Lohan AJ, Loftus B, Graciet E, Wellmer F. Control of reproductive floral organ identity specification in Arabidopsis by the C function regulator AGAMOUS. Plant Cell. 2013;25:2482–503.
Goncalves B, Hasson A, Belcram K, Cortizo M, Morin H, Nikovics K, Vialette-Guiraud A, Takeda S, Aida M, Laufs P, Arnaud N. A conserved role for CUP-SHAPED COTYLEDON genes during ovule development. Plant J. 2015;83:732–42.
Krizek BA, Prost V, Macias A. AINTEGUMENTA promotes petal identity and acts as a negative regulator of AGAMOUS. Plant Cell. 2000;12:1357–66.
Balasubramanian S, Schneitz K. NOZZLE links proximal-distal and adaxial-abaxial pattern formation during ovule development in Arabidopsis thaliana. Development. 2002;129:4291–300.
Gross-Hardt R, Lenhard M, Laux T. WUSCHEL signaling functions in interregional communication during Arabidopsis ovule development. Genes Dev. 2002;16:1129–38.
Bencivenga S, Simonini S, Benková E, Colombo L. The transcription factors BEL1 and SPL are required for cytokinin and auxin signaling during ovule development in Arabidopsis. Plant Cell. 2012;24:2886–97.
Schneitz K, Baker SC, Gasser CS, Redweik A. Pattern formation and growth during floral organogenesis, HUELLENLOS and AINTEGUMENTA are required for the formation of the proximal region of the ovule primordium in Arabidopsis thaliana. Development. 1998;125:2555–63.
Villanueva JM, Broadhvest J, Hauser BA, Meister RJ, Schneitz K, Gasser CS. INNER NO OUTER regulates abaxial-adaxial patterning in Arabidopsis ovules. Genes Dev. 1999;13:3160–9.
Skinner DJ, Hill TA, Gasser CS. Regulation of ovule development. Plant Cell. 2004;16:S32–45.
Brown RH, Nickrent DL, Gasser CS. Expression of ovule and integument-associated genes in reduced ovules of Santalales. Evol Dev. 2010;12:231–40.
Lang JD, Ray S, Ray A. sin1, a mutation affecting female fertility in Arabidopsis, interacts with mod1, its recessive modifier. Genetics. 1994;137:1101–10.
Gaiser JC, Robinson-Beers K, Gasser CS. The Arabidopsis SUPERMAN gene mediates asymmetric growth of the outer integument of ovules. Plant Cell. 1995;7:333–45.
Schneitz K. The molecular and genetic control of ovule development. Curr Opin Plant Biol. 1999;2:13–7.
Gifford ML, Dean S, Ingram GC. The Arabidopsis ACR4 gene plays a role in cell layer organisation during ovule integument and sepal margin development. Development. 2003;130:4249–58.
McAbee JM, Hill TA, Skinner DJ, Izhaki A, Hauser BA, Meister RJ, Venugopala Reddy G, Meyerowitz EM, Bowman JL, Gasser CS. ABERRANT TESTASHAPE encodes a KANADI family member, linking polarity determination to separation and growth of Arabidopsis ovule integuments. Plant J. 2006;46:522–31.
Huang SN, Liu ZY, Li CY, Yao RP, Li DY, Liu WJ, Feng H. Transcriptome analysis of a female-sterile mutant (fsm) in Chinese cabbage (Brassica campestris ssp. pekinensis). Front. Plant Sci. 2017;8:546.
Byzova MV, Franken J, Aarts MGM, de Almeida-Engler J, Engler G, Mariani C, Van Lookeren Campaqne MM, Angenent GC. Arabidopsis STERILE APETALA, a multifunctional gene regulating inflorescence, flower, and ovule development. Genes Dev. 1999;13:1002–14.
Wang Z, Li N, Jiang S, Gonzalez N, Huang X, Wang Y, Inzé D, Li Y. SCFSAP controls organ size by targeting PPD proteins for degradation in Arabidopsis thaliana. Nat Commun. 2016;7:11192.
