
Abstract
Background
Plants can suffer ammonium (NH4 +) toxicity, particularly when NH4 + is supplied as the sole nitrogen source. However, our knowledge about the underlying mechanisms of NH4 + toxicity is still largely unknown. Lemna minor, a model duckweed species, can grow well in high NH4 + environment but to some extent can also suffer toxic effects. The transcriptomic and physiological analysis of L. minor responding to high NH4 + may provide us some interesting and useful information not only in toxic processes, but also in tolerance mechanisms.
Results
The L. minor cultured in the Hoagland solution were used as the control (NC), and in two NH4 + concentrations (NH4 + was the sole nitrogen source), 84 mg/L (A84) and 840 mg/L (A840) were used as stress treatments. The NH4 + toxicity could inhibit the growth of L. minor. Reactive oxygen species (ROS) and cell death were studied using stained fronds under toxic levels of NH4 +. The malondialdehyde content and the activities of superoxide dismutase and peroxidase increased from NC to A840, rather than catalase and ascorbate peroxidase. A total of 6.62G nucleotides were generated from the three distinct libraries. A total of 14,207 differentially expressed genes (DEGs) among 70,728 unigenes were obtained. All the DEGs could be clustered into 7 profiles. Most DEGs were down-regulated under NH4 + toxicity. The genes required for lignin biosynthesis in phenylpropanoid biosynthesis pathway were up-regulated. ROS oxidative-related genes and programmed cell death (PCD)-related genes were also analyzed and indicated oxidative damage and PCD occurring under NH4 + toxicity.
Conclusions
The first large transcriptome study in L. minor responses to NH4 + toxicity was reported in this work. NH4 + toxicity could induce ROS accumulation that causes oxidative damage and thus induce cell death in L. minor. The antioxidant enzyme system was activated under NH4 + toxicity for ROS scavenging. The phenylpropanoid pathway was stimulated under NH4 + toxicity. The increased lignin biosynthesis might play an important role in NH4 + toxicity resistance.
Similar content being viewed by others

Background
Ammonium (NH4 +) and nitrate (NO3 −) are the two inorganic nitrogen (N) forms that can be directly absorbed by plants [1]. Compared to NO3 −, NH4 + is more easily absorbed by plants as its assimilation requires less energy. But in fact, only few plants are known to be NH4 + specialists, most of high plants are usually sensitive to NH4 + [2, 3]. Non-specialists could display toxicity symptoms such as leaf chlorosis, growth suppression, yield depressions, and even mortality in high NH4 + conditions, particularly when NH4 + is supplied as the sole N source [3]. At the ecosystem level, some studies have even shown that increased NH4 + in soil and water environment was associated with reduced crop yield, and decline of forest and macrophyte abundances [4–6].
NH4 + toxicity is not only a significant ecological issue, but also an important plant physiological process [7]. Plant scientists have been trying to reveal its occurrence, signal transmission and physiological targets in plants [3, 7, 8]. Usually, for most plants, the root bears the brunt of NH4 + toxicity [7, 9–11]. The root often accumulates high levels of NH4 + in high NH4 + condition, and the root cells could experience a futile transmembrane NH4 + cycling that could carry high energetic cost resulting to decline plant growth [9, 12–14]. Associated enzymes involved in the regulation of NH4 + influx, a signaling pathway model under NH4 + toxicity in Arabidopsis thaliana has been described [7, 8]. Additionally, recent studies also revealed that the NH4 + toxicity could break the intracellular pH balance and C/N balance [3, 7, 15], and cause oxidative damage [16, 17].
The knowledge on NH4 + toxicity has greatly expanded in recent years, but the underlying mechanism are still largely unclear, further researches, especially in the subcellular level, using more advanced -omics approaches to follow up NH4 +-induced global changes in plants are also required [8, 18]. Transcriptome analysis is an effective method for global expression profiling of genes involved in stresses and toxicity in living organisms [19, 20]. For example, transcriptomic profiling using microarrays have been used in Arabidopsis to identify molecular changes involved in NH4 + toxicity [21]. With the rapid development of high-throughput sequencing, the next-generation transcriptome profiling approach or RNA sequencing (RNA-seq) has been gaining wide attention and use. RNA-seq could provide more information at a more affordable cost compared with the microarray and now an emerging powerful tool for transcriptome analysis [22].
Duckweeds are simple floating aquatic plants composed by frond and root. It has been considered to be a model species for aquatic plants and has been greatly used previously especially in the fields of toxicity studies, phytoremediation and biofuels production [23]. Lemna minor L. is one of the most widely distributed duckweed species and gains increasing interests due to its better adaptability to varying environmental conditions including high NH4 + stress [24, 25]. L. minor could grow well in high NH4 + environment and has been even recognized as ‘NH4 + specialist’, but has been shown to still suffer toxicity in very high NH4 + levels [15]. On the other hand, mechanisms and processes of toxicity in duckweeds however are a bit different from the terrestrial plant. Such as in Arabidopsis, most of the NH4 + contact happens mainly in roots, thus the roots firstly suffer NH4 + toxicity [7, 26]. While for the floating duckweeds, the frond and root are all directly exposed to the toxic environment. This may lead to some different responses from the terrestrial plant. In this study, we use RNA-seq to investigate the global changes in common duckweed Lemna minor under NH4 + toxicity. Those transcriptome analyses may provide some interesting insights and useful information not only in intoxication processes, but also on its potential tolerance mechanisms.
Methods
Sample preparation
Samples were prepared as described in Wang et al. [15]. L. minor was collected from a eutrophic pond in Chengdu, Sichuan, China (location: 30° 38.86′N, 104° 18.01′ E; elevation 499 m), and no specific permissions were required for specimen collection. To guarantee genetic uniformity, all of the L. minor materials originated from single colony and cultivated in Hoagland solution with 84 mg/L NO3 −. The L. minor cultured in the Hoagland solution were used as the control (NC). For the treatments, cultures were grown in two NH4 + concentrations, 84 mg/L (A84) and 840 mg/L (A840) in improved Hoagland solution, in which NH4Cl was used to provide NH4 +, and KCl and CaCl2 were used to replace KNO3 and Ca(NO3)2 to avoid the impact of nitrate. All the solutions used in this study were adjusted to pH 5.5 with 1 M HCl.
Before inoculation, the fronds collected from Hoagland were washed five times with deionized water. Then, 0.2 g (fresh weight) of plant materials was cultivated in plastic basins with water depth of 2 cm. The plants were grown for one week in incubator at 23 ± 1 °C with a photon flux density of 50–60 μmol · m−2 · s−1 provided by cool white fluorescent bulbs in a 16 h light/8 h dark cycle. The medium in each container was replaced every day.
Growth and physiological analysis
The relative growth rate (RGR) based on fronds number was used to evaluate the duckweed growth in different treatments as previously described in Wang et al. [15]. A total of 0.5 g fronds homogenized in 5 ml 0.1 % trichloroacetic acid was used for malondialdehyde (MDA) estimation by the thiobarbituric reaction following Dhindsa and Matowe [27]. Superoxide dismutase (SOD) was measured using a kit from Nanjing Jiancheng Bioengineering Institute (Jiangsu, China). Peroxidase (POD) and catalase (CAT) were measured by absorption photometry using a spectrophotometer as described by Bestwick et al. and Aebi [28, 29], respectively. Ascorbate peroxidase (APX) activity assays were according to the method of Chen and Asada where the extinction coefficient of ascorbate at 290 nm was used for calculating APX enzyme activity [30].
Fronds of L. minor from the three treatments were stained by 3,3′-diaminobenzidine (DAB) or nitroblue tetrazolium (NBT) for measuring H2O2 or O2 − level, respectively [31]. Cell death was examined by Evans blue staining as described by Kim et al. [32].
RNA extraction, cDNA library preparation and sequencing
The whole plants with fronds and roots were ground in liquid nitrogen and total RNA was extracted using RNeasy® Plant Mini Kit (Qiagen) as per manufacturer’s protocol. The integrity of RNA was assessed by formaldehyde agarose gel electrophoresis. A total of 30 μg mixed RNA from three biological replicates detected by 2100 Bioanalyzer (Agilent, USA) was digested with DNase I (TAKARA), and then purified by Dynabeads® Oligo (dT)25 (Life, USA). 100 ng derived mRNAs were fragmented and reverse transcribed into first-strand cDNAs with random hexamer and then the second-strand cDNAs were synthesized by using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina (NEB). The double-stranded cDNAs were purified and ligated to adaptors for Illumina paired-end sequencing. The cDNA library was sequenced using the Illumina HiSeq2500 system by Shanghai Hanyu Biotech lab (Shanghai, China).
De novo assembly of RNA-seq reads and quantifying gene expression
For the assembly library, raw reads were filtered using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) to remove adapters and low-quality reads (base quality < 20, read length < 40 bp). The obtained quality-filtered reads were de novo assembled into contigs by the Trinity Program [33]. Unigenes were defined after removing redundancy and short contigs from the assembly. The unigenes were predicted by “GetORF” in the EMBOSS package [34] and aligned to the protein sequence database NCBI NR (non-redundant protein database), Swiss-Prot (Annotated protein sequence database), KEGG (Kyoto encyclopedia of genes and genomes) and COG (Clusters of orthologous groups of protein) by Blastp with an E-value threshold of 1 × 10 −5.
The number of unique-match reads was calculated and normalized to RPKM (reads per kb per million reads) for gene expression analysis. Comparison of unigene expression between treatments was according to DESeq as described by Abders and Huber [35]. The differentially expressed genes (DEGs) between NC and A84, or between NC and A840, or between A84 and A840 were restricted with FDR (false discovery rate) ≤ 0.001 and the absolute value of log2 Ratio ≥1.
To examine the expression profile of DEGs, the expression data υ (from NC, A84 and A840 treatment) were normalized to 0, log2 (υA84/υNC), log2 (υA840/υNC), and then clustered by Short Time-series Expression Miner software (STEM) [36]. The clustered profiles with p-value ≤ 0.05 were considered as significantly expressed. Then the DEGs in all or in each profile were subjected to gene ontology (GO) classifications using WEGO [37], and KEGG pathway enrichment analysis.
Validation of differential expression using qRT-PCR
The cDNA was generated from 1 μg total RNA isolated from the fronds using a Prime-Script™ 1st Strand cDNA Synthesis Kit (TAKARA, Japan). Primers for quantitative real time PCR (qRT-PCR) were designed using Primer Premier 5.0 software (Premier, Canada) and synthesized by Sangon Biotech (Shanghai) Co., Ltd. The 18S (GenBank accession number: KJ400889) was selected as reference. All the primers are shown in Additional file 1: Table S1. qRT-PCR was performed on a Bio-Rad iQ5 Optical System Real Time PCR System (Bio-Rad, USA). Each reaction mixture was 20 μL containing 10 μL of SYBR Green PCR Master Mix (TaKaRa, Japan), 250 nM of each primer, and 6 μL of diluted first-strand cDNAs. The qRT-PCRs were run as follows: 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s in 96-well optical reaction plates. The Ct values were determined for three biological replicates, with three technical replicates for each value. Expression levels of the tested reference genes were determined by Ct values and calculated by 2-△△Ct.
Statistical analysis
All data were statistically analyzed by means of the SPSS with LSD to identify differences. Significant differences (P < 0.05) between treatments are indicated by different letters.
Results
Phenotypic and physiological responses to NH4 + toxicity in L. minor
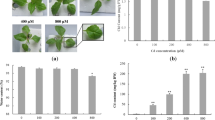
Figure 1 a-c shows changes in the appearance of L. minor fronds at the end of experiment. The fronds in NC looked green and healthy, as well as in A84. But in A840, some mother fronds looked greensick (Fig. 1 c, shown by arrow). The RGR based on fronds number showed a downward trend from NC to A840 (Fig. 1 e). This could indicate that the NH4 + concentrations of 84 mg/L affected the propagation of L. minor, and the much higher concentration of 840 mg/L significantly inhibited the growth and could cause some damage.

Phenotypic and physiological responses of Lemna minor in NC, A84 and A840. a-c, the appearance of L. minor in NC, A84 and A840, respectively, red arrows showed the greensick fronds, scale bar 5 mm; d Histochemically staining of cell death, O2 − and H2O2 by Evans blue, nitroblue tetrazolium (NBT) and 3,3′-diaminobenzidine (DAB), respectively; e relative growth rate (RGR) based on fronds number; f MDA contents; g-j, enzyme activity of catalase (CAT), superoxide dismutase (SOD), peroxidase (POD) and ascorbate peroxidase (APX), respectively
Evans blue was used to determine the high NH4 +-stress induced cell death (Fig. 1d). Almost no dead cell was stained in the fronds cultured in NC. Dead cells were however detected in both mother and newborn fronds in the plants grown in both tested NH4 + concentrations, especially in A840.
Fronds of L. minor were stained with DAB or NBT to reveal in situ accumulation of two main reactive oxygen species (ROS), H2O2 and O2 −, respectively (Fig. 1d). Histochemically stained cells showed that the H2O2 and O2 − significantly accumulated in both the mother and newborn fronds in A840 after seven days. The fronds in A84 were also found to have some ROS accumulation. For the fronds in NC, the ROS was just slightly accumulated in some mother fronds that might be induced by the normal ageing.
The MDA content was used to detect the lipid peroxidation and membrane damage induced by oxidative stress. The contents of MDA in L. minor in A84 and A840 were higher than in NC, and the highest MDA content reached 4.4 nmol/g in A840 (Fig. 1f). The activity of the antioxidant defense system was also analyzed (Fig. 1g-j). Like the change of MDA, the activities of SOD and POD all increased from NC to A840, and the values all increased almost doubled. In contrast, the CAT decreased from NC (6.5 u/g) to A840 (3.8 u/g). For the APX, the highest activity was in A84 (45.28 u/g) and the lowest one was in A840 (8.96 u/g).
Overview of three libraries data sets by RNA-seq
As shown in Table 1, a total of 6.62G nucleotides, equivalent to 33,136,337 raw reads and 32,403,455 quality filtered (clean) reads were generated from the three separate libraries from NC, A84, and A840. The RNA-seq generated clean reads ranged from 10.4 to 11.1 million on each sample. The Q20 percentages of the three libraries were from 97.21 to 97.44 %, and the GC contents ranged from 50.68 to 51.69 %. All clean reads were pooled together and then de novo assembled by Trinity. Based on chosen criteria, an average of 79.91 % of the clean reads was mapped, with perfect matches were from 47.03 to 47.09 %. In each library, the scales of clean reads uniquely mapped to the database were 76.87, 80.09 and 79.91 %, respectively. There were still approximately 20.09 % of clean reads that cannot be mapped back to any references, which could be due to the limited reference gene database of L. minor.
The final assembly of L. minor had 71,094 contigs with length ≥ 200 bp and after further removal of redundant sequences, 70,728 unigenes were obtained. The length of unigenes ranged from 201 b to 14,857 b, with a mean size of 620 bp and N50 number of 988 bp (Table 2). In NC library, the number of genes identified increased with the number of reads until above 6 million, but 4 million in the other two libraries (Additional file 2: Figure S1A). The unigene coverage analyzed as a means of evaluating the quality of the RNA-seq data was mostly >50 %. More than half of the sequences have coverage more than 80 % (Additional file 2: Figure S1B).
The amino acid sequences predicted by ‘Getorf’ were searched by BLASTP. A final number of 29,171, 28,476, 17,209 and 14,172 unigenes (E-value < 1e−5) had significant matches in NR, KEGG and COG databases, respectively. As shown in Additional file 3: Figure S2, the sequences matched with the species in NR were determined as following: 27 % Vitis vinifera, 16 % Ricinus communis, 14 % Populus trichocarpa, 8 % Glycine max and 4 % Oryza sativa etc. The COG matched L. minor unigenes dataset were categorized into 25 functional COG clusters (Additional file 4: Figure S3). The five largest functional categories in which sequences were identified to include 1) Posttranslational modification, protein turnover, chaperones; 2) Signal transduction mechanisms; 3) General function prediction only; 4) Translation, ribosomal structure and biogenesis; and 5) Intracellular trafficking, secretion, and vesicular transport.
Identification and overview of the differentially expressed genes
We performed a pairwise comparison using NC as the control, and A84, or A840 as the treatments (Fig. 2a). Most of genes were down-regulated in the two treatments but the up-regulated genes in A840 were slightly higher than the down-regulated genes.

Unigene expression pairwise comparison (a) and three main DEGs expression profiles (p-value ≤ 0.05) (b-d) in three libraries (NC, A84 and A840)
Results from FDR identification showed that 14,207 unigenes were classified as DEGs, which were then used for the subsequent analysis. All the 14,207 DEGs could be clustered into 7 profiles by STEM (Additional file 5: Figure S4; Additional file 6), in which 12,959 DEGs were further clustered into 3 profiles (p-value ≤ 0.05), including two down-regulated patterns (Profile 1 and Profile 0) and one up-regulated pattern (Profile 7) (Fig. 2b-d). Profile 1 and Profile 0 contained 11,625 and 954 DEGs, respectively, while Profile 7 contained 380 DEGs.
Next, the DEGs within the three profiles were subjected to GO-term analysis (Fig. 3). The DEGs were classified into three main categories including cellular component, biological process, and molecular function. Cell and cell parts under cellular component category were the two top abundant subcategories of the two down-regulated patterns (Profile 1 and Profile 0). For the up-regulated pattern of Profile 7, the metabolic process under molecular function was the top subcategories.

GO classification of profile 1 (a), profile 0 (b) and profile 7 (c) in three libraries (NC, A84 and A840)
All DEGs were subjected to KEGG pathway enrichment analysis, and 36.26 % (5151/14,207) of the DEGs could be annotated. The 20 top KEGG pathways with the highest representation of the DEGs are shown in Table 3. The ribosome (ko03010), plant hormone signal transduction (ko04075), glycolysis/gluconeogenesis (ko00010), starch and sucrose metabolism (ko00500), purine metabolism (ko00230), phenylpropanoid biosynthesis (ko00940), pyrimidine metabolism (ko00240), pyruvate metabolism (ko00620), DNA replication (ko03030) and plantpathogen interaction (ko04626) pathways are significantly enriched. The 372 unigenes among 4366 DEGs (8.52 %) in profile 1, and 122 unigenes accounting for 32.28 % of 378 in profile 0 were annotated to ribosome pathway as the most enriched one, whereas in Profile 7, only 1 unigene accounting for 1.41 % of 71 DEGs, was annotated to this pathway.
For the up-regulated pattern of Profile 7, the ten significantly enriched pathways were Phenylpropanoid biosynthesis (ko00940), Metabolic pathways (ko01100), Phenylalanine metabolism (ko00360), Biosynthesis of secondary metabolites (ko01110), Isoquinoline alkaloid biosynthesis (ko00950), Photosynthesis (ko00195), Tyrosine metabolism (ko00350), Plant-pathogen interaction (ko04626), RNA polymerase (ko03020), Oxidative phosphorylation (ko00190) (Table 4). The Metabolic pathways had the largest DEGs number (39), but the Phenylpropanoid biosynthesis has the biggest P-value.
Analysis of phenylpropanoid biosynthesis pathway genes from L. minor unigenes
In plants, the phenylpropanoid biosynthesis pathway contributes to multiple biosynthetic branches, such as lignin and flavonoid biosynthesis. The expression of transcripts encoding for key enzymes for lignin and flavonoid biosynthesis were analyzed in this study (Fig. 4). The results showed that most of lignin biosynthesis related genes were up-regulated, but not for the expression of transcripts encoding for key enzymes for flavonoid synthesis.

The expression of phenylpropanoid biosynthesis pathway genes from L. minor unigenes. a lignin and flavonoid biosynthesis in phenylpropanoid biosynthesis pathway (the numbers in the box are the RPKMs of NC, A84 and A840, respectively); b qRT-PCR analysis. PAL, Phenylalanine ammonia lyase; C4H, Cinnamic acid 4-hydroxylase; 4CL, 4-hydroxycinnamoyl-CoA ligase; HCT, Hydroxycinnamoyl-CoA: shikimate/quinate hydroxycinnamoyl transferase; CCR, Cinnamoyl-CoA reductase; C3H, p-coumaroyl shikimate 30-hydroxylase; CCoAOMT, Caffeoyl-CoA O-methyltransferase; F5H, Coniferaldehyde/ferulate 5-hydroxylase; COMT, Caffeic acid O-methyltransferase; CAD, Cinnamyl alcohol dehydrogenase; LAC, laccase; POD, Peroxidase; CHS, Chalcone synthase; CHI, Chalcone isomerase; F3H, Flavanone 3-hydroxylas; FLS, Flavonol synthase
As shown in Fig. 4, in lignin biosynthesis pathway, the genes of PAL (Phenylalanine ammonia-lyase), 4CL (4-hydroxycinnamoyl-CoA ligase), COMT (caffeic acid O-methyltransferase), C3H (p-coumaroyl shikimate 3′-hydroxylase), F5H (coniferaldehyde/ferulate 5-hydroxylase), CCR (cinnamoyl-CoA reductase), LAC (laccase), POD were all up-regulated. HCT (hydroxycinnamoyl-CoA: shikimate/quinate hydroxycinnamoyl transferase) and C4H (cinnamic acid 4-hydroxylase) had almost no change but the gene of CCoAOMT (caffeoyl-CoA O-methyltransferase) showed significant downward trend. This indicated that under NH4 + stress, shift from caffeoyl CoA to feruloy CoA might be difficult during the biosynthesis of G and S type lignin, which are the main components of monocot lignin [38]. To get feruloy CoA, another way might be potentially utilized which involves the caffeic acid, which can be changed into ferulic acid by COMT, and subsequently changed into feruloy CoA. This mechanism has also been shown to be similar to other monocotyledon like switchgrass [39].
Expression profiles of oxidative-related and PCD-related genes
Expression of the 14 ROS oxidative-related genes including six oxidative marker genes, six ROS-scavenging genes and two ROS-producing genes are summarized in Fig. 5. The oxidative marker genes included a trypsin/chymotrypsin inhibitor, a DNAJ heat shock protein, a FAD-binding protein and three cytochrome P450 genes, which are regarded as hallmarks for the general oxidative stress response [40, 41]. The ROS-scavenging genes consisted of genes of CAT, SOD and POD.

Heatmap (a) and qRT-PCR (b) analysis of the expression levels of oxidative-related and PCD-related genes. TI, Trypsin/chymotrypsin inhibitor; DNAJ, DNAJ heat shock family protein; FAD-B, FAD-binding domain-containing protein; P450 77A3, Cytochrome P450 77A3; P450 78, Cytochrome P450 85A-like; P450 85A, Cytochrome P450 85A-like; CAT, catalase; SOD-Mn, Superoxide dismutase [Mn]; SOD-Fe, Superoxide dismutase [Fe]; SOD- Cu/Zn, Cu/Zn superoxide dismutase; POD, Peroxidase; APX, L-ascorbate peroxidase; Rboh A, Respiratory burst oxidase homolog protein A; Rboh D, Respiratory burst oxidase protein D; NR, Nitrate reductase; NSHB1, Hemoglobin 1; AOX, Alternative oxidase 1; MAC4, Metacaspase 4; DAD1, Defender against cell death 1; BAXI1, BAX inhibitor 1
NADPH oxidases play a key role in generating ROS [42] and have been shown that RbohD and RbohF genes in Arabidopsis and RbohA of Hordeum vulgare could indeed induce ROS production [43, 44]. In this study, two Rboh-like protein genes, the RbohA and RbohD in L. minor were up-regulated under high NH4 + stress according to the RNA-seq and qRT-PCR results.
On the other hand, three genes indirectly related to oxidative stress were also detected. Under high NH4 + stress, the nitrate reductase (NR) and non-symbiotic hemoglobin 1 (NSHB1) genes were all down-regulated, but the alternative oxidase 1 (AOX1) gene was up-regulated (Fig. 5).
Metacaspases act as initiators and regulators for programmed cell death (PCD) in plants [45]. In DEGs, a metacaspase gene of MAC4 (metacaspase 4) was significantly up-regulated under NH4 + stress. Conversely, two inhibitor of PCD, namely BAX inhibitor 1 and DAD1 (defender against cell death 1), were significantly down-regulated.
Discussion
NH4 + toxicity-induced global changes in gene expression in L. minor
RNA-seq is a powerful tool that can provide a global overview of gene expression at the transcriptome level. Despite great potentials for both bioenergy applications and environmental studies, only 135 gene sequences of L. minor were currently deposited in public databases such as Genbank (accessed 12/10/2015). It is a challenge to do analysis and characterization of L. minor RNA-seq dataset without a sequenced genome, and in fact a lack of a sequenced genome in the Lemna. Until now, only the chloroplast genome of L. minor has been generated [46]. Despite such limitations, careful curation of the sequences and assembly using the robust Trinity Program allowed us to still identify 70,728 unigenes in L. minor, and more than 40 % of them were annotated.
The differential expression analysis of RNA-seq data revealed that most of the DEGs were down-regulated under NH4 + stress. GO and KEGG enrichment analysis revealed that the down-regulated genes (profile 1 and profile 0) mainly categorized into cellular structure and function, metabolic process and gene transcription indicating that some important physiological or “housekeeping” functions might have been inhibited. The same response has been observed in Arabidopsis under NH4 + stress [21]. On the other hand, the up-regulated genes were mainly associated with the metabolic processes, especially the secondary metabolism, such as the Phenylpropanoid biosynthetic pathway (Table 2). It has been suggested that some secondary metabolites play an important role in defenses of abiotic stress [47]. For example, some flavonoids and lignin precursors have been reported to accumulate in response to various abiotic stresses [48].
NH4 + toxicity caused oxidative stress and induced cell death
ROS is usually detected in overproduction under abiotic stresses, where it can cause some damages which ultimately results to oxidative stress [49]. Previous studies have shown that L. minor could suffer NH4 + toxicity in high NH4 + concentrations [16], and that NH4 + concentration of 56 mg/L could induce oxidative stress [50]. In this study, we further explored the effects of two higher NH4 + concentrations (84 and 840 mg/L) on oxidative damage of L. minor. Like other aquatic plants [51], the accumulated ROS and increased MDA content in L. minor in the two NH4 + treatments indicated that oxidative damage occurring. This is because the increased ROS could induce oxidative stress that contributes to lipid peroxidation and membrane damage, and the MDA has been considered as the indicator of the damage [52]. In addition to the physical evidence, some molecular evidences on NH4 + toxicity induced damage were also presented produced from the RNA-seq and qRT-PCR analysis (Fig. 5). The expression of some oxidative marker genes like trypsin/chymotrypsin inhibitor, DNAJ heat shock protein and cytochrome P450 genes were enhanced under high NH4 + stress. Furthermore, the up-regulated ROS-producing genes of RbohA and RbohD also indicated oxidative stress occurring in L. minor under NH4 + toxicity.
The ROS scavenging enzymes play an important role in the plant’s defense system in response to the generation of ROS. Among the enzymatic antioxidants, the enzyme SOD represents the first line of antioxidant defense by transforming O2 − into H2O2, and then the APX, POD, and CAT subsequently metabolize H2O2 [53]. Under NH4 + toxicity, the SOD activity of L. minor increased but not the gene expression of the three types of SOD, indicating a lag from the gene transcription to enzyme action in this 7-day stress treatment. According to Huang’s observations, the activity of SOD decreased until the 14th day under NH4 + toxicity [50]. The activated SOD could transform O2 − into H2O2 but its removal only relies on POD for the gene expression and activities of CAT and APX which all decreased in A840.
Like ROS, nitric oxide (NO) also plays an important role in plant responses to environmental stress, and there are complex networks of interactions between ROS and NO when plants suffer oxidative stress [54]. In plants, NR could reduce nitrate to produce nitrite, as well as reduce nitrite to produce NO, which possesses antioxidant properties and likely to act as a signal in activating ROS-scavenging enzyme activities under oxidative stresses [55]. The non-symbiotic hemoglobin could scavenge NO, thus building a futile cycle with NR [56]. The alternative oxidase can scavenge NO with ROS as the substrates, as well as prevent the production of excess ROS by stabilizing the redox state of the mitochondrial ubiquinone pool [56, 57]. In this study, the NH4 + is the sole N source in the two stress treatments, the down-regulated L. minor NR gene in A84 and A840 indicated the gene might not be activated without nitrate. The NR-mediated NO production might also be suppressed, even though the NSHB1 gene was also down-regulated. The slightly up-regulated AOX1 gene in L. minor may be involved in preventing ROS excessive increase under high NH4 + stress.
ROS is one of the key regulators of PCD that is an active and genetically controlled form of cell death [58]. In this study, except for the ROS, the cell death was also detected in L. minor suffering from NH4 + toxicity by staining. RNA-seq results further showed that a metacaspase gene, MAC4, was significantly up-regulated in the two NH4 + treatments. In plants, the metacaspase is a discovered gene family that has distant caspase homologs closely related to PCD [59]. The MAC4 of Arabidopsis plays a positive regulatory role in abiotic stress-induced PCD [60]. In addition, our results also showed that the PCD inhibiters, like BAX inhibitor 1 [61] and DAD1 [62], significantly decreased in their gene expression. Thus, we can speculate that the NH4 + toxicity induced PCD of L. minor, and that the ROS might play as an intermediate signaling molecule.
Lignin biosynthesis plays an important role in NH4 + toxicity resistance
Lignin is the major components of cell wall and the main structure in plant mechanical support and defense system [63]. There are two pathways for lignin biosynthesis in plants, namely of monolignol and phenylpropanoid pathways [64]. And the stimulation of the phenylpropanoid pathway has been considered as a common feature of some abiotic stress response such as drought, salinity, ozone intoxication and heavy metals [63, 65]. Previous studies also showed that both nitrogen deficiency and fertilization (NH4NO3) could induce a set of genes required for phenylpropanoid metabolism [66, 67]. In this study, the RNA-seq and qRT-PCR results also showed enhanced expression of some key enzyme genes in phenylpropanoid pathway under high NH4 + stress in L. minor. In addition, all up-regulated genes were lignin biosynthesis-related, rather than flavonoid synthesis, which could be due to the antagonistic relationships of the two biosynthetic pathways [68]. However, it could still be suggested that the NH4 + toxicity could stimulate the phenylpropanoid pathway of L. minor, and lead to a shift of metabolism towards lignin.
G and S type lignin are the main components of monocot lignin [39]. In L. minor, a series of genes required for the biosynthesis of two types of lignin were up-regulated, including the rate-limiting enzymatic genes in lignin biosynthesis, like PAL [69] and F5H [70]. The increased lignin synthesis would result to higher lignin content, which together with other antioxidants, could play an important role in limiting ROS production in the apoplast [63]. This mechanism could be one of the reasons why L. minor could resist high NH4 + stress.
Conclusions
In this study we report the first large transcriptome study carried out in L. minor where we have compared physiological and transcriptional responses to NH4 + toxicity. Evidence from physiological observations, transcriptome and qRT-PCR analysis indicated that NH4 + toxicity could induce ROS accumulation which in turn results to oxidative damage and later to cell death in L. minor. The antioxidant enzyme system was activated under NH4 + toxicity for ROS scavenging. We also identified that the phenylpropanoid pathway was stimulated under NH4 + toxicity, and the lignin biosynthesis was also up-regulated in this metabolic pathway. The increased lignin biosynthesis might play an important role in NH4 + toxicity resistance.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data
The dataset is available from the NCBI Sequence Read Archive (SRA). The BioProject and SRA accession are PRJNA302233 and SRP066224, respectively.
Abbreviations
- 4CL:
-
4-hydroxycinnamoyl-CoA ligase
- AOX:
-
alternative oxidase
- APX:
-
ascorbate peroxidase
- C3H:
-
p-coumaroyl shikimate 3′-hydroxylase
- C4H:
-
cinnamic acid 4-hydroxylase
- CAT:
-
catalase
- CCoAOMT:
-
caffeoyl-CoA O-methyltransferase
- CCR:
-
cinnamoyl-CoA reductase
- COG:
-
clusters of orthologous groups of protein
- COMT:
-
caffeic acid O-methyltransferase
- DAB:
-
3,3′-diaminobenzidine
- DAD1:
-
defender against cell death 1
- DEG:
-
differentially expressed gene
- F5H:
-
coniferaldehyde/ferulate 5-hydroxylase
- GO:
-
gene ontology
- HCT:
-
hydroxycinnamoyl-CoA: shikimate/quinate hydroxycinnamoyl transferase
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LAC:
-
laccase
- MAC4:
-
metacaspase 4
- MDA:
-
malondialdehyde
- NBT:
-
nitroblue tetrazolium (NBT)
- NO:
-
nitric oxide
- NR:
-
non-redundant protein database
- NR:
-
nitrate reductase
- NSHB1:
-
hemoglobin 1
- PAL:
-
phenylalanine ammonia-lyase
- PCD:
-
programmed cell death
- POD:
-
peroxidase
- qRT-PCR:
-
quantitative real time PCR
- Rboh:
-
respiratory burst oxidase homolog
- RNA-seq:
-
RNA sequencing
- ROS:
-
reactive oxygen species
- RPKM:
-
reads per kb per million reads
- SOD:
-
superoxide dismutase
- SRA:
-
sequence read archive
- STEM:
-
short time-series expression miner software
References
Nordin A, Hogberg P, Nasholm T. Soil nitrogen form and plant nitrogen uptake along a boreal forest productivity gradient. Oecologia. 2001;129:125–32.
Kronzucker HJ, Siddiqi YM, Glass ADM. Conifer root discrimination against soil nitrate and the ecology of forest succession. Nature. 1997;385:59–61.
Britto DT, Kronzucker HJ. NH4 + toxicity in higher plants: a critical review. J Plant Physiol. 2002;159:567–84.
van Breemen N, Burrough PA, Velthorst EJ, van Dobben HF, de Wit T, Ridder TB, et al. Soil acidification from atmospheric ammonium sulphate in forest canopy throughfall. Nature. 1982;299:548–50.
Stevens CJ, Manning P, van den Berg LJL, de Graaf MCC, Wamelink GWW, Boxman AW, et al. Ecosystem responses to reduced and oxidised nitrogen inputs in European terrestrial habitats. Environ Pollut. 2011;159:665–76.
Cao T, Xie P, Ni LY, Wu AP, Zhang M, Wu SK, Smolders AJP. The role of NH4 + toxicity in the decline of the submersed macrophyte Vallisneria natans in lakes of the Yangtze River basin, China. Mar Freshw Res. 2007;58:581–7.
Li BH, Li GJ, Kronzucker HJ, Baluska F, Shi WM. Ammonium stress in Arabidopsis: signaling, genetic loci, and physiological targets. Trends Plant Sci. 2014;19:107–14.
Bittsánszky A, Pilinszky K, Gyulai G, Komives T. Overcoming ammonium toxicity. Plant Sci. 2015;231:184–90.
Li Q, Li BH, Kronzucker HJ, Shi WM. Root growth inhibition by NH4 + in Arabidopsis is mediated by the root tip and is linked to NH4 + efflux and GMPase activity. Plant Cell Environ. 2010;33:1529–42.
Zou N, Li BH, Dong GQ, Kronzucker HJ, Shi WM. Ammonium-induced loss of root gravitropism is related to auxin distribution and TRH1 function, and is uncoupled from the inhibition of root elongation in Arabidopsis. J Exp Bot. 2012;63:3777–88.
Zhou Y, Bai L, Song CP. Ammonium homeostasis and signaling in plant cells. Chin Sci Bull. 2015;60:741–7.
Kronzucker HJ, Britto DT, Davenport RJ, Tester M. Ammonium toxicity and the real cost of transport. Trends Plant Sci. 2001;6:335–7.
Britto DT, Siddiqi MY, Glass ADM, Kronzucker HJ. Futile transmembrane NH4 + cycling: A cellular hypothesis to explain ammonium toxicity in plants. Proc Natl Acad Sci U S A. 2001;98:4255–8.
Barth C, Gouzd ZA, Steelt HP, Imperio RM. A mutation in GDP-mannose pyrophosphorylase causes conditional hypersensitivity to ammonium, resulting in Arabidopsis root growth inhibition, altered ammonium metabolism, and hormone homeostasis. J Exp Bot. 2010;61:379–94.
Wang WG, Yang C, Tang XY, Zhu QL, Pan K, Hu QC, et al. Effects of high ammonium level on biomass accumulation of common duckweed Lemna minor L. Environ Sci Pollut Res. 2014;21:14202–10.
Nimptsch J, Pflugmacher S. Ammonia triggers the promotion of oxidative stresss in the aquatic macrophyte Myriophyllum mattogrossense. Chemosphere. 2007;66:708–14.
Wang C, Zhang SH, Wang PF, Li W, Lu J. Effects of ammonium on the antioxidative response in Hydrilla verticillata (L.f.) Royle plants. Ecotoxicol Environ Saf. 2009;73:189–95.
Zheng X, He K, Kleist T, Chen F, Luan S. Anion channel SLAH3 functions in nitrate-dependent alleviation of ammonium toxicity in Arabidopsis. Plant Cell Environ. 2015;38:474–86.
Kreps JA, Wu Y, Chang HS, Zhu T, Wang X, Harper JF. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002;130:2129–41.
Deshmukh R, Sonah H, Patil G, Chen W, Prince S, Mutava R, et al. Integrating omic approaches for abiotic stress tolerance in soybean. Front Plant Sci. 2014;5:244.
Yang HY, von der Fecht-Bartenbach J, Friml J, Lohmann JU, Neuhauser B, Ludewig U. Auxin-modulated root growth inhibition in Arabidopsis thaliana seedlings with ammonium as the sole nitrogen source. Funct Plant Biol. 2015;42:239–51.
Vikman P, Fadista J, Oskolkov N. RNA sequencing: current and prospective uses in metabolic research. J Mol Endocrinol. 2014;53:R93–101.
Wang W, Messing J. Status of duckweed genomics and transcriptomics. Plant Biol. 2015;17(SI):10–5.
Cheng JJ, Stomp AM. Growing duckweed to recover nutrients from wastewaters and for production of fuel ethanol and animal feed. Clean-Soil Air Water. 2009;37:17–26.
Radić S, Stipaničev D, Cvjetko P, Marijanović Rajčić M, Sirac S, Pevalek-Kozlina B, et al. Duckweed Lemna minor as a tool for testing toxicity and genotoxicity of surface waters. Ecotoxicol Environ Saf. 2011;74:182–7.
Loque D, Yuan L, Kojima S, Gojon A, Wirth J, Gazzarrini S, et al. Additive contribution of AMT1;1 and AMT1;3 to high-affinity ammonium uptake across the plasma membrane of nitrogen-deficient Arabidopsis roots. Plant J. 2006;48:522–34.
Dhindsa RS, Matowe W. Drought tolerance in two mosses: correlated with enzymatic defence against lipid peroxidation. J Exp Bot. 1981;32:79–91.
Bestwick CS, Brown IR, Mansfield JW. Localized change in peroxidase activity accompanies hydrogen peroxide generation during the development of a non-host hypersensitive reaction in lettuce. Plant Physiol. 1998;118:1067–78.
Aebi H. Catalase in vitro. Method Enzymol. 1984;105:121–6.
Chen GX, Asada K. Ascorbate peroxidase in tea leaves - occurrence of 2 isozymes and the differences in their enzymatic and molecular-properties. Plant Cell Physiol. 1989;30:987–98.
Lv WT, Lin B, Zhang M, Hua XJ. Proline accumulation is inhibitory to Arabidopsis seedlings during heat stress. Plant Physiol. 2011;156:1921–33.
Kim M, Ahn JW, Jin UH, Choi D, Paek KH, Pai HS. Activation of the programmed cell death pathway by inhibition of proteasome function in plants. J Biol Chem. 2003;278:19406–15.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–7.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Boil. 2010;11:R106.
Ernst J, Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7:191.
Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang ZJ, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–7.
Poovaiah CR, Nageswara-Rao M, Soneji JR, Baxter HL, Stewart Jr CN. Altered lignin biosynthesis using biotechnology to improve lignocellulosic biofuel feedstocks. Plant Biotechnol J. 2014;12:1163–73.
Li QZ, Song J, Peng SB, Wang JP, Qu GZ, Sederoff RR, et al. Plant biotechnology for lignocellulosic biofuel production. Plant Biotechnol J. 2014;12:1174–92.
Gadjev I, Vanderauwera S, Gechev TS, Laloi C, Minkov IN, Shulaev V, et al. Transcriptomic footprints disclose specificity of reactive oxygen species signaling in Arabidopsis. Plant Physiol. 2006;141:436–45.
Sugimoto M, Oono Y, Gusev O, Matsumoto T, Yazawa T, Levinskikh MA, et al. Genome-wide expression analysis of reactive oxygen species gene network in Mizuna plants grown in long-term spaceflight. BMC Plant Biol. 2014;14:4.
Marino D, Dunand C, Puppo A, Pauly N. A burst of plant NADPH oxidases. Trends Plant Sci. 2011;17:9–15.
Torres MA, Dangl JL, Jones JD. Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc Natl Acad Sci U S A. 2002;99:517–22.
Trujillo M, Altschmied L, Schweizer P, Kogel KH, Hückelhoven R. Respiratory burst oxidase homologue A of barley contributes to penetration by the powdery mildew fungus Blumeria graminis f. sp. Hordei. J Exp Bot. 2006;57:3781–91.
Lam E, Zhang Y. Regulating the reapers: activating metacaspases for programmed cell death. Trends Plant Sci. 2012; 17: 487–94.
Mardanov AV, Ravin NV, Kuznetsov BB, Samigullin TH, Antonov AS, Kolganova TV, et al. Complete sequence of the duckweed (Lemna minor) chloroplast genome: Structural organization and phylogenetic relationships to other angiosperms. J Mol Evol. 2008;66:555–64.
Chen F, Liu CJ, Tschaplinski TJ, Zhao N. Genomics of secondary metabolism in populus: interactions with biotic and abiotic environments. Crit Rev Plant Sci. 2009;28:375–92.
Bartwal A, Mall R, Lohani P, Guru SK, Arora S. Role of secondary metabolites and brassinosteroids in plant defense against environmental stresses. J Plant Growth Regul. 2013;32:216–32.
Gill SS, Tuteja N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol Biochem. 2010;48:909–30.
Huang L, Lu YY, Gao X, Du G, Ma XX, Liu M, et al. Ammonium-induced oxidative stress on plant growth and antioxidative response of duckweed (Lemna minor L.). Ecol Eng. 2013;58:355–62.
Wang C, Zhang SH, Wang PF, Li W, Lu J. Effects of ammonium on the antioxidative response in Hydrilla verticillata (L.f.) Royle plants. Ecotox Environ Safe. 2010;73:189–95.
Farmer EE, Mueller MJ. ROS-mediated lipid peroxidation and RES-activated signaling. Annu Rev Plant Biol. 2013;64:429–50.
Apel K, Hirt H. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol. 2004;55:373–99.
Sandalio LM, Romero-Puertas MC. Peroxisomes sense and respond to environmental cues by regulating ROS and RNS signalling networks. Ann Bot. 2015;116:475–85.
Siddiqui MH, Al-Whaibi MH, Basalah MO. Role of nitric oxide in tolerance of plants to abiotic stress. Protoplasma. 2011;248:447–55.
Gupta Kapuganti J, Fernie Alisdair R, Kaiser WM. On the origins of nitric oxide. Trends Plant Sci. 2011;16:160–8.
Millenaar FF, Lambers H. The alternative oxidase: in vivo regulation and function. Plant Biol. 2003;5:2–15.
Serrano I, Romero-Puertas MC, Sandalio LM, Olmedilla A. The role of reactive oxygen species and nitric oxide in programmed cell death associated with self-incompatibility. J Exp Bot. 2015;66:2869–76.
Kwon SI, Hwang DJ. Expression analysis of the metacaspase gene family in Arabidopsis. J Plant Biol. 2013;56:391–8.
Watanabe N, Lam E. Arabidopsis metacaspase 2d is a positive mediator of cell death induced during biotic and abiotic stresses. Plant J. 2011;66:969–82.
Ihara-Ohori Y, Nagano M, Muto S, Uchimiya H, Kawai-Yamada M. Cell death suppressor Arabidopsis Bax inhibitor-1 is associated with calmodulin binding and ion homeostasis. Plant Physiol. 2007;143:650–60.
Yamada T, Takatsu Y, Kasumi M, Marubashi W, Ichimura K. A homolog of the defender against apoptotic death gene (DAD1) in senescing gladiolus petals is down-regulated prior to the onset of programmed cell death. J Plant Physiol. 2004;161:1281–3.
Cabane M, Afif D, Hawkins S. Lignins and abiotic stresses. In: Jouann L, Lapierre C, editors. Lignins: biosynthesis, biodegradation and bioengineering. San Diego: Elsevier Academic Press Inc; 2012.
Vanholme R, Morreel K, Darrah C, Oyarce P, Grabber JH, Ralph J, et al. Metabolic engineering of novel lignin in biomass crops. New Phytol. 2012;196:978–1000.
Vogt T. Phenylpropanoid biosynthesis. Mol Plant. 2010;3:2–20.
Fritz C, Palacios-Rojas N, Feil R, Stitt M. Regulation of secondary metabolism by the carbon-nitrogen status in tobacco: Nitrate inhibits large sectors of phenylpropanoid metabolism. Plant J. 2006;46:533–48.
Pitre FE, Lafarguette F, Boyle B, Pavy N, Caron S, Dallaire N, et al. High nitrogen fertilization and stem leaning have overlapping effects on wood formation in poplar but invoke largely distinct molecular pathways. Tree Physiol. 2010;30:1273–89.
Besseau S, Hoffmann L, Geoffroy P, Lapierre C, Pollet B, Legrand M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell. 2007;19:148–62.
Zhang XB, Gou MY, Liu CJ. Arabidopsis Kelch repeat F-box proteins regulate phenylpropanoid biosynthesis via controlling the turnover of phenylalanine ammonia-lyase. Plant Cell. 2013;25:4994–5010.
Meyer K, Shirley AM, Cusumano JC, Bell-Lelong DA, Chapple C. Lignin monomer composition is determined by the expression of a cytochrome P450-dependent monooxygenase in Arabidopsis. Proc Natl Acad Sci USA. 1998;95:6619–23.
Acknowledgements
We thank the Gene Denovo at Guangzhou for its assistance in related bioinformatics analysis.
Funding
This research was supported by the NSFC project (51108239) and Applied Basic Research Program of Sichuan Province (2013JY0005).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: WGW, QZ. Performed the experