
Abstract
Background
The Growth-regulating factor (GRF) family encodes plant-specific transcription factors which contain two conserved domains, QLQ and WRC. Members of this family play vital roles in plant development and stress response processes. Although GRFs have been identified in various plant species, we still know little about the GRF family in soybean (Glycine max).
Results
In the present study, 22 GmGRFs distributed on 14 chromosomes and one scaffold were identified by searching soybean genome database and were clustered into five subgroups according to their phylogenetic relationships. GmGRFs belonging to the same subgroup shared a similar motif composition and gene structure. Synteny analysis revealed that large-scale duplications played key roles in the expansion of the GmGRF family. Tissue-specific expression data showed that GmGRFs were strongly expressed in growing tissues, including the shoot apical meristems, developing seeds and flowers, indicating that GmGRFs play critical roles in plant growth and development. On the basis of expression analysis of GmGRFs under shade conditions, we found that all GmGRFs responded to shade stress. Most GmGRFs were down-regulated in soybean leaves after shade treatment.
Conclusions
Taken together, this research systematically analyzed the characterization of the GmGRF family and its primary roles in soybean development and shade stress response. Further studies of the function of the GmGRFs in the growth, development and stress tolerance of soybean, especially under shade stress, will be valuable.
Similar content being viewed by others


Background
Growth-regulating factors (GRFs) are plant-specific transcription factors which regulate plant growth, development and abiotic stress response [1,2,3,4,5]. The first GRF, named OsGRF1, was identified from deepwater rice (Oryza sativa); expression of OsGRF1 was induced by gibberellin (GA), and it mediated stem elongation in a GA-dependent manner [6]. Numerous studies demonstrated that there are two conserved domains, QLQ and WRC, in the N-terminal region of GRF proteins. The QLQ (Gln, Leu, Gln) domain serves as a protein-protein interaction feature which could interact with the GRF-interacting factor (GIF) [7], while the plant-specific WRC (Trp, Arg, Cys) domain comprises a C3H motif for DNA binding and a nuclear localization signal (NLS) [8]. Compared with the conserved nature of the amino acid residue sequence in the N-terminal region, the C-terminal region of the GRFs is variable, with some investigations indicating that the C-terminal region possessed transactivation activity [7,8,9]. In addition, some less-conserved motifs, such as TQL and FFD, are usually present in the C- terminal region of GRFs [10].
As they are development-related transcription factors, it is not strange that GRFs mediate the shape and size of leaves by regulating cell proliferation [11, 12]. Overexpression of AtGRF1, AtGRF2 and AtGRF5 resulted in larger leaves than in wild-type (WT) Arabidopsis, while the leaves of grf mutants, such as grf3–1, grf5–1, grf1–1/grf2, grf2/grf3 and grf1/2/3, were much smaller than the WT [1, 2, 11, 12]. Furthermore, GRFs also regulate root growth, floral development and seed size [9, 13,14,15]. In addition to GRF, the GIFs, are also involved in the control of leaf size and architecture. Compared to the WT, the leaves of gif1 were narrower and smaller [7, 16]. Biochemical analysis showed that GRF and GIF combine to form a transcriptional complex in vivo to modulate cell proliferation and ultimately to control leaf size [5, 16]. Furthermore, it has been shown that the microRNA miR396 directly inhibits the expression of GRFs through post-transcriptional regulation [11, 14]. Constitutive overexpression of Arabidopsis miR396a and miR396b, or heterologous expression of ptc-miR396c (from Populus trichocarpa) and ath-miR396a (from Arabidopsis thaliana) in tobacco (Nicotiana tabacum) significantly reduce mRNA levels of GRFs and lead to narrower and smaller leaves, which mimic Arabidopsis grf1/2/3 [17,18,19,20]. Thus, the transcription levels of GRFs are regulated strictly and quantitatively by the miRNA-GRF-GIF cascade.
Light is an important environmental factor that plays critical roles in plant growth and development, and ultimately determines crop yield [21]. However, in maize (Zea mays)-soybean (Glycine max) relay strip intercropping systems, the light environment in the soybean canopy changes due to it being lower than the maize canopy, so that soybean was under shade stress (resulting from reductions in light quantity and in the red: far-red light ratio) [22, 23]. Furthermore, dense-planting patterns in crops also lead to shade stress among neighboring seedlings [24]. Previous studies had demonstrated that soybean morphological traits changed markedly under shade conditions, resulting in increased plant height, decreased yield, and reduced root length [23, 25, 26]. Notably, leaf expansion is also suppressed when soybean responds to shade stress [27, 28]. However, the specific regulatory mechanisms underlying leaf development under shade conditions are still largely unknown, especially with respect to the GRF family-mediated pathways.
In the present study, 22 GmGRFs were dissected from the soybean genome. Their sequence characteristics, chromosome distribution, phylogenetic relationships, gene structures, conserved motif compositions and synteny were then systematically characterized. Based on these findings, the expression profiles of GmGRFs in various vegetative and reproductive tissues were documented. Furthermore, we also analyzed the expression patterns of GmGRFs under shade stress conditions. These results will not only help us to better understand the functions of the GmGRF family, but will also provide a foundation for improving crops, especially soybean, through genetic modification.
Results
Identification of GmGRFs
Based on the Hidden Markov Model (HMM) of WRC and QLQ domains, a total of 22 GmGRFs were identified from soybean genome and named as GmGRF1 – GmGRF22 according to their locations on chromosomes (Table 1). The length of the coding sequences (CDS) of GmGRFs varied from 927 bp (GmGRF10) to 1830 bp (GmGRF19). Accordingly, GmGRF10, with 308 amino acid residues, was the smallest GmGRF, whereas the largest GmGRF was GmGRF19 (609 amino acid residues). The theoretical molecular weight (MW) of these putative GmGRFs ranged from 34.58 to 66.90 kDa and the isoelectric point (pI) ranged from 6.35 (GmGRF13) to 9.10 (GmGRF1) (Table 1).
