
Abstract
Background
Frequently occurring drought stress negatively affects the production of maize worldwide. Numerous efforts have been made to develop drought-tolerant maize lines and to explore drought tolerant mechanisms in maize. However, there is a lack of comparative studies on transcriptomic changes between drought-tolerant and control maize lines.
Results
In the present study, we have developed a drought-tolerant maize mutant (C7–2t) by irradiating the seeds of maize inbred line ChangC7–2 (C7–2) with 60Co-γ. Compared to its wild type C7–2, C7–2t exhibited a significantly delayed wilting and higher drought tolerance under both the controlled and field conditions, indicating its high water-holding ability. Transcriptomic profiling was performed to identify differentially expressed genes (DEGs) between C7–2 and C7–2t during drought. As a result, a total of 4552 DEGs were implied in drought tolerance of C7-2 and C7-2t. In particular, the expression of photosynthesis-related genes in C7–2 was inhibited, whereas these genes in C7–2t were almost unaffected under drought. Moreover, a specific set of the DEGs were involved in phenylpropanoid biosynthesis and taurine (hypotaurine) metabolism in C7–2t; these DEGs were enriched in cell components associated with membrane systems and cell wall biosynthesis.
Conclusions
The drought tolerance of C7–2t was largely due to its high water-holding ability, stable photosynthesis (for supporting osmoregulation) and strengthened biosynthesis of cell walls under drought conditions.
Similar content being viewed by others


Background
Frequently occurring drought stress negatively affects the production of maize worldwide [1, 2]. Enhancing maize resistance to drought is an effective way to address the problem of yield loss caused by drought stress [3, 4]. Breeders and agronomists are interested in developing drought-tolerant lines and uncovering their drought-tolerant characteristics [5]. Therefore, it is of considerable significance to clarify the mechanisms of maize response to drought stress and adaptation to meet the increasing food demands [6].
The drought tolerance in plants is usually evaluated according to differences in phenotypes and physiological and biochemical responses [7, 8]. Drought-tolerant plants can maintain stable morphological structures during prolonged periods of drought stress by high water-holding ability [9, 10]. On the one hand, drought-tolerant lines need to enhance the ability of their roots to absorb water from soil [9]. By reducing the number and increasing the volume of individual cortical cells in maize, the root system can reduce its exploration of the surface soil and grow into deeper soil, which allows maize to obtain more water from soil [11, 12]. On the other hand, drought tolerance can be enhanced by reducing the density and controlling the opening of stomata in leaves [13, 14]. Moreover, plants can synthesize proline and soluble sugars to reduce the water potential in cells and maintain cell homeostasis by osmoregulation under drought conditions [15, 16]. Drought stress also activates the peroxide-scavenging enzyme system to remove excess reactive oxygen species (ROS) induced by drought, which can damage the cell membrane systems and eventually cause cell death [17, 18]. The activity of the antioxidant (enzymatic and nonenzymatic) system represents an effective index to evaluate drought tolerance in maize [19, 20]. Drought-tolerant maize lines have been identified by comparing physiological and biochemical changes among hybrids, inbred lines and transgenic lines [21,22,23]. However, these changes could not fully explain the drought tolerance in maize due to variations in genetic backgrounds. It has been well documented that drought response of plants involves a complex regulatory network; therefore, omic studies are particularly required for functional characterization of key drought-response genes to improve drought-tolerant traits in crop plants [24].
To date only a few drought-tolerant genes such as ZmVPP1 and ZmPP2C in maize were identified by traditional sequencing methods and functionally characterized [22, 25, 26]. Many key genes were implied in drought tolerance in maize [22, 26,27,28,29]; however, a relative long period was required to clarify the functions of these drought-response genes [25].
With the advantages of low cost, high throughput and high sensitivity, RNA-seq is a powerful tool for the large-scale identification of drought-responsive genes and can facilitate the mining of key drought tolerance genes in plants, e.g., maize [30,31,32,33,34,35,36,37]. For example, RNA-seq studies have shown that the upregulation of cell wall biosynthesis/aquaporin-related genes allows maize recombinant inbred lines to gain drought adaptability under drought conditions [27]; the genes related to cell wall remodeling are involved in drought-response processes in a drought-tolerant maize line, and the syntheses of amino acids and carbohydrates are related to drought tolerance [36].
In the present study, we created a drought-tolerant line (C7–2t) by irradiating seeds of maize inbred line Chang7–2 (C7–2). To explore the mechanisms of drought resistance of C7–2t, we have compared physiological, biochemical and transcriptomic changes between C7–2 and C7–2t. Our results would highlight the drought tolerance signatures of C7–2t and contribute to the identification and functional studies of novel drought tolerance genes in maize.
Results
Creation and screening of drought-tolerant maize mutants
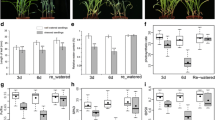
The 60Co-γ radiated maize seeds at 200 Gy for 1 h were used for screening of drought-tolerant lines because the fatality rate of mutagenesis was 50%, i.e. about half of the treated seeds lost their germination abilities. Then, the promising drought-tolerant mutants were obtained by continuous self-crossing for five years. The laboratory study further verified that one of the mutants (C7–2t) showed an excellent drought-tolerant performance at the early seedling stage (Fig. 1a; Fig. 1b). The drought-tolerance index of the mutant C7–2t was significantly higher compared to its wild-type C7–2 (Fig. S1a). Clearly, the leaves of the 35-day-old C7–2 plants curled earlier than those of C7–2t plants, which maintained the normal leaf morphology under water defect conditions in the field (Fig. 1c; Fig. 1d). Moreover, the anthesis-silk interval (ASI) of C7–2t was significantly shorter than that of C7–2 (Fig. S1b; Fig. S1c). In addition, there was no significant difference in plant height, ear height and biomass in the fields between both lines (Fig. S2). Therefore, drought-tolerant C7–2t showed an improved performance in the field. Next, physiological, biochemical and transcriptomic analyses were performed to explore the mechanisms underlying the drought tolerance in C7–2t.

Evaluation of drought tolerance of C7–2 and C7–2t. a Normal watering conditions (control), 16-day-old seedlings. b Water withheld for 8 days (drought), 16-day-old seedlings. c and d, seedlings in the field at 35 days after sowing
Differential drought responses between both maize lines
Under the normal conditions, there was no significant difference in relative water content (RWC), proline content, malondialdehyde (MDA) content, or chlorophyll (Chl) fluorescence parameters between C7–2t and C7–2, whereas the catalase (CAT) activity and soluble sugar content (SSC), in the leaves of C7–2t was higher than that of C7–2 (Fig. 2).