Marsch-Martínez N, de Folter S. Hormonal control of the development of the gynoecium. Curr Opin Plant Biol. 2016;29:104–14.
Gomez MD, Barro-Trastoy D, Escoms E, Saura-Sanchez M, Sanchez I, Briones-Moreno A, Vera-Sirera F, Carrera E, Ripoll J, Yanofsky MF, Lopez-Diaz I, Alonso JM, Perez-Amador MA. Gibberellins negatively modulate ovule number in plants. Development. 2018;145:163865.
Zhang CW, Chen FY, Zhao ZY, Hu LL, Liu HQ, Cheng ZH, Weng YQ, Chen P, Li YH. Mutations in CsPID encoding a Ser/Thr protein kinase are responsible for round leaf shape in cucumber (Cucumis sativus L.). Theor Appl Genet. 2018;131:1379–89.
Lee G, Piao R, Lee Y, Kim B, Seo J, Lee D, Jang S, Jin Z, Lee C, Chin JH, Koh HJ. Identification and characterization of LARGE EMBRYO, a new gene controlling embryo size in rice (Oryza sativa L.). Rice. 2019;12:22.
Li N, Liu Z, Wang Z, Ru L, Gonzalez N, Baekelandt A, Pauwels L, Goossens A, Xu R, Zhu Z, Inzé D, Li Y. STERILE APETALA modulates the stability of a repressor protein complex to control organ size in Arabidopsis thaliana. PLoS Genet. 2018;14:e1007218.
Sicard A, Kappela C, Lee YW, Woźniak NJ, Marona C, Stinchcombe JR, Wright SI, Lenhard M. Standing genetic variation in a tissue-specific enhancer underlies selfing-syndrome evolution in Capsella. Proc Natl Acad Sci U S A. 2016;113:13911–6.
Yang LM, Liu HQ, Zhao JY, Pan YP, Cheng SY, Lietzow CD, Wen CL, Zhang XL, Weng YQ. LITTLELEAF (LL) encodes a WD40 repeat domain-containing protein associated with organ size variation in cucumber. Plant J. 2018;95:834–47.
Lu HY, Cao JS, Yu XL. Gene regulation involved in gynoecium development. Chin J Cell Biol. 2009;30:63–8.
Sun HL, Song J, Gao ZH, Ni ZJ, Zhang Z. Isolation and expression analysis of pm KNAT2 gene from Japanese apricot. Sci Agri Sin. 2014;47:3444–52.
Bowman JL, Drews GN, Meyerowitz EM. Expression of the Arabidopsis floral homeotic gene AGAMOUS is restricted to specific cell types late in flower development. Plant Cell. 1991;3:749–58.
Dreni L, Kater MM. MADS reloaded: evolution of the AGAMOUS subfamily genes. New Phytol. 2014;201:717–32.
Dreni L, Osnato M, Kater MM. The ins and outs of the rice AGAMOUS subfamily. Mol Plant. 2013;6:650–64.
Alvarez-Buylla ER, Liljegren SJ, Pelaz S. MADS-box gene evolution beyond flowers: expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. 2000;24:457–66.
Bertoni G. What the nucellus can tell us. Plant Cell. 2016;28:1234.
Mizukami Y, Fischer RL. Plant organ size control: AINTEGUMENTA regulates growth and cell numbers during organogenesis. Proc Natl Acad Sci U S A. 2000;97:942–7.
Cucinotta M, Colombo L, Roig-Villanova I. Ovule development, a new model for lateral organ formation. Front Plant Sci. 2014;5:117.
Elliott RC, Betzner AS, Huttner E, Oakes MP, Tucker WQ, Gerentes D, Perez P, Smyth DR. AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell. 1996;8:155–68.
Klucher KM, Chow H, Reiser L, Fischer RL. The AINTEGUMENTA gene of Arabidopsis required for ovule and female gametophyte development is related to the floral homeotic gene APETALA2. Plant Cell. 1996;8:137–53.
Brambilla V, Battaglia R, Colombo M, Masiero S, Bencivenga S, Kater MM, Colombo L. Genetic and molecular interactions between BELL1 and MADS box factors support ovule development in Arabidopsis. Plant Cell. 2007;19:2544–56.
Galbiati F, Roy DS, Simonini S, Cucinotta M, Ceccato L, Cuesta C, Simaskova M, Benkova E, Kamiuchi Y, Aida M, Weijers D, Simon R, Masiero S, Colombo L. An integrative model of the control of ovule primordia formation. Plant J. 2013;76:446–55.
Bowman JL, Smyth DR. CRABS CLAW, a gene that regulates carpel and nectary development in Arabidopsis, encodes a novel protein with zinc finger and helix-loop-helix domains. Development. 1999;126:2387–96.
Horiguchi G, Ferjani A, Fujikura U, Tsukaya H. Coordination of cell proliferation and cell expansion in the control of leaf size in Arabidopsis thaliana. J Plant Res. 2006;119:37–42.
Randall RS, Sornay E, Dewitte W, Murray JAH. AINTEGUMENTA and the D-type cyclin CYCD3; 1 independently contribute to petal size control in Arabidopsis: evidence for organ size compensation being an emergent rather than a determined property. J Exp Bot. 2015;66:3991–4000.
Krizek BA. Ectopic expression of AINTEGUMENTA in Arabidopsis plants results in increased growth of floral organs. Dev Genet. 1999;25:224–36.
Czesnick H, Lenhard M. Size control in plants-lessons from leaves and flowers. Cold Spring Harb Perspect Biol. 2015;7:a019190.
Hu Y, Xie A, Chua NH. The Arabidopsis auxin-inducible gene ARGOS controls lateral organ size. Plant Cell. 2003;15:1951–61.
Krizek BA. AINTEGUMENTA and AINTEGUMENTA-LIKE6 act redundantly to regulate Arabidopsis floral growth and patterning. Plant Physiol. 2009;150:1916–29.
Krizek BA, Bequette CJ, Xu K, Blakley IC, Fu ZQ, Stratmann J, Loraine AE. RNA-Seq links AINTEGUMENTA and AINTEGUMENTA-LIKE6 to cell wall remodeling and plant defense pathways in Arabidopsis. Plant Physiol. 2016;171:2069–84.
Nole-Wilson S, Tranby T, Krizek BA. AINTEGUMENTA-like (AIL) genes are expressed in young tissues and may specify meristematic or division-competent states. Plant Mol Biol. 2005;57:613–28.
Nicolas M, Cubas P. Chapter 16: the role of TCP transcription factors in shaping flower structure, leaf morphology, and plant architecture. In: Gonzalez DN, editor. Plant Transcription Factors-evolutionary, Structural, and Functional Aspects. Cambridge: Academic Press, England; 2016. p. 249–67.
Wu MF, Tian Q, Reed JW. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression and regulates both female and male reproduction. Development. 2006;133:4211–8.
Boualem A, Fergany M, Fernandez R, Troadec C, Martin A, Morin H. A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science. 2008;321:836–8.
Nole-Wilson S, Rueschhoff EE, Bhatti H, Franks RG. Synergistic disruptions in seuss cyp85A2 double mutants reveal a role for brassinolide synthesis during gynoecium and ovule development. BMC Plant Biol. 2010;10:198.
Marsch-Martínez N, Ramos-Cruz D, Reyes-Olalde JI, Lozano-Sotomayor P, Zúñiga-Mayo VM, de Folter S. The role of cytokinin during Arabidopsis gynoecia and fruit morphogenesis and patterning. Plant J. 2012;72:222–34.
Pagnussat GC, Alandete-Saez M, Bowman JL, Sundaresan V. Auxin-dependent patterning and gamete specification in the Arabidopsis female gametophyte. Science. 2009;324:1684–9.
Song YL, Wang L, Xiong LZ. Comprehensive expression profiling analysis of OsIAA gene family in developmental processes and in response to phytohormone and stress treatments. Planta. 2009;229:577–91.
Hutchison CE, Li J, Argueso C, Gonzalez M, Lee E, Lewis MW, Maxwell BB, Perdue TD, Schaller GE, Alonso JM, Ecker JR, Kieber JJ. The Arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell. 2006;18:3073–87.
Bartrina I, Otto E, Strnad M, Werner T, Schmülling T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell. 2011;23:69–80.
Kinoshita-Tsujimura K, Kakimoto T. Cytokinin receptors in sporophytes are essential for male and female functions in Arabidopsis thaliana. Plant Signal Behav. 2011;6:66–71.
Zúñiga-Mayo VM, Reyes-Olalde JI, Marsch-Martinez N, de Folter S. Cytokinin treatments affect the apical-basal patterning of the Arabidopsis gynoecium and resemble the effects of polar auxin transport inhibition. Front Plant Sci. 2014;5:191.
Yamasaki S, Fujii N, Takahashi H. Characterization of ethylene effects on sex determination in cucumber plants. Sex Plant Reprod. 2003;16:103–11.
De Martinis D, Mariani C. Silencing gene expression of the ethylene-forming enzyme results in a reversible inhibition of ovule development in transgenic tobacco plants. Plant Cell. 1999;11:1061–71.
Licausi F, Ohme-Takagi M, Perata P. APETALA2/ethylene responsive factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol. 2013;199:639–49.
Gomez MD, Ventimilla D, Sacristan R, Perez-Amador MA. Gibberellins regulate ovule integument development by interfering with the transcription factor ATS. Plant Physiol. 2016;172:2403–15.
Huang SN, Liu ZY, Li DY, Yao RP, Meng Q, Feng H. Screening of Chinese cabbage mutant produced by 60Co-ray mutagenesis of isolated microspore cultures. Plant Breed. 2014;133:480–8.
Huang SN, Liu ZY, Yao RP, Li DY, Zhang T, Li X, Hou L, Wang YH, Tang XY, Feng H. Candidate gene prediction for a petal degeneration mutant, pdm, of the Chinese cabbage (Brassica campestris, ssp. pekinensis) by using fine mapping and transcriptome analysis. Mol Breed. 2016;36:26.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25:402–8.
Huang SN, Peng SL, Liu ZY, Li CY, Tan C, Yao RP, Li DY, Li X, Hou L, Feng H. Investigation of the genes associated with a male sterility mutant (msm) in Chinese cabbage (Brassica campestris ssp. pekinensis) using RNA-Seq. Mol Gen Genomics. 2020;295:233–49.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Funding
This research was supported by grants from the and the National Natural Science Foundation of China (31801854) and the Open Project of Key laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, P.R. China (IVF201803). The funding played roles in the design of the study and collection, analysis, and interpretation of data.
Author information
Authors and Affiliations
Contributions
HF and SH designed the experiments and supervised this study. SH and WL conducted the research and wrote the manuscript. JX, ZL, and CL participated in the research and analyzed the data. All authors approved the manuscript.
Authors’ information
Department of Horticulture, Shenyang Agricultural University, Shenyang 110866, China.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Summary of all expressed genes detected in the ‘FT’ and fsm1 mutant libraries.
Additional file 2: Table S2.
List of DEGs identified by comparing the fsm1 mutants and ‘FT’ plants.
Additional file 3: Table S3.
List of specifically expressed genes identified in fsm1 mutants compared with those in the ‘FT’ plants.
Additional file 4: Figure S1.
GO functional classification of DEGs between the fsm1 mutants and ‘FT’ plants.
Additional file 5: Table S4.
Significantly enriched GO terms identified in the fsm1 mutants compared with those in the ‘FT’ plants.
Additional file 6: Table S5.
Twenty most significantly enriched KEGG metabolic pathways.
Additional file 7: Table S6.
DEGs involved in the plant hormone signal transduction pathways.
Additional file 8: Table S7.
Primers for BraA04g009730.3C coding sequences.
Additional file 9: Table S8.
Primer sequences used for the RT-qPCR analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, S., Liu, W., Xu, J. et al. The SAP function in pistil development was proved by two allelic mutations in Chinese cabbage (Brassica rapa L. ssp. pekinensis). BMC Plant Biol 20, 538 (2020). https://doi.org/10.1186/s12870-020-02741-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-020-02741-5