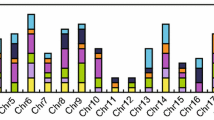
GmGRFs were unevenly distributed across the chromosomes. Twenty GmGRFs were distributed on 14 chromosomes and two GmGRFs were located on scaffold_28 (Fig.1). Of the 14 chromosomes, chromosomes 11 and 17 contained the largest number of GmGRFs, each with three genes. Both chromosomes 1 and 9 carried two GmGRFs each, while only one GmGRF was observed on each of chromosomes 3, 4, 6, 7, 10, 12, 13, 15, 16 and 19 (Fig.1).

Distribution of GmGRFs on soybean chromosomes or scaffold
Sequence and phylogenetic analysis of GmGRFs
All GmGRFs contained conserved QLQ and WRC domains in their N-terminal regions (Additional file 1: Figure S1). To gain an insight into the evolutionary relationships among GRFs from soybean (22), rice (12) and Arabidopsis (9), MEGA 7.0 software was used to construct a neighbor-joining phylogenetic tree. As shown in Fig. 2, a total of 43 GRFs from different species were clustered into six subgroups (I–VI). Five of the six subgroups contained GmGRFs, whereas subgroup VI had only AtGRF and OsGRF members. Among the six subgroups, subgroups IV and VI were relatively small, with only four GRFs each. By contrast, subgroups I and V contained the largest number of GRFs (ten each), followed by subgroups II (nine), III (six). The phylogenetic tree suggested that the GmGRFs showed a closer relationship with AtGRFs than with OsGRFs, which may be partly because both soybean and Arabidopsis are dicotyledonous plants.

Phylogenetic analysis of GRFs from Glycine max (Gm), Oryza sativa (Os) and Arabidopsis thaliana (At). Clustal W was used to align 43 GRFs, namely nine AtGRF, 12 OsGRF, and 22 GmGRF, while MEGA 7.0 software was employed to construct a neighbor-joining phylogenetic tree with 1000 bootstrap replications
Structural analysis of GmGRFs
To further explore the evolutionary relationship among GmGRFs, we constructed a phylogenetic tree and analyzed the gene structures and motif characteristics of GmGRFs (Fig. 3). According to Fig. 2, GmGRFs could be clustered into five groups. It was obvious that the 22 GmGRFs contained two to four introns (six genes with two introns, twelve with three introns, and four with four introns) (Fig. 3c). The conserved structure of GmGRFs was similar to those from other plant species, in which most genes contained three introns [29,30,31]. The lengths of individual GmGRFs were variable in intron length could partly reflect the length of different genes. For instance, the longest gene, GmGRF2, with a size of 5.7 kb, was due mainly to the fact that it contained a total intron length of 4.8 kb.

Phylogenetic analysis, gene structures and conserved motifs of GmGRFs. a The phylogenetic relationship of GmGRFs. A neighbor-joining phylogenetic tree was constructed by MEGA7.0 software with the Poisson model and 1000 bootstrap replications. b Conserved motif arrangements of GmGRFs. Ten conserved motifs labeled with different colors were found in the GmGRF sequences using the MEME program. Among them, motif 1 and motif 2 are the QLQ and WRC conserved domains. c Exon-intron organizations of GmGRFs. The green boxes represent 5’or 3′ untranslated regions, yellow boxes represent the coding sequences, and black lines represent the introns. The lengths of the exons and introns can be determined by the scale at the bottom
The Multiple EM for Motif Elicitation (MEME) web server was employed to identify the conserved motifs of GmGRFs (Fig. 3b and Additional file 2: Table S1). All GmGRFs contained motif 1 and motif 2 which were annotated as the GRF specific domains, QLQ and WRC, in their N-terminal regions. Each GmGRF has between four and ten conserved motifs, with the GRFs of subgroup V (GmGRF18, GmGRF19, GmGRF21 and GmGRF22) containing the largest number of motifs. The GmGRFs belonging to the same subgroup have a similar motif composition (e.g. GmGRF4 and GmGRF5, GmGRF18 and GmGRF21). In addition, some motifs appeared in only certain specific subgroups. For example, motif 3 is unique to subgroup I and II, while motifs 7, 8, 9 and 10 are specific to subgroup V. Overall, gene structures and motif characteristics strongly supported the phylogenetic relationships of GmGRFs.
Synteny analysis of GmGRFs
Gene duplication plays an important role in increasing the numbers of genes and their subsequent evolution. For instance, over 90% of regulatory and developmental genes in the Arabidopsis genome had been duplicated [32]. In order to analyze the duplication events of GmGRFs, Multiple Collinearity Scan toolkit X (MCScanX) software was employed. Ultimately, we found that 19 of the 22 GmGRFs (86.36%) were distributed in the duplication regions, suggesting that these genes were generated by large-scale duplication events, whole-genome duplication (WGD) or segmental duplication (Fig. 4 and Additional file 3: Table S2). Additionally, according to a previous methodology [33], GmGRF18-GmGRF19 and GmGRF21-GmGRF22 belonging to chromosome 17 and scaffold_28, respectively, were identified as tandem duplication genes. Further, we calculated the nonsynonymous substitution rate (Ka) and synonymous substitution rate (Ks) of these duplicated gene pairs. The results showed that the Ka/Ks ratios of most GmGRF pairs were less than 1, indicating that these GmGRFs had undergone purifying selection processes.

The syntenic relationships among GmGRFs. The scaffold_28 was not shown in the Circos map
Expression profiles of GmGRFs
Based on the analysis of the results from Figs. 2, 3 and 4, we selected eight GmGRFs belonging to different subgroups for GA response and tissue-specific expression analysis. After GA3 treatment, the expression levels of all selected eight GmGRFs were down-regulated (Additional file 4: Figure S2). Then we investigated the expression patterns of these GmGRFs in seven soybean tissues (roots, flowers, stems, pods, leaves, shoot apical meristem, and developing seeds). As shown in Fig. 5, although all GmGRFs were expressed in all seven tissues, the transcription levels of the different genes varied greatly among the different tissues. In general, GmGRFs were highly expressed in growing tissues, such as developing seeds, flowers and shoot apical meristems. Of the GmGRFs, the expression levels of GmGRF1, GmGRF6 and GmGRF18 were highest in developing seeds (Fig. 5a, c and g). Among these three genes, GmGRF6 was more preferentially expressed in developing seeds, indicating 100-fold higher than that in root. However, in flowers, the transcription levels of GmGRF5, GmGRF11 and GmGRF20 were highest (Fig. 5b, e and h). Furthermore, GmGRF9 was abundantly expressed in roots, while GmGRF17 showed the highest transcription level in shoot apical meristems (Fig. 5d and f). The results demonstrated multiple potential functions of GmGRFs in regulating growth and development of distinct soybean tissues.

Expression profiles of GmGRFs in seven tissues. Expression levels of eight selected GmGRFs were examined by qRT-PCR. The housekeeping GmTubulin was used as an endogenous reference gene. R, root; S, stem; L, leaf; F, flower; P, pod; SM, shoot apical meristem; DS, developing seed. Error bars represent standard errors
Expression profiles of GmGRFs in response to shade stress
Soybean plant morphology changed greatly in response to shade stress, dubbed as shade avoidance syndrome (SAS), including decreased leaf area and weight, and excessive elongation of stems (Fig. 6 and Additional file 5: Figure S3). Previous studies have demonstrated that GRFs play important roles in regulating leaf size and plant response to abiotic stresses, including abscisic acid (ABA) and osmotic stresses [4, 34,35,36]. Therefore, in order to further explore the roles of GmGRFs under shade conditions, we measured the transcription levels of eight GmGRFs by qRT-PCR. The results showed that the transcription of all the GmGRFs tested was influenced by shade. Expression of almost all GmGRFs was significantly down-regulated under shade stress, albeit to different degrees (Fig. 7). Among these genes, the transcription levels of GmGRF9 and GmGRF17 were more strongly suppressed, while GmGRF20 showed the weakest transcription repression. On the other hand, the expression level of GmGRF5 was up-regulated under shade stress conditions (Fig. 7b). Based on the expression patterns of GmGRFs, GmGRF5, GmGRF9 and GmGRF17 may play important roles in regulating leaf growth under shade conditions.

Soybean leaf area and weight decreased under shade conditions. a Representative photographs of soybean leaves under white light and shade conditions. Bar = 10 mm. Leaf fresh and dry weight (b, c), leaf area (d) and leaf mass per area (LMA; e) were analyzed after shade stress. Ten soybean leaves were measured under each condition. Error bars represent standard errors. The asterisk (*) indicates the significant difference at P < 0.05 by Student’s t-test analysis. L, shade; S, shade

Time course of expression patterns of GmGRFs under shade conditions. The housekeeping GmTubulin was used as an internal control. Error bars represent standard errors. The asterisk (*) indicates the significant difference at P < 0.05 by Student’s t-test analysis
Discussion
As members of plant-specific gene family, GRFs play important roles in plant growth and development, especially in regulating organ size [5, 11, 15]. Recent studies in rice have shown that OsGRF4 can improve rice yield by promoting cell division and nitrogen uptake efficiency [37,38,39]. Therefore, it is necessary to understand the mechanisms regulating levels of GRFs during plant development and response to stress. To date, GRF family has been identified in various plants (Additional file 6: Table S3), such as Arabidopsis, rice, Zea mays, Brassica napus, Citrus sinensis, Camellia sinensis, Populus trichocarpa and Pyrus×bretschneideri [1, 8, 40,41,42,43]. However, little information on the function of GRFs in soybean is available.
In order to better understand the characteristics and functions of GmGRFs, we studied members of the GmGRF family by bioinformatics assay, qRT-PCR assay, and plant morphology analysis. In the current study, a total of 22 GmGRFs was identified by searching soybean genome database, and all putative GmGRFs were shown to contain QLQ and WRC domains (Table 1 and Additional file 1: Figure S1). Phylogenetic analysis showed that 22 GmGRFs could be clustered into five subgroups according to their evolutionary relationships (Fig. 1). The exon-intron organizations and motif arrangements were consistent with the phylogenetic analysis, while the structure of GmGRFs belonging to different subgroups displayed lower identities.
Numerous studies have shown that gene duplication can not only increase the number of GRFs, but is also an avenue for generating novel genes, a phenomenon which is conducive to enabling the plant to adapt to various environments [44,45,46,47]. Indeed, the expansion of the GRF family occurred mainly through gene duplication, especially large-scale duplications (WGD or segmental duplications) [5, 30, 43]. Consistently, in the present study, most GmGRFs were distributed in duplication blocks, indicating that WGD or segmental duplications had played major roles in the expansion of the GmGRF family. In addition, soybean contains more GRFs than does another legume, Medicago truncatula, a model plant (Additional file 6: Table S3 and Additional file 7: Table S4). This may mainly be due to two WGD events which occurred during the evolution of the soybean genome (58 and 13 million years ago), while the Medicago genome experienced only a single WGD event at 58 million years ago [48, 49]. It is noted that this phenomenon also exists in other soybean gene families, such as the homeodomain-leucine zipper and homeobox gene families [50, 51]. Altogether, these results suggested that large-scale duplications were universal during the expansion process of the GmGRF family.
Previous research had reported that GRFs are pivotal regulators of plant growth and development [5, 34]. For example, overexpression of AtGRF5 leads to leaf enlargement, while the leaves of the Arabidopsis grf5 mutant are narrower than those of the WT [12]. Generally, the expression levels of GRFs in actively growing tissues was higher than that in mature tissues [29]. Indeed, several studies had revealed that, with the aging of organs, the transcription levels of GRFs decreased [19, 41]. In the present study, we found that GmGRFs are highly expressed in shoot apical meristems, developing seeds and flowers (Fig. 5). Interestingly, among all eight selected GmGRFs, GmGRF1, GmGRF6 and GmGRF18 were highly expressed in developing seeds, suggesting that these genes may play important roles in seed development. In addition to developing seeds, GRFs are also important in flower development and root growth [9, 52,53,54]. Correspondingly, we found that some GmGRFs, such as GmGRF9, GmGRF11and GmGRF20, were expressed abundantly in flowers and roots. The gene expression analysis indicated that GmGRFs may play important roles in the growth and development of soybean tissues.
Light not only provides energy for plants, but also regulates plant growth and development [55, 56]. Plants can sense changes in the light environment through photoreceptor systems, such as phytochromes, cryptochromes and phototropins [57,58,59]. As sessile photoautotrophs, plants need to compete with neighbors for light, nutrients and other resources. For example, when exposed to shade stress from neighboring plants, plants adapted to open ranges (e.g. Arabidopsis, rice and soybean) can develop shade avoidance response, leading to shade avoidance syndrome [60,61,62]. Shade avoidance response is conducive to survival of the plant under shade conditions [61, 63]. In the context of agricultural production, however, shade avoidance can cause a decline in crop yield [63]. In maize-soybean intercropping systems, for example, soybean yield decreased significantly, because of the shade stress arising from the taller neighboring maize plants [23]. In the present investigation, the transcription of almost all GmGRF genes tested decreased under shade treatment (Fig. 7), suggesting that GmGRFs possess the veiled functions with regard to plant shade response.
The leaf is the primary tissue of photosynthesis and its size directly affects photosynthetic efficiency [64, 65]. Numerous studies have shown that leaf area is regulated by many genes related to cell division and expansion [36]. Among them, GRFs can regulate leaf area by controlling cell proliferation [65]. In addition, UV-B radiation inhibits leaf growth by decreasing the expression of GRFs in Arabidopsis and maize [66, 67]. Morphological analysis showed that soybean leaf size, leaf area and dry and fresh weight decreased significantly under shade stress (Fig. 6), and, in line with this, the expression levels of almost all GmGRF genes were down-regulated under shade conditions (Fig. 7). Given the positive regulatory effect of GRFs on plant cell proliferation in diverse plant species [68], these results were consistent (Figs. 6 and 7). Interestingly, under shade conditions, the expression levels of most GmGRFs were down-regulated while GmGRF5 transcription was up-regulated under these conditions. Therefore, further study of the function of GmGRF5 under shade stress may be helpful in increasing the leaf area of intercropped soybean, thereby potentially increasing the yield of soybean.
Conclusions
In this study, we systematically analyzed the basic characteristics and functions of GmGRFs. A total of 22 GmGRFs were identified from the soybean genome with the help of the HMM of two conserved domains specific to GRF, namely QLQ and WRC. These GmGRFs were distributed on 14 chromosomes and one scaffold, and could be clustered into five subgroups according to their phylogenetic relationships. By further analysis, we found that GmGRFs belonging to the same subgroup had similar gene structures and motif compositions. Gene duplication analysis suggested that both large-scale and tandem duplications, especially the former, contributed to the expansion of the GmGRF family. The result of qRT-PCR studies showed that GmGRFs were involved in the growth and development of soybean, as well as in shade response. Importantly, we identified some potentially useful genes, such as GmGRF1, GmGRF5 and GmGRF6 by analyzing the GmGRF family. These results will provide a foundation for future research on the GRFs in soybean.
Methods
Plant materials and shade treatment
Soybean cultivar Nandou-12, a prevailing cultivar in southwestern China, was employed in this study. The seeds of Nandou-12 were originally obtained from Nanchong Institute of Agricultural Sciences, Sichuan Province, China. Soybean plants were grown in a phytotron with 65% relative humidity, a 12-h light (25 °C)/12-h dark (20 °C) photoperiod and a light intensity of 380 μmol m− 2 s− 1. In order to analyze the expression patterns of GmGRFs in different tissues of soybean, we collected roots (16 d after sowing), stems (16 d after sowing), leaves (16 d after sowing), shoot apical meristems (16 d after sowing), flowers (36 d after sowing), pods (44 d after sowing) and developing seeds (60 d after sowing). For the GA treatment, six-day soybean seedlings were sprayed with 100 μM GA3 and the hypocotyls of soybean seedlings were harvested after 0, 3, 6 and 9 h. Furthermore, to explore the transcription profiles of GmGRFs under shade stress, black nylon net and far-red light-emitting diode (LED) were employed to adjust the light environment (light intensity and quality) [26]. 10-day soybean seedlings were transferred to the shade environment with 113 μmol m− 2 s− 1 of photosynthetically active radiation (PAR) and 0.4 of red: far-red light ratio (R/FR). The first compound leaves of soybean seedlings were collected at 0, 3, 6 and 9 h after shade treatment. All samples were frozen immediately in liquid nitrogen and then stored at − 80 °C for further analysis.
Seven days after shade treatment, ten soybean seedlings from different pots were used for analyzing morphological characteristics. Leaf area of first compound leaves was measured by ImageJ software. Seeding height, stem diameter, leaf and root fresh weight were also determined. Subsequently, leaves, stems and roots were exposed to 105 °C for 0.5 h and then dried at 80 °C until constant weight. Finally, dry weight, root-shoot ratio and leaf mass per area (LMA) were also calculated.
Identification of GmGRFs
Soybean protein sequences and genome annotation were downloaded from Phytozome database (http://www.phytozome.net/). The HMM of WRC (PF08879) and QLQ (PF08880) domains were obtained from PFAM database (http://pfam.xfam.org/) and used to predict GmGRFs with HMMER software. Then, these protein sequences (E-value ≤1e− 10) were used to construct soybean-specific HMM for identifying GmGRFs [69]. Finally, predicted proteins were considered as GmGRFs only if they contained QLQ and WRC conserved domains verified by SMART software (http://smart.embl-heidelberg.de/) and PFAM databases. All putative GmGRFs were drawn on chromosomes using MapGene2Chrom web v2 (http://mg2c.iask.in/mg2c_v2.0/).
Sequence and phylogenetic analysis
The exon-intron distribution patterns of GmGRFs were analyzed using Dual Systeny Plotter software (https://github.com/CJ-Chen/TBtools) [70, 71]. To predict MW and pI of GmGRFs, the ExPASy proteomics server (https://web.expasy.org/protparam/) was used [72]. Conserved motifs of GmGRFs were predicted by MEME online program (http://meme-suite.org/tools/meme) [73]. Multiple sequence alignments of GmGRFs were analyzed by Clustal W [74]. MEGA 7.0 software was employed to construct phylogenetic trees using the neighbor-joining method with Poisson model, pairwise deletion, and 1000 bootstrap replications [75].
Gene duplication and evolution analysis
MCScanX program with the default parameters was used to analyze the duplication events of GmGRFs [76]. According to the results of MCScanX, the nonsynonymous substitution rate (Ka) and synonymous substitution rate (Ks) of duplicated genes were calculated by KaKs_Calculator 2.0 software [77].
Gene expression analysis
Total RNA preparation, first-strand cDNA synthesis and the qRT-PCR assay were performed as previously described [78]. According to the manufacturer’s protocol, total RNA was treated with DNase I and then 2 μg total RNA was reverse-transcribed using Moloney murine leukemia virus reverse transcriptase (200 units per reaction; Promega Corporation). The qRT-PCR was performed using Vazyme™ AceQ qPCR SYBR Green Master mix on a QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific, USA) [79]. The soybean housekeeping GmTubulin was used as an endogenous reference gene and each reaction had three repetitions. 10 μL reaction mixture included 5 μL Vazyme™ AceQ qPCR SYBR Green Master mix, 0.2 μL forward primer, 0.2 μL reverse primer, 1 μL cDNA template and 3.6 μL Dnase-free ddH2O. The qRT-PCR reaction procedure was set as follows: 95 °C for 30s, and then 40 cycles of 95 °C for 15 s, 60 °C for 30 s. The expression levels of GmGRFs were calculated by the comparative CT method [80]. Sequences of the primers for qRT-PCR were listed in Additional file 8: Table S5.
Statistical analysis
In this study, the Student’s t-test in Microsoft Excel software was used to analyze the differences between different treatments. P < 0.05 was considered as statistical significance.
Availability of data and materials
All data used and analyzed in this study are included in the article and its additional file.
Abbreviations
- ABA:
-
Abscisic acid
- GA:
-
Gibberellin
- GIF:
-
GRF-interacting factor
- GRF:
-
Growth-regulating factor
- HMM:
-
Hidden Markov Model
- Ka:
-
Nonsynonymous substitution rate
- Ks:
-
Synonymous substitution rate
- LED:
-
Light-emitting diode
- LMA:
-
Leaf mass per area
- MCScanX:
-
Multiple Collinearity Scan toolkit X
- MEME:
-
Multiple EM for Motif Elicitation
- MW:
-
Molecular weight
- NLS:
-
Nuclear localization signal
- PAR:
-
Photosynthetically active radiation
- pI:
-
Isoelectric point
- R/FR:
-
Red: far-red light ratio
- SAS:
-
Shade avoidance syndrome
- WGD:
-
Whole-genome duplication
- WT:
-
Wild-type
References
Kim JH, Choi D, Kende H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003;36(1):94–104.
Kim JH, Lee BH. GROWTH-REGULATING FACTOR4 of Arabidopsis thaliana is required for development of leaves, cotyledons, and shoot apical meristem. J Plant Biol. 2006;49(6):463–8.
Baucher M, Moussawi J, Vandeputte OM, Monteyne D, Mol A, Pérez-Morga D, et al. A role for the miR396/GRF network in specification of organ type during flower development, as supported by ectopic expression of Populus trichocarpa miR396c in transgenic tobacco. Plant Biol. 2013;15(5):892–8.
Kim JS, Mizoi J, Kidokoro S, Maruyama K, Nakajima J, Nakashima K, et al. Arabidopsis GROWTH-REGULATING FACTOR7 functions as a transcriptional repressor of abscisic acid- and osmotic stress-responsive genes, including DREB2A. Plant Cell. 2012;24(8):3393–405.
Omidbakhshfard MA, Proost S, Fujikura U, Mueller-Roeber B. Growth-regulating factors (GRFs): a small transcription factor family with important functions in plant biology. Mol Plant. 2015;8(7):998–1010.
Van dKE, Kim JH, Kende H. A novel gibberellin-induced gene from rice and its potential regulatory role in stem growth. Plant Physiol. 2000;122(3):695–704.
Kim JH, Kende H. A transcriptional coactivator, AtGIF1, is involved in regulating leaf growth and morphology in Arabidopsis. Proc Natl Acad Sci U S A. 2004;101(36):13374–9.
Choi D, Kim JH, Kende H. Whole genome analysis of the OsGRF gene family encoding plant-specific putative transcription activators in rice (Oryza sativa L.). Plant Cell Physiol. 2004;45(7):897–904.
Liu H, Guo S, Xu Y, Li C, Zhang Z, Zhang D, et al. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 2014;165(1):160–74.
Zhang DF, Bo L, Jia GQ, Zhang TF, Dai JR, Li JS, et al. Isolation and characterization of genes encoding GRF transcription factors and GIF transcriptional coactivators in maize (Zea mays L.). Plant Sci. 2008;175(6):809–17.
Debernardi JM, Mecchia MA, Vercruyssen L, Smaczniak C, Kaufmann K, Inze D, et al. Post-transcriptional control of GRF transcription factors by microRNA miR396 and GIF co-activator affects leaf size and longevity. Plant J. 2014;79(3):413–26.
Horiguchi G, Kim G-T, Tsukaya H. The transcription factor AtGRF5 and the transcription coactivator AN3 regulate cell proliferation in leaf primordia of Arabidopsis thaliana. Plant J. 2005;43(1):68–78.
Hewezi T, Maier TR, Nettleton D, Baum TJ. The Arabidopsis microRNA396-GRF1/GRF3 regulatory module acts as a developmental regulator in the reprogramming of root cells during cyst nematode infection. Plant Physiol. 2012;159(1):321–35.
Liang G, He H, Li Y, Wang F, Yu D. Molecular mechanism of microRNA396 mediating pistil development in Arabidopsis. Plant Physiol. 2014;164(1):249–58.
Li S, Gao F, Xie K, Zeng X, Cao Y, Zeng J, et al. The OsmiR396c-OsGRF4-OsGIF1 regulatory module determines grain size and yield in Rice. Plant Biotechnol J. 2016;14(11):2134–46.
Lee BH, Ko JH, Lee S, Yi L, Pak JH, Kim JH. The Arabidopsis GRF-INTERACTING FACTOR gene family performs an overlapping function in determining organ size as well as multiple developmental properties. Plant Physiol. 2009;151(2):655–68.
Liu D, Song Y, Chen Z, Yu D. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol Plant. 2009;136(2):223–36.
Yang F, Liang G, Liu D, Yu D. Arabidopsis MiR396 mediates the development of leaves and flowers in transgenic tobacco. J Plant Biol. 2009;52(5):475–81.
Rodriguez RE, Mecchia MA, Debernardi JM, Schommer C, Weigel D, Palatnik JF. Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development. 2010;137(1):103–12.
Kim JH. Biological roles and an evolutionary sketch of the GRF-GIF transcriptional complex in plants. BMB Rep. 2019;52(4):227–38.
Valladares F, Niinemets Ü. Shade tolerance, a key plant feature of complex nature and consequences. Annu Rev Ecol Evol Syst. 2008;39:237–57.
Yang F, Huang S, Gao R, Liu W, Yong T, Wang X, et al. Growth of soybean seedlings in relay strip intercropping systems in relation to light quantity and red:far-red ratio. Field Crops Res. 2014;155(155):245–53.
Liu X, Rahman T, Song C, Su B, Yang F, Yong T, et al. Changes in light environment, morphology, growth and yield of soybean in maize-soybean intercropping systems. Field Crops Res. 2017;200:38–46.
Pierik R, Testerink C. The art of being flexible: how to escape from shade, salt, and drought. Plant Physiol. 2014;166(1):5–22.
Wu Y, Gong W, Yang F, Wang X, Yong T, Yang W. Responses to shade and subsequent recovery of soya bean in maize-soya bean relay strip intercropping. Plant Prod Sci. 2016;19(2):206–14.
Yang F, Fan Y, Wu X, Cheng Y, Liu Q, Feng L, et al. Auxin-to-gibberellin ratio as a signal for light intensity and quality in regulating soybean growth and matter partitioning. Front Plant Sci. 2018;9:56.
Kozuka T, Horiguchi G, Kim GT, Ohgishi M, Sakai T, Tsukaya H. The different growth responses of the Arabidopsis thaliana leaf blade and the petiole during shade avoidance are regulated by photoreceptors and sugar. Plant Cell Physiol. 2005;46(1):213–23.
Gong W, Qi P, Du J, Sun X, Wu X, Song C, et al. Transcriptome analysis of shade-induced inhibition on leaf size in relay intercropped soybean. PLoS One. 2014;9(6):e98465.
Wang F, Qiu N, Qian D, Li J, Zhang Y, Li H, et al. Genome-wide identification and analysis of the growth-regulating factor family in Chinese cabbage ( Brassica rapa L. ssp. pekinensis ). BMC Genomics. 2014;15:807.
Zhang J, Li Z, Jin J, Xie X, Zhang H, Chen Q, et al. Genome-wide identification and analysis of the growth-regulating factor family in tobacco (Nicotiana tabacum). Gene. 2018;639:117–27.
Shang S, Wu C, Huang C, Tie W, Yan Y, Ding Z, et al. Genome-wide analysis of the GRF family reveals their involvement in abiotic stress response in cassava. Genes. 2018;9(2):110.
Maere S, De Bodt S, Raes J, Casneuf T, Van Montagu M, Kuiper M, et al. Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci U S A. 2005;102(15):5454–9.
Liu C, Xie T, Chen C, Luan A, Long J, Li C, et al. Genome-wide organization and expression profiling of the R2R3-MYB transcription factor family in pineapple ( Ananas comosus ). BMC Genomics. 2017;18(1):503.
Hoe Kim J, Tsukaya H. Regulation of plant growth and development by the GROWTH-REGULATING FACTOR and GRF-INTERACTING FACTOR duo. J Exp Bot. 2015;66(20):6093–107.
Khatun K, Ahk R, Park JI, Nath UK, Kim CK, Lim KB, et al. Molecular characterization and expression profiling of tomato GRF transcription factor family genes in response to abiotic stresses and phytohormones. Int J Mol Sci. 2017;18(5):1056.
Gonzalez N, Vanhaeren H, Inzé D. Leaf size control: complex coordination of cell division and expansion. Trends Plant Sci. 2012;17(6):332–40.
Li S, Tian Y, Wu K, Ye Y, Yu J, Zhang J, et al. Modulating plant growth-metabolism coordination for sustainable agriculture. Nature. 2018;560(7720):595–600.
Sun P, Zhang W, Wang Y, He Q, Shu F, Liu H, et al. OsGRF4 controls grain shape, panicle length and seed shattering in rice. J Integr Plant Biol. 2016;58(10):836–47.
Hu J, Wang Y, Fang Y, Zeng L, Xu J, Yu H, et al. A rare allele of GS2 enhances grain size and grain yield in rice. Mol Plant. 2015;8(10):1455–65.
Liu X, Guo LX, Jin LF, Liu YZ, Liu T, Fan YH, et al. Identification and transcript profiles of citrus growth-regulating factor genes involved in the regulation of leaf and fruit development. Mol Biol Rep. 2016;43(10):1059–67.
Wu ZJ, Wang WL, Zhuang J. Developmental processes and responses to hormonal stimuli in tea plant ( Camellia sinensis ) leaves are controlled by GRF and GIF gene families. Funct Integr Genomics. 2017;17(5):503–12.
Ma JQ, Jian HJ, Yang B, Lu K, Zhang AX, Liu P, et al. Genome-wide analysis and expression profiling of the GRF gene family in oilseed rape ( Brassica napus L.). Gene. 2017;620:36–45.
Cao Y, Han Y, Jin Q, Lin Y, Cai Y. Comparative genomic analysis of the GRF genes in Chinese pear (Pyrus bretschneideri Rehd), poplar (Populous), grape (Vitis vinifera), Arabidopsis and rice (Oryza sativa). Front Plant Sci. 2016;7:1750.
Ghosh A, Islam T. Genome-wide analysis and expression profiling of glyoxalase gene families in soybean ( Glycine max ) indicate their development and abiotic stress specific response. BMC Plant Biol. 2016;16:87.
Chen X, Chen Z, Zhao H, Zhao Y, Cheng B, Xiang Y. Genome-wide analysis of soybean HD-zip gene family and expression profiling under salinity and drought treatments. PLoS One. 2014;9(2):e87156.
Kim S, Park M, Yeom SI, Kim YM, Lee JM, Lee HA, et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet. 2014;46(3):270–8.
Anguraj Vadivel AK, Krysiak K, Tian G, Dhaubhadel S. Genome-wide identification and localization of chalcone synthase family in soybean (Glycine max [L]Merr). BMC Plant Biol. 2018;18:325.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463(7278):178–83.
Young ND, Debelle F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480(7378):520–4.
Belamkar V, Weeks NT, Bharti AK, Farmer AD, Graham MA, Cannon SB. Comprehensive characterization and RNA-Seq profiling of the HD-zip transcription factor family in soybean (Glycine max) during dehydration and salt stress. BMC Genomics. 2014;15:950.
Annapurna B, Rajesh G, Rohini G, Mukesh J. Genome-wide analysis of homeobox gene family in legumes: identification, gene duplication and expression profiling. PLoS One. 2015;10(3):e0119198.
Liu J, Hua W, Yang HL, Zhan GM, Li RJ, Deng LB, et al. The BnGRF2 gene (GRF2-like gene from Brassica napus) enhances seed oil production through regulating cell number and plant photosynthesis. J Exp Bot. 2012;63(10):3727–40.
Jérémie B, Ghazanfar Abbas K, Jean-Philippe C, Pilar BS, Juan Manuel D, Ramiro R, et al. miR396 affects mycorrhization and root meristem activity in the legume Medicago truncatula. Plant J. 2013;74(6):920–34.
Kuijt SJ, Greco R, Agalou A, Shao J. T Hoen CC, Overnas E, et al. Interaction between the GROWTH-REGULATING FACTOR and KNOTTED1-LIKE HOMEOBOX families of transcription factors. Plant Physiol. 2014;164(4):1952–66.
Yuan M, Zhao YQ, Zhang ZW, Chen YE, Ding CB, Yuan S. Light regulates transcription of chlorophyll biosynthetic genes during chloroplast biogenesis. Crit Rev Plant Sci. 2017;36(1):35–54.
van Gelderen K, Kang C, Pierik R. Light signaling, root development, and plasticity. Plant Physiol. 2018;176(2):1049–60.
Keuskamp DH, Keller MM, Ballaré CL, Ronald P. Blue light regulated shade avoidance. Plant Signal Behav. 2012;7(4):514–7.
Fraser DP, Hayes S, Franklin KA. Photoreceptor crosstalk in shade avoidance. Curr Opin Plant Biol. 2016;33:1–7.
Xu F, He S, Zhang J, Mao Z, Wang W, Li T, et al. Photoactivated CRY1 and phyB interact directly with AUX/IAA proteins to inhibit auxin signaling in Arabidopsis. Mol Plant. 2018;11(4):523–41.
Carriedo LG, Maloof JN, Brady SM. Molecular control of crop shade avoidance. Curr Opin Plant Biol. 2016;30:151–8.
Ruberti I, Sessa G, Ciolfi A, Possenti M, Carabelli M, Morelli G. Plant adaptation to dynamically changing environment: the shade avoidance response. Biotechnol Adv. 2012;30(5):1047–58.
Fan Y, Chen J, Wang Z, Tan T, Li S, Li J, et al. Soybean (Glycine max L. Merr.) seedlings response to shading: leaf structure, photosynthesis and proteomic analysis. BMC Plant Biol. 2019;19:34.
Wille W, Pipper CB, Rosenqvist E, Andersen SB, Weiner J. Reducing shade avoidance responses in a cereal crop. AoB Plants. 2017;9(5):plx039.
Michael M. A role for leaf epidermis in the control of leaf size and the rate and extent of mesophyll cell division. Am J Bot. 2010;97(2):224–33.
Omidbakhshfard MA, Fujikura U, Olas JJ, Xue GP, Balazadeh S, Mueller-Roeber B. GROWTH-REGULATING FACTOR 9 negatively regulates Arabidopsis leaf growth by controlling ORG3 and restricting cell proliferation in leaf primordia. PLoS Genet. 2018;14(7):e1007484.
Romina C, Rodriguez RE, Debernardi JM, Palatnik JF, Paula C. Repression of growth regulating factors by the microRNA396 inhibits cell proliferation by UV-B radiation in Arabidopsis leaves. Plant Cell. 2013;25(9):3570–83.
Fina J, Casadevall R, Abdelgawad H, Prinsen E, Markakis MN, Beemster GT, et al. UV-B inhibits leaf growth through changes in growth regulating factors and gibberellin levels. Plant Physiol. 2017;174(2):1110–26.
Wu Y, Gong W, Yang W. Shade inhibits leaf size by controlling cell proliferation and enlargement in soybean. Sci Rep. 2017;7(1):9259.
Lozano R, Hamblin MT, Prochnik S, Jannink JL. Identification and distribution of the NBS-LRR gene family in the cassava genome. BMC Genomics. 2015;16:360.
Chen C, Xia R, Chen H, He Y. TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. bioRxiv. 2018. https://doi.org/10.1101/289660.
Xie T, Chen C, Li C, Liu J, Liu C, He Y. Genome-wide investigation of WRKY gene family in pineapple: evolution and expression profiles during development and stress. BMC Genomics. 2018;19:490.
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics Protocols Handbook: Humana Press; 2005. p. 571–607. https://doi.org/10.1385/1-59259-890-0:571.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Zhang Z, Li J, Zhao X-Q, Wang J, Wong GK-S, Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genom Proteom Bioinf. 2006;4(4):259–63.
Shu K, Chen Q, Wu Y, Liu R, Zhang H, Wang P, et al. ABI4 mediates antagonistic effects of abscisic acid and gibberellins at transcript and protein levels. Plant J. 2016;85(3):348–61.
Zhou W, Chen F, Zhao S, Yang C, Meng Y, Shuai H, et al. DA-6 promotes germination and seedling establishment from aged soybean seeds by mediating fatty acid metabolism and glycometabolism. J Exp Bot. 2019;70(1):101–14.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8.
Acknowledgments
We would like to thank all members in our lab for the valuable discussion during preparation of this manuscript. The authors are very grateful to Professor Qi Xie and his research group from Institute of Genetics and Developmental Biology, Chinese Academy of Sciences.
Funding
This work was supported by the grants from National Natural Science Foundation of China (31872804, 31701064), National Key Research and Development Program of China (2017YFD0201300), and Sichuan Science and Technology Program (2018JZ0060) to Dr. Kai Shu.
Author information
Authors and Affiliations
Contributions
KS and FC designed the research. FC and Y-ZY performed most of the data analysis and experiments. X-FL, W-GZ, Y-JD, CZ and W-GL performed part of the experiments. KS, FC and Y-WY analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Sequence alignment of two conserved domains, QLQ and WRC, of the GmGRFs. (PDF 228 kb)
Additional file 2:
Table S1. Characterization of ten conserved motifs in GmGRF sequences. (PDF 23 kb)
Additional file 3:
Table S2. Duplication events of GmGRFs. (PDF 44 kb)
Additional file 4:
Figure S2. The transcription levels of GmGRFs in response to GA3. The housekeeping GmTubulin was used as an internal control. Error bars represent standard errors. The asterisk (*) indicates the significant difference at P < 0.05 by Student’s t-test analysis. (PDF 46 kb)
Additional file 5:
Figure S3. Shade stress induces shade avoidance response in soybean. (A) Representative photographs of soybean seedlings under white light and shade conditions. Bar = 100 mm. Seeding height (B), stem diameter (C), above-ground tissue fresh and dry weight (D, F), root fresh and dry weight (E, G) and root-shoot ratio (H). Ten soybean seedlings were measured under each condition. Error bars represent standard errors. The asterisk (*) indicates the significant difference at P < 0.05 by Student’s t-test analysis. L, shade; S, shade. (PDF 134 kb)
Additional file 6:
Table S3. Genome size and GRF number in plants. (PDF 32 kb)
Additional file 7:
Table S4. MtGRFs in Medicago. (PDF 20 kb)
Additional file 8:
Table S5. Primers sequence used in this study. (PDF 40 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, F., Yang, Y., Luo, X. et al. Genome-wide identification of GRF transcription factors in soybean and expression analysis of GmGRF family under shade stress. BMC Plant Biol 19, 269 (2019). https://doi.org/10.1186/s12870-019-1861-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1861-4