Comparison of the physiological indexes of C7–2 and C7–2t under the control and drought conditions. Different letters indicate significant difference at 0.05 significance level
During a 7-day drought treatment, C7–2 showed an increasingly severe wilting, whereas C7–2t did not show any signs of wilting (Fig. 1b); RWC of both lines decreased, with a more significant reduction in C7–2 than C7–2t (Fig. 2a), and the MDA content in leaves under drought was lower in C7–2t than C7–2 (Fig. 2b), suggesting C7–2t suffered less damage that C7–2 under drought stress. Moreover, the contents of SSC and proline, and the activity of CAT in C7–2t were significantly higher than those in C7–2 (Fig. 2c, d, e), implying that C7–2t was likely to enhance drought tolerance by modulation the contents of SSC and proline, and the activity of CAT.
Under drought total Chl content in the leaves of both lines decreased. It needs to note that Chl content in C7–2t was also lower than that of C7–2 under normal conditions (Fig. 2f). Clearly, the mutation has affected the Chl synthesis in the C7–2t. However, the maximum photochemical quantum yield (Fv/Fm) and effective photochemical quantum yield (Y(II)) of photosystem II (PS II) in C7–2t were all significantly higher than those in C7–2 under drought stress (Fig. 2g, h).
Gene sequence assembly, annotation and categorization
For a comparison of the transcript profiles of C7–2 and C7–2t under drought stress, 12 samples from three independent biological repeats were collected for RNA-seq analysis. Raw reads of 142,060,004, 140,113,794, 134,927,492 and 141,943,752 were generated from plants in C7–2 control (CC), C7–2 drought treatment (CD), C7–2t control (TC) and C7–2t drought treatment (TD), respectively. After removal of the adapters, low-quality sequences and ambiguous reads, 138,139,372 (CC), 135,923,246 (CD), 131,095,380 (TC), and 138,366,112 (TD) clean paired-end reads were obtained, respectively (Table 1). The expression levels of the genes were evaluated using FPKM (fragments per kilobase of exon model per million reads mapped) values. The percentage of genes with FPKM ≥1 in the 12 samples ranged from 56.19 to 59.91%. There were fewer genes with FPKM > 1 in C7–2t than in C7–2. The percentage of genes with FPKM ≥100 in the 12 samples ranged from 2.09 to 2.38% (Table S1).
Five public databases (NCBInr, NCBInt, UniProtKB, KEGG, and COG) were referred for the annotation of all the detected genes. Approximately 98.78% of detected sequences (31,536) had at least one significant match (E < 1e− 6) in one of the five databases. One or more gene ontology (GO) terms were assigned to 23,231 genes (72.77%), with 7178 identified GO items belonging to biological process, cellular component and molecular function categories (Table S1). In the biological process category, 7.97, 5.23, and 2.26% of the detected genes were annotated to DNA-templated transcription (GO:0006351), regulation of DNA-templated transcription (GO:0006355) and defense response (GO:0006952), respectively. In the cellular component category, 17.48, 14.88, and 9.75% of genes were annotated as nucleus (GO:0005634), integral component of membrane (GO:0016021), and cytoplasm (GO:0005737), respectively. In the molecular function category, ATP binding (GO:0005524; 11.75%), metal ion binding (GO:0046872; 7.51%) and DNA binding (GO:0003677; 6.11%) ranked highest in GO terms of the detected genes.
GO classification of the detected genes in both lines under drought stress displayed a notably high degree of similarity (Fig. 3). In the biological process group, the top three GO categories were cellular process, metabolic process and biological regulation. In the cellular component group, a large number of genes were enriched in cell part, organelle, and membrane. In the molecular function group, the top three GO categories were binding, catalytic activity, and transporter activity.

GO functional classification of the DEGs in C7–2 and C7–2t regarding biological processes, cellular components and molecular function. The abscissa stands for the number of genes annotated into GO terms, and the ordinate stands for the GO classification
Identification of differentially expressed genes (DEGs)
In total, 4353 and 1034 drought-responsive DEGs were identified in C7–2 (CD vs CC) and C7–2t (TD vs TC), respectively (Fig. 4; Table S2). Among the DEGs in C7–2, 2214 were upregulated and 2139 were downregulated. C7–2t, 634 DEGs were upregulated, and 400 were downregulated. A total of 835 genes were shared by the two comparisons (CD vs CC and TD vs TC), including 520 upregulated genes and 315 downregulated genes (Table S3). These DEGs mainly respond to water stress-related stimuli dramatically changed, e.g., the upregulated DEGs dehydrins (DHN1, Zm00001d037894; COR410, Zm00001d017547, Zm00001d051420) and universal stress protein A (Zm00001d034027).

A Venn diagram showing the expression patterns of the DEGs in C7–2 and C7–2t. Red and blue numbers indicate the numbers of the up- and down-regulated genes, respectively
After excluding the shared DEGs, 3518 and 199 DEGs were detected in C7–2 and C7–2t, respectively (Fig. 4; Table S4), suggesting differential drought-responsive pathways between the two lines. Among the 199 DEGs specific to C7–2t, 114 were upregulated and 85 downregulated, which may be related to the drought tolerance of C7–2t. Among the 3518 DEGs specific to C7–2, 1694 were upregulated, and 1824 genes were downregulated. Clearly, more gene expressions were affected by drought stress in C7–2 than in C7–2t. Consequently, the growth of C7–2 plants was significantly inhibited by drought stress compared to C7–2t, suggesting that C7–2t had an active drought adaptation strategy.
GO and KEGG enrichment of the DEGs
GO enrichment analysis was conducted to investigate the variability in biological processes under drought stress. A total of 182 and 90 GO terms were found as overrepresentations (FDR < 0.05) under drought in C7–2 and C7–2t, respectively (Table S5).
These altered genes in C7–2 primarily affect chloroplast part (GO:0044434) and photosynthesis. The DEGs related to the photosynthetic membrane (GO:0034357) and chloroplast thylakoid membrane (GO:0009535) were significantly enriched. Under drought stress, Zinc transporter 4 (Zm00001d036965) and ATP-dependent zinc metalloprotease FTSH 6 (Zm00001d037232) were upregulated; Photosystem II reaction center PSB28 protein (Zm00001d000409), photosynthetic NDH subunit of lumenal location 2 (Zm00001d016943), and photosynthetic NDH subunit of lumenal location 3 (Zm00001d018623) were downregulated. Moreover, the genes associated with chlorophyll binding (GO:0016168) were significantly suppressed, e.g. chlorophyll a/b binding protein (Zm00001d044401; Zm00001d044402; Zm00001d005814; Zm00001d048998) which mainly take part in light harvesting (GO:0009765). Furthermore, the electron transfer processes of photosynthesis were affected (GO:0009773, GO:0009767). Downregulation of these genes mentioned above could explain the decline in photosynthetic efficiency in C7–2 under drought stress (Fig. 2).
With respect to the drought-induced responses in C7–2t, 90 biological processes were enriched under drought (Table S5), much less compared with C7–2, suggesting that C7–2t was relatively less affected by drought stress. It is noteworthy that GO terms related to cell wall were significantly enriched, including plant-type secondary cell wall biogenesis (GO:0009834), plant-type cell wall organization or biogenesis (GO:0071669), cell wall organization or biogenesis (GO:0071554) and plant-type cell wall biogenesis (GO:0009832). Many of the genes involved in these processes were specifically detected in C7–2t, such as expansin-like A2 (Zm00001d029783), xyloglucan glycosyltransferase (Zm00001d038676, Zm00001d020560). In addition, GO terms related to secondary metabolism were also significantly enriched in C7–2t under drought stress, e.g., flavonoid 3′,5′-hydroxylase activity (GO:0033772) and tyrosine ammonia-lyase activity (GO:0052883), which were required in cell wall synthesis.
A KEGG enrichment analysis was performed to compare the effects of drought on metabolic processes between C7–2t and C7–2 via calculations of the q-value. In C7–2t, seven pathways were significantly enriched (Fig. 5; Table S6): plant hormone signal transduction (26 genes), phenylpropanoid biosynthesis (19), phenylalanine metabolism (6), starch and sucrose metabolism (15), taurine and hypotaurine metabolism (4), benzoxazinoid biosynthesis (5), and inositol phosphate metabolism (9). In C7–2, 24 pathways were significantly enriched (Fig. 4; Table S6), of which the top five pathways significantly enriched were starch and sucrose metabolism (50), photosynthesis-antenna proteins (13), benzoxazinoid biosynthesis (14), plant hormone signal transduction (66), and amino sugar and nucleotide sugar metabolism (41). Compared with C7–2, phenylpropanoid biosynthesis and taurine and hypotaurine metabolism were particularly enriched pathways in C7–2t, with eight upregulated and 11 downregulated DEGs involved in the phenylpropanoid biosynthesis pathway and four upregulated genes involved in the taurine and hypotaurine metabolism pathway.

KEGG heatmap of the DEGs in C7–2 and C7–2t. a C7–2, drought vs control; b C7–2 vs C7–2t under drought stress; c C7–2t, drought vs contro0l; d C7–2 vs C7–2t under the control conditions. The enrichment degree of each pathway is inversely proportional to q-value
Cell wall biosynthesis under drought
GO analysis showed that the cell wall (GO:0005618) genes in C7–2t were significantly enriched under drought stress. A total of 23 DEGs in C7–2t were enriched into the entry of plant type cell wall organization or biogenesis (GO:0071669) in the biological process classification, including five downregulated and 18 upregulated. Particularly, the genes encoding expansin proteins participating in cell wall biosynthesis were significantly upregulated, e.g., expansin-like A2, expansin-like A1, expansin-B4, expansin-B11, expansin-B12, with log2 fold change from 1.1 to 3.29. In addition, the genes (Zm00001d020531; Zm00001d005775; Zm00001d032776; Zm00001d043477; Zm00001d005478; Zm00001d005451) that induced by drought stress encode cellulose synthase A catalytic subunit. These results suggest that C7–2t could reduce the negative effects of drought stress through strengthening the synthesis of cell walls. Although the DEGs encoding expansins were also detected in C7–2, no significant enrichment results were obtained under drought stress.
Stable sugar synthesis under drought
The DEGs encoding bidirectional sugar transporter (Zm00001d016590; Zm00001d040656; Zm00001d010440; Zm00001d015914; Zm00001d044421) and sucrose synthase (Zm00001d029091; Zm00001d029087; Zm00001d045042) were all up-regulated in C7–2 and C7–2t. The upregulation of sugar synthesis and transport under drought requires a steady supply of energy from photosynthesis.
In C7–2, drought-responsive 69 genes were involved in photosynthesis (GO:0015979), photosynthesis and dark reaction (GO:0019685), photosynthesis, light harvesting, photosynthesis (GO:0009765), light harvesting in photosystem I (GO:0009768), and other biological process. Among them, 66 DEGs were inhibited by drought, but only three were induced by drought (Zm00001d042211, Zm00001d001820, protochlorophyllide reductase A; Zm00001d016166, phosphoenolpyruvate carboxylase).
In C7–2, the expressions of the DEGs encoding triose phosphate/phosphate translocator under drought were inconsistent: one up-regulated (Zm00001d005542), two down-regulated (Zm00001d032383; Zm00001d039258). A gene encoding sucrose synthase (Zm00001d051837) and several genes encoding sugar transporter were down-regulated (Zm00001d029251, Zm00001d009603, Zm00001d029254, Zm00001d009605). The downregulation expression of these genes was not detected in C7–2t. These results suggested that the stable photosynthesis of C7–2t under drought stress may provide a better guarantee for the synthesis and metabolism of sugars.
qRT-PCR verification of the DEGs
A total of 18 DEGs of interest were selected to evaluate the accuracy of the RNA-seq results (Table S7) with qRT-PCR (Fig. 6). In particular, the expression level of sucrose synthase 6 was decreased in C7–2 under drought; the expression levels of peroxidase 17, peroxidase 42, superoxide dismutase 2 and other five DEGs involved in photosynthesis were higher in C7–2t than those in C7–2. The results of qRT-PCR were consistent with those of the transcriptomic analysis.

qRT-PCR comparison of the expression of 18 drought-responsive DEGs between C7–2 and C7–2t
Discussion
Differentially physiological and biochemical responses between C7–2t and C7–2
In the present study, a drought-tolerant maize mutant C7–2t by 60Co-γ irradiation was developed. Compared to its wild type C7–2, C7–2t exhibited a higher drought tolerance under drought stress. There were significant differences in physiological and biochemical indexes between C7–2t and C7–2 (Fig. 2).
RWC is widely used to identify the drought tolerance of various plant species, including maize [20, 38]. Plants with higher RWC can increase their adaptability by reducing the drought-caused damage to their antioxidant systems [39]. C7–2t showed a higher RWC in leaves, i.e. a high moisture retention ability, than C7–2 under drought. Proline and soluble sugar plays important roles in osmotic regulation [40], correlating with drought tolerance [41, 42]. Under drought, C7–2t accumulated more proline and soluble sugars in the leaves than those in C7–2.
An improved efficiency of the antioxidative defense system could protect photosynthetic pigments, proteins, and DNA from excess ROS damage [40, 43]. CAT activity is inversely proportional to MDA content under severe drought stress [20]. The assay of CAT activity and MDA content indicated that C7–2t suffered less ROS damage than C7–2 under drought stress.
In the field, light distribution in plant canopy can be improved by reducing Chl content [44, 45]. On the other hand, nitrogen and energy saved by reducing Chl synthesis would enhance the specific responses to drought stress [46]. Thus, low Chl content in the leaves of C7–2t might be a part of its drought adaptation strategy. Taken together, higher levels in RWC, osmolyte accumulation, antioxidant activities and photosynthetic efficiency and a less level of MDA contributed to higher drought tolerance of C7–2t than C7–2.
The contribution of osmoregulation of soluble sugars to drought tolerance of C7–2t
Soluble sugars, including glucose, sucrose, maltose, and trehalose, play active roles in osmoregulation under drought stress [47,48,49]. In a drought-resistant wheat, the increased expression of soluble sugar synthesis-related genes explained its drought tolerance [50]. In the present study, the soluble sugar content in C7–2t was higher than that in C7–2 under the control and drought stress conditions (Fig. 2). Clearly, C7–2t has an efficient drought adaptation strategy partly based on osmoregulation of soluble sugars. In the present study, the expression of photosynthesis-related genes in C7–2t was almost unaffected, thus C7–2t could synthesize soluble sugar more efficiently under drought conditions.
C7–2t regulated the specific component of soluble sugar at the transcriptome level. Glucose could form trehalose with a 1,1-glycosidic linkage and reduce the injury caused by high concentrations of trehalose-6-phosphate under drought stress [48]. In the present study, the expression of trehalose 6-phosphate synthase/phosphatase and trehalose 6-phosphate phosphatase showed a sharp fluctuation in C7–2 (log2 fold changes from − 3.46 to 7.63) under drought stress, whereas their expressions were inhibited in C7–2t, implying that the elevated content of soluble sugars in C7–2t might not be due to the enhanced trehalose synthesis.
Moreover, the expression of β-amylase (Zm00001d014083) was activated under drought in both C7–2 and C7–2t, indicating that starch in the leaves could be decomposed into maltose. However, several genes encoding β-amylase in C7–2 were downregulated under drought (Zm00001d009016, Zm00001d029983, Zm00001d047077), possibly resulting in an decreased content of maltose and an increased sensitivity of photosynthetic system to drought stress [51]. In summary, C7–2t could synthesize more soluble sugars in its leaves and maintain the stability of photosynthesis under drought stress, thereby making it more drought-tolerant than C7–2.
The contribution of cell wall biosynthesis-related genes to water retention in C7–2t
The involvement of an organized multienzyme complex in polysaccharide biosynthesis provided a strong guarantee for cell wall synthesis [52]. The elevated expression of glycosyltransferase contributed to drought resistance of Arabidopsis thaliana cytokinin-deficient mutants [53]. In the present study, several glucuronosyltransferases in C7–2t (Zm00001d039231, Zm00001d007231) and C7–2 (Zm00001d002064, Zm00001d007231, Zm00001d008250) were upregulated under drought stress. GO enrichment analysis revealed that in C7–2t many DEGs were related to cell wall organization, especially the expansin family [54] that was induced by various abiotic stresses and ABA [55, 56]. Stomatal density was shown to decrease by overexpression of RhEXPA4 [57]. Furthermore, expansins could reduce water loss by discontinuing cell wall activity and stiffening cell structures [27].
The lignin content and intermediate product of the phenylpropanoid synthesis pathway were positively correlated with drought tolerance in maize [58, 59]. The accumulation of caffeic acid and p-coumaric acid in the xylem sap regulated the growth of maize leaves by influencing lignin biosynthesis under drought stress [60, 61]. p-Coumaric acid could rapidly lignify tissues under stress and improve adaptability under drought stress [62, 63]. In the present study, KEGG and GO enrichment analyses showed that lignin-related metabolic activities, such as phenylpropanoid synthesis pathway, were enhanced in C7–2t under drought. Most possibly, phenylpropanoid biosynthesis pathway and its intermediates, caffeic acid and p-coumaric acid, were involved in the response of C7–2t to drought stress.
The role of ABA in drought adapation strategy in C7–2t
9-cis Epoxycarotenoid dioxygenase 1 was a key enzyme involved in ABA synthesis [64, 65]. In the present study, the log2 fold change of the genes encoding 9-cis epoxycarotenoid dioxygenase 1 (Zm00001d018819, Zm00001d033222) reached 4.48 and 3.15 in C7–2t, whereas the corresponding changes were 7.03 and 4.57 in C7–2, suggesting that the sensitivity of C7–2 to drought stress may result from more endogenous ABA produced under drought stress.
PP2C participates in the ABA signal transduction pathway as a negative regulator under stress [26]. A recent study proved that PP2C-A10 (localized on chromosome 6) was related to ER stress [26]. In the present study, 29 PP2C genes (25 upregulated and 4 downregulated) and 14 (all upregulated) were detected in C7–2 and C7–2t, respectively. Especially, the log2 fold change of PP2C-A10 was significantly lower in C7–2t than in C7–2. These results that ABA-dependent upregulated genes provided transcriptional support for drought tolerance in plants [6], including C7–2t.
Conclusions
In the present study, we developed a more drought-tolerance inbred line C7–2t compared with C7–2 in both controlled and field conditions. The differences in physiological, biochemical and transcriptomic changes between C7–2 and C7–2t could explain the drought tolerance mechanisms in C7–2t (Fig. 7). The drought-tolerant mutant C7–2t and the drought-responsive DEGs identified here will be useful for basic research and drought tolerance breeding in maize.

Possible drought-adaptation mechanisms in the drought-tolerant mutant C7–2t
Methods
Creation of drought-tolerant maize mutants
The seeds of maize (Zea mays L.) C7–2 were purchased from Henan Qiule Seed Industry Science and Technology Company, Ltd. (Zhengzhou, China). The seeds were artificially mutated by 60Co-γ irradiation for 1 h with three doses of 150, 200 and 250 Gy. The 200 Gy radiation had a fatality rate of approximately 50%. Thus, the 200 Gy-treated seeds were planted in the field in 2013 to produce the M0 generation. Through reducing watering times to maintain soil drought, the drought-tolerant plants were selected and self-pollinated. Field selection process was continuously performed for five years (mainly by QBZ and WW). Since M4 generation, the promising drought-tolerant lines (such as C7–2t) that performed better than C7–2 in the field were collected and preserved in seed storage room (College of Life Sciences, Henan Agricultural University, China). The drought tolerance of C7–2t was further evaluated in pot experiments, based on phenotypes, physiological and biochemical changes during drought [66].
Maize materials and growth conditions
The seeds were washed with 2% sodium hypochlorite for 10 min and rinsed with distilled water three times. The samples were then planted in a plastic box filled with 1100 g of the growing medium (Pindstrup Substrate, 0–6 mm, pH 6.0, Pindstrup Mosebrug A/S, Denmark) and 1000 ml of water. The boxes were placed into a growth chamber with 27 °C, 60% humidity and 14 h of light and 10 h of darkness. Seven-day-old seedlings were divided into two groups: (a) control plants with normal watering and (b) drought-treated plants that were subjected to stopping watering for seven days. Afterwards, the expanded third leaves were collected for physiological, biochemical and RNA-seq analysis. Leaves from three individual plants represented a sample and were stored at − 80 °C after being frozen in liquid N2. All experiments were performed in at least three independent biological replicates.
Physiological and biochemical assays
Fresh leaves were divided into two equal groups: one was dried at 80 °C for 12 h to determine dry weight, and the other was placed in distilled water in the dark for 12 h to determine the turgid weight. RWC was calculated by the following equation: RWC = (fresh weight - dry weight) / (turgid weight - dry weight) · 100% [67]. CAT activity and Chl, free proline, SSC and MDA content were assayed by the established methods [68,69,70,71,72]. Chl fluorescence was determined by a MINI-PAM-II (Walz, Germany). The maximum photochemical quantum yield (Fv/Fm) and effective photochemical quantum yield (Y(II)) of PS II were calculated [73, 74]. All assays were performed in three biological experiments.
RNA extraction and detection
Twelve samples in three biological replicates were used to extract RNA using an Ultrapure RNA Kit (CoWin Biotech Co., China) according to the manufacturer’s instructions. The RNA was treated with RNase-free DNase I (Takara, Japan) to remove any possible DNA. The integrity was then checked by gel electrophoresis and an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The concentrations of total RNA were determined using a NanoDrop 8000 spectrophotometer (NanoDrop, Wilmington, DE). Total RNA with a RIN value ≥7.3 and a 28S:18S ratio ≥ 1.2 was subjected to RNA-seq analysis by Annoroad Gene Technology Co., Ltd. (Beijing, China).
RNA-seq analysis
About 2 μg of RNA per sample was used as input material for RNA-seq analysis. Sequencing libraries were generated using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (#E7530L, NEB, USA) following the manufacturer’s recommendations. Index codes were added to attribute the sequences in each sample. Briefly, mRNA was purified from the total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out by divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). RNase H and polymerase I were used for catalytic first-strand cDNA and second-strand cDNA syntheses, respectively. A-tailing and adapters were implemented after purification and terminal reparation. The purified cDNA template was enriched by PCR, and then the library was completed. Cleaned RNA-seq reads were obtained from the raw reads after removing the contaminated reads, low-quality reads and reads whose N base was greater than 5% for the total bases.
The reference genomes and the annotation file were downloaded from the ENSEMBL database (http://www.ensembl.org/index.html). Bowtie2 was used to construct the genome index. The clean data were aligned to the reference genome by HISAT2 [75, 76], and gene expression was calculated by FPKM [77]. DEGs were estimated by the software DESeq2, after which p-values were calculated according to the Wald test [78]. The p-values were corrected by the BH method. Genes with q ≤ 0.05 and |log2_ratio| ≥ 1 were identified as DEGs.
Functional enrichment analysis
GO terms with FDR < 0.05 were considered to be significantly enriched. Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/) enrichment of the DEGs was implemented by hypergeometric test, in which p-values were adjusted by multiple comparisons as q-values. KEGG terms with q < 0.05 were considered to be significantly enriched.
Quantitative qRT-PCR analysis
Twelve total RNA samples were extracted from three independent groups for each line under drought and control conditions. First-strand cDNA synthesis were performed with SuperReal PreMix Plus (Tiangen, Beijing, China).
A total of 18 genes were selected, and gene-specific primers were designed with the online tool Primer3 (http://primer3.ut.ee). The ubiquitin gene was chosen as a loading control in conjunction with primers Ubi 5′ (5′-TAAGCTGCCGATGTGCCTGCG-3′) and Ubi 3′ (5′-CTGAAAGACAGAACATAATGAGCACAG-3′). Each PCR (20 μL) contained 10 μL of 2× SuperReal PreMix Plus, primers at 0.6 μM and appropriately diluted cDNA. qRT-PCR was performed using Thermo Fisher Scientific StepOnePlus™ Real-Time PCR Instrument according to the manufacturer’s instructions. The thermal cycling conditions were 95 °C for 15 min followed by 40 cycles of 10 s at 95 °C and 32 s at 60 °C. At the second dissociation stage, 95 °C for 10 s followed by 65–95 °C with increments of 0.5 °C for 0.05 s were used. All reactions were performed in triplicates. The relative expression levels were calculated by the 2−ΔΔCT method [79].
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. The raw datasets of transcriptomic analysis are available from the corresponding author on reasonable request after the publication of the work.
Abbreviations
- ASI:
-
Anthesis-silk interval
- C7–2:
-
Inbred line Chang7–2
- C7–2t:
-
A drought-tolerant inbred line
- CAT:
-
Catalase
- CC:
-
C7–2 control
- CD:
-
C7–2 drought treatment
- Chl:
-
Chlorophyll
- DEGs:
-
Differentially expressed genes
- FPKM:
-
Fragments per kilobase of exon model per million reads mapped
- GO:
-
Gene ontology
- MDA:
-
Malondialdehyde
- ROS:
-
Reactive oxygen species
- RWC:
-
Relative water content
- SSC:
-
Soluble sugar content
- TC:
-
C7–2t control
- TD:
-
C7–2t drought treatment
References
Feller U, Vaseva II. Extreme climatic events: impacts of drought and high temperature on physiological processes in agronomically important plants. Front Environ Sci. 2014;2:39. https://doi.org/10.3389/fenvs.2014.00039.
Wu S, Ning F, Zhang Q, Wu X, Wang W. Enhancing omics research of crop responses to drought under field conditions. Front Plant Sci. 2017;8:174. https://doi.org/10.3389/fpls.2017.00174.
Boyer JS. Plant productivity and environment. Science. 1982;218:443–8. https://doi.org/10.1126/science.218.4571.443.
Salekdeh GH, Reynolds M, Bennett JM, Boyer J. Conceptual framework for drought phenotyping during molecular breeding. Trends Plant Sci. 2009;14:488–96. https://doi.org/10.1016/j.tplants.2009.07.007.
Avramova V, Nagel KA, Abdelgawad H, Bustos D, Duplessis M, Fiorani F, Beemster GTS. Screening for drought tolerance of maize hybrids by multi-scale analysis of root and shoot traits at the seedling stage. J Exp Bot. 2016;67:2453. https://doi.org/10.1093/jxb/erw055.
Zhu JK. Abiotic stress signaling and responses in plants. Cell. 2016;167:313–24. https://doi.org/10.1016/j.cell.2016.08.029.
Ribaut JM, Ragot M. Marker-assisted selection to improve drought adaptation in maize: the backcross approach, perspectives, limitations, and alternatives. J Exp Bot. 2007;58:351–60. https://doi.org/10.1093/jxb/erl214.
Reynolds M, Langridge P. Physiological breeding. Curr Opin Plant Biol. 2016;31:162–71. https://doi.org/10.1016/j.pbi.2016.04.005.
Comas LH, Becker SR, Cruz VMV, Byrne PF, Dierig DA. Root traits contributing to plant productivity under drought. Front Plant Sci. 2013;4:442. https://doi.org/10.3389/fpls.2013.00442.
Kumar A, Sharma KD. Leaf water content - a simple indicator of drought tolerance in crop plants. Indian J Agr Sci. 2010;80:1095–7.
Chimungu JG, Brown KM, Lynch JP. Reduced root cortical cell file number improves drought tolerance in maize. Plant Physiol. 2014;166:1943–55. https://doi.org/10.1104/pp.114.249037.
Chimungu JG, Brown KM, Lynch JP. Large root cortical cell size improves drought tolerance in maize. Plant Physiol. 2014;166:2166–78. https://doi.org/10.1104/pp.114.250449.
Boccalandro HE, Rugnone ML, Moreno JE, Ploschuk EL, Serna L, Yanovsky MJ, Casal JJ. Phytochrome B enhances photosynthesis at the expense of water-use efficiency in Arabidopsis. Plant Physiol. 2009;150:1083–92. https://doi.org/10.1104/pp.109.135509.
Liu J, Zhang F, Zhou J, Chen F, Wang B, Xie X. Phytochrome B control of total leaf area and stomatal density affects drought tolerance in rice. Plant Mol Biol. 2012;78:289–300. https://doi.org/10.1007/s11103-011-9860-3.
Mattioni C, Lacerenza NG, Troccoli A, Leonardis AMD, Fonzo ND. Water and salt stress-induced alterations in proline metabolism of Triticum durum seedling. Physiol Plant. 1997;101:787–92. https://doi.org/10.1111/j.1399-3054.1997.tb01064.x.
Kumar SG, Reddy AM, Sudhakar C. NaCl effects on proline metabolism in two high yielding genotypes of mulberry (Morus alba L.) with contrasting salt tolerance. Plant Sci. 2003;165:1245–51. https://doi.org/10.1016/s0168-9452(03)00332-7.
Munné-Bosch S, Peñuelas J. Photo- and antioxidative protection, and a role for salicylic acid during drought and recovery in field-grown Phillyrea angustifolia plants. Planta. 2003;217:758–66. https://doi.org/10.2307/23388121.
Anjum SA, Wang LC, Farooq M, Hussain M, Xue LL, Zou CM. Brassinolide application improves the drought tolerance in maize through modulation of enzymatic antioxidants and leaf gas exchange. J Agron Crop Sci. 2011;197:177–85. https://doi.org/10.1111/j.1439-037X.2010.00459.x.
Adebayo MA, Menkir A. Assessment of hybrids of drought tolerant maize ( Zea mays L.) inbred lines for grain yield and other traits under stress managed conditions. Nigerian J Genet. 2014;28:19–23. https://doi.org/10.1016/j.nigjg.2015.06.004.
Anjum SA, Ashraf U, Tanveer M, Khan I, Hussain S, Shahzad B, Zohaib A, Abbas F, Saleem MF, Ali I, Wang LC. Drought induced changes in growth, osmolyte accumulation and antioxidant metabolism of three maize hybrids. Front Plant Sci. 2017;8:69. https://doi.org/10.3389/fpls.2017.00069.
Ali F, Ahsan M, Ali Q, Kanwal N. Phenotypic stability of Zea mays grain yield and its attributing traits under drought stress. Front Plant Sci. 2017;8:1397. https://doi.org/10.3389/fpls.2017.01397.
Wang XL, Wang HW, Liu SX, Ferjani A, Li JS, Yan JB, Yang XH, Qin F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat Genet. 2016;48:1233. https://doi.org/10.1038/ng.3636.
Beyene Y, Mugo S, Semagn K, Asea G, Trevisan W, Tarekegne A, Tefera T, Gethi J, Kiula B, Gakunga J, Karaya H, Chavangi A. Genetic distance among doubled haploid maize lines and their testcross;performance under drought stress and non-stress conditions. Euphytica. 2013;192:379–92. https://doi.org/10.1007/s10681-013-0867-5.
Garrido-Cardenas JA, Mesa-Valle C, Manzano-Agugliaro F. Trends in plant research using molecular markers. Planta. 2018;247:543–57. https://doi.org/10.1007/s00425-017-2829-y.
Yang X, Gao S, Xu S, Zhang Z, Prasanna BM, Li L, Li J, Yan J. Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol Breeding. 2011;28:511–26. https://doi.org/10.1007/s11032-010-9500-7.
Xiang Y, Shan X, Qin F, Dai M. Deletion of an endoplasmic reticulum stress response element in a ZmPP2C-A gene facilitates drought tolerance of maize seedlings. Mol Plant. 2017;10:456–69. https://doi.org/10.1016/j.molp.2016.10.003.
Min H, Chen C, Wei S, Shang X, Sun M, Xia R, Liu X, Hao D, Chen H, Xie Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front Plant Sci. 2016;7:1080. https://doi.org/10.3389/fpls.2016.01080.
Shi J, Gao H, Wang H, Lafitte HR, Archibald RL, Yang M, Hakimi SM, Mo H, Habben JE. ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol J. 2017;15:207–16. https://doi.org/10.1111/pbi.12603.
Semagn K, Beyene Y, Dan M, Mugo S, Prasanna BM, Magorokosho C, Atlin G. Quality control genotyping for assessment of genetic identity and purity in diverse tropical maize inbred lines. Theor Appl Genet. 2012;125:1487–501. https://doi.org/10.1007/s00122-012-1928-1.
Lu X, Zhou X, Cao Y, Zhou M, McNeil D, Liang S, Yang C. RNA-seq analysis of cold and drought responsive transcriptomes of Zea mays ssp mexicana L. Front Plant Sci. 2017;8:136. https://doi.org/10.3389/fpls.2017.00136.
Zhang X, Liu X, Zhang D, Tang H, Sun B, Li C, Hao L, Liu C, Li Y, Shi Y. Genome-wide identification of gene expression in contrasting maize inbred lines under field drought conditions reveals the significance of transcription factors in drought tolerance. PLoS One. 2017;12:e0179477. https://doi.org/10.1371/journal.pone.0179477.
Li P, Cao W, Fang H, Xu S, Yin S, Zhang Y, Lin D, Wang J, Chen Y, Xu C. Transcriptomic profiling of the maize (Zea mays L.) leaf response to abiotic stresses at the seedling stage. Front. Plant Sci. 2017;8:290. https://doi.org/10.3389/fpls.2017.00290.
Song K, Kim HC, Shin S, Kim KH, Moon JC, Kim JY, Lee BM. Transcriptome analysis of flowering time genes under drought stress in maize leaves. Front Plant Sci. 2017;8:267. https://doi.org/10.3389/fpls.2017.00267.
Opitz N, Marcon C, Paschold A, Malik WA, Lithio A, Brandt R, Piepho HP, Nettleton D, Hochholdinger F. Extensive tissue-specific transcriptomic plasticity in maize primary roots upon water deficit. J Exp Bot. 2015;67:1095. https://doi.org/10.1093/jxb/erv453.
Kakumanu A, Ambavaram MMR, Curtis K, Arjun K, Utlwang B, Elijah M, Ruth G, Andy P. Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-Seq. Plant Physiol. 2012;160:846–67. https://doi.org/10.1104/pp.112.200444.
Zenda T, Liu S, Wang X, Liu G, Jin H, Dong A, Yang Y, Duan H. Key maize drought-responsive genes and pathways revealed by comparative transcriptome and physiological analyses of contrasting inbred lines. Int J Mol Sci. 2019;20:1268. https://doi.org/10.3390/ijms20061268.
Li Y, Sun C, Huang Z, Pan J, Wang L, Fan X. Mechanisms of progressive water deficit tolerance and growth recovery of Chinese maize foundation genotypes Huangzao 4 and Chang 7-2, which are proposed on the basis of comparison of physiological and transcriptomic responses. Plant Cell Physiol. 2009;50:2092–111. https://doi.org/10.1093/pcp/pcp145.
Gao Y, Lynch JP. Reduced crown root number improves water acquisition under water deficit stress in maize (Zea mays L.). J Exp Bot. 2016;67:4545–57. https://doi.org/10.1093/jxb/erw243.
Anjum SA, Tanveer M, Ashraf U, Hussain S, Shahzad B, Khan I, Wang L. Effect of progressive drought stress on growth, leaf gas exchange, and antioxidant production in two maize cultivars. Environ Sci Pollut Res Int. 2016;23:17132–41. https://doi.org/10.1007/s11356-016-6894-8.
Farooq M, Wahid A, Kobayashi N, Fujita D, Basra SMA. Plant drought stress: effects, mechanisms and management. Agron Sustain Dev. 2009;29:185–212. https://doi.org/10.1051/agro:2008021.
Altuntaş C, Sezgin A, Demiralay M, Terzi R, Sağlam A, Kadioğlu A. Application of sucrose modulates the expressions of genes involved in proline and polyamine metabolism in maize seedlings exposed to drought. Biol Plantarum. 2019;63:247–52. https://doi.org/10.32615/bp.2019.028.
Vendruscolo ECG, Ivan S, Marcos P, Carlos Alberto S, Molinari HBC, Celso Jamil M, Vieira LGE. Stress-induced synthesis of proline confers tolerance to water deficit in transgenic wheat. J Plant Physiol. 2007;164:1367–76. https://doi.org/10.1016/j.jplph.2007.05.001.
Blokhina O, Virolainen E, Fagerstedt KV. Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann Bot. 2003;91:179–94. https://doi.org/10.1093/aob/mcf118.
Ort RD, Zhu X, Melis A. Optimizing antenna size to maximize photosynthetic efficiency. Plant Physiol. 2011;155:79–85. https://doi.org/10.1104/pp.110.165886.
Zhu X, Long SP, Ort RD. Improving photosynthetic efficiency for greater yield. Annu Rev Plant Biol. 2010;61:235–61. https://doi.org/10.1146/annurev-arplant-042809-112206.
Slattery RA, Vanloocke A, Bernacchi CJ, Zhu XG, Ort DR. Photosynthesis, light use efficiency, and yield of reduced-chlorophyll soybean mutants in field conditions. Front Plant Sci. 2017;8:549. https://doi.org/10.3389/fpls.2017.00549.
Ibrahim HA, Abdellatif YMR. Effect of maltose and trehalose on growth, yield and some biochemical components of wheat plant under water stress. Ann Agric Sci. 2016;61:267–74. https://doi.org/10.1016/j.aoas.2016.05.002.
Redillas MCFR, Park SH, Lee JW, Kim YS, Jeong JS, Jung H, Bang SW, Hahn TR, Kim JK. Accumulation of trehalose increases soluble sugar contents in rice plants conferring tolerance to drought and salt stress. Plant Biotechnol Rep. 2012;6:89–96. https://doi.org/10.1007/s11816-011-0210-3.
Hershkovitz N, Oren A, Cohen Y. Accumulation of trehalose and sucrose in cyanobacteria exposed to matric water stress. Appl Environ Microbiol. 1991;57:645–8. https://doi.org/10.1002/bit.260370511.
Hou J, Huang X, Sun W, Du C, Wang C, Xie Y, Ma Y, Ma D. Accumulation of water-soluble carbohydrates and gene expression in wheat stems correlates with drought resistance. J Plant Physiol. 2018;231:182–91. https://doi.org/10.1016/j.jplph.2018.09.017.
Kaplan F, Guy CL. RNA interference of Arabidopsis beta-amylase8 prevents maltose accumulation upon cold shock and increases sensitivity of PSII photochemical efficiency to freezing stress. Plant J. 2005;44:730–43. https://doi.org/10.1111/j.1365-313x.2005.02565.x.
Culbertson AT, Ehrlich JJ, Choe JY, Honzatko RB, Zabotina OA. Structure of xyloglucan xylosyltransferase 1 reveals simple steric rules that define biological patterns of xyloglucan polymers. P Natl Acad Sci USA. 2018;115:6064–9. https://doi.org/10.1073/pnas.1801105115.
Li P, Li YJ, Zhang FJ, Zhang GZ, Jiang XY, Yu HM, Hou BK. The Arabidopsis UDP-glycosyltransferases UGT79B2 and UGT79B3, contribute to cold, salt and drought stress tolerance via modulating anthocyanin accumulation. Plant J. 2017;89:85–103. https://doi.org/10.1111/tpj.13324.
Marowa P, Ding A, Kong Y. Expansins: roles in plant growth and potential applications in crop improvement. Plant Cell Rep. 2016;35:949–65. https://doi.org/10.1007/s00299-016-1948-4.
Abuqamar S, Ajeb S, Sham A, Enan MR, Iratni R. A mutation in the expansin-like A2 gene enhances resistance to necrotrophic fungi and hypersensitivity to abiotic stress in Arabidopsis thaliana. Mol Plant Pathol. 2013;14:813–27. https://doi.org/10.1111/mpp.12049.
Zhao MR, Han YY, Feng YN, Li F, Wang W. Expansins are involved in cell growth mediated by abscisic acid and indole-3-acetic acid under drought stress in wheat. Plant Cell Rep. 2012;31:671–85. https://doi.org/10.1007/s00299-011-1185-9.
Lü P, Kang M, Jiang X, Dai F, Gao J, Zhang C. RhEXPA4, a rose expansin gene, modulates leaf growth and confers drought and salt tolerance to Arabidopsis. Planta. 2013;237:1547–59. https://doi.org/10.1007/s00425-013-1867-3.
Hu Y, Li WC, Xu YQ, Li GJ, Liao Y, Fu F-L. Differential expression of candidate genes for lignin biosynthesis under drought stress in maize leaves. J Appl Genet. 2009;50:213–23. https://doi.org/10.1007/bf03195675.
Yang L, Fountain JC, Ji P, Ni X, Chen S, Lee RD, Kemerait RC, Guo B. Deciphering drought-induced metabolic responses and regulation in developing maize kernels. Plant Biotechnol J. 2018;16:1616–28. https://doi.org/10.1111/pbi.12899.
Alvarez S, Marsh EL, Schroeder SG, Schachtman DP. Metabolomic and proteomic changes in the xylem sap of maize under drought. Plant Cell Environ. 2010;31:325–40. https://doi.org/10.1111/j.1365-3040.2007.01770.x.
Vincent D, Lapierre C, Pollet B, Cornic G, Negroni L, Zivy M. Water deficits affect caffeate O-methyltransferase, lignification, and related enzymes in maize leaves. A proteomic investigation. Plant Physiol. 2005;137:949–60. https://doi.org/10.1104/pp.104.050815.
Lin JS, Huang XX, Li Q, Cao Y, Bao Y, Meng XF, Li YJ, Fu C, Hou BK. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in Arabidopsis thaliana. Plant J. 2016;88:26–42. https://doi.org/10.1111/tpj.13229.
Varbanova M. Molecular and biochemical basis for stress-induced accumulation of free and bound p-coumaraldehyde in cucumber. Plant Physiol. 2011;157:1056–66. https://doi.org/10.1104/pp.111.184358.
Raghavendra AS, Gonugunta VK, Christmann A, Grill E. ABA perception and signalling. Trends Plant Sci. 2010;15:395–401. https://doi.org/10.1016/j.tplants.2010.04.006.
Babak B, Satoshi I, Miki F, Yasunari F, Hironori T, Yuriko O, Kazuko YS, Masatomo K, Kazuo S. Characterization of the promoter region of an Arabidopsis gene for 9-cis-epoxycarotenoid dioxygenase involved in dehydration-inducible transcription. DNA Res. 2013;20:315–24. https://doi.org/10.1093/dnares/dst012.
Chen D, Wang S, Cao B, Cao D, Leng G, Li H, Yin L, Shan L, Deng X. Genotypic variation in growth and physiological response to drought stress and re-watering reveals the critical role of recovery in drought adaptation in maize seedlings. Front Plant Sci. 2016;6:1241. https://doi.org/10.3389/fpls.2015.01241.
Galmés J, Flexas J, Savé R, Medrano H. Water relations and stomatal characteristics of Mediterranean plants with different growth forms and leaf habits: responses to water stress and recovery. Plant Soil. 2007;290:139–55. https://doi.org/10.1007/s11104-006-9148-6.
Bates LS, Waldren RP, Teare ID. Rapid determination of free proline for water-stress studies. Plant Soil. 1973;39:205–7. https://doi.org/10.1007/BF00018060.
Fales FW. The assimilation and degradation of carbohydrates by yeast cells. J Biol Chem. 1951;193:113–24. https://doi.org/10.1515/bchm2.1951.286.1-6.270.
Dhindsa RS, Plumb-Dhindsa P, Thorpe TA. Leaf senescence: correlated with increased levels of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J Exp Bot. 1981;32:93–101. https://doi.org/10.1093/jxb/32.1.93.
Zheng G, Fan C, Di S, Wang X, Xiang C, Pang Y. Over-expression of arabidopsis EDT1 gene confers drought tolerance in alfalfa (Medicago sativa L.). Front Plant Sci. 2017;8:2125. https://doi.org/10.3389/fpls.2017.02125.
Sartory DP, Grobbelaar JU. Extraction of chlorophyll a from freshwater phytoplankton for spectrophotometric analysis. Hydrobiologia. 1984;114:177–87. https://doi.org/10.1007/bf00031869.
Kitajima M, Butler WL. Quenching of chlorophyll fluorescence and primary photochemistry in chloroplasts by dibromothymoquinone. Biochim Biophys Acta. 1975;376:105–15. https://doi.org/10.1016/0005-2728(75)90209-1.
Genty B, Briantais JM, Baker NR. The relationship between the quantum yield of photosynthetic electron transport and quenching of chlorophyll fluorescence. Biochim Biophys Acta. 1989;990:87–92. https://doi.org/10.1016/S0304-4165(89)80016-9.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. https://doi.org/10.1038/nmeth.3317.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. https://doi.org/10.1186/gb-2009-10-3-r25.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. https://doi.org/10.1038/nbt.1621.
Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–8. https://doi.org/10.1093/bioinformatics/btp612.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8. https://doi.org/10.1006/meth.2001.1262.
Acknowledgments
We thank Zhaokun Wu, Liangjie Niu, and Hang Zhang for their valuable inputs on creation and selection of the maize mutant C7-2t.
Funding
The study was supported by the National Natural Science Foundation of China (Grant No. U1904107, to WW) and the Key Project and Special Foundation of Research, Development and Promotion in Henan Province, China (Grant No.192102110014, to HL). We declare that all funding bodies have no roles of in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
QBZ conducted the experiments. WW and HL conceived and designed the experiments. QBZ, HL, XLW and WW analyzed the data. QBZ and WW drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
: Table S1 FPKM value and annotation of all the genes detected from RNA sequencing.
Additional file 2
: Table S2 The DEGs detected in C7–2 and C7–2t.
Additional file 3
: Table S3 The common DEGs shared by C7–2 and C7–2t.
Additional file 4
: Table S4 The DEGs specific to C7–2 and C7–2t.
Additional file 5
: Table S5 GO enrichment analysis of the DEGs in C7–2 and C7–2t.
Additional file 6
: Table S6 KEGG enrichment analysis of the DEGs in C7–2 and C7–2t.
Additional file 7
: Table S7 Primers used for qRT-PCR.
Additional file 8
: Fig. S1 Comparison of drought tolerance index, barren ear tip distance and ASI between C7–2 and C7–2t. (a) Evaluation via pot experiments in third-leaf stage of seedlings. (b) and (c) Showing the difference in ear traits and the abscissa standing for ASI.
Additional file 9
: Fig. S2 Plant height, ear height and biomass of C7–2 and C7–2t at harvest in the field.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Q., Liu, H., Wu, X. et al. Identification of drought tolerant mechanisms in a drought-tolerant maize mutant based on physiological, biochemical and transcriptomic analyses. BMC Plant Biol 20, 315 (2020). https://doi.org/10.1186/s12870-020-02526-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-020-02526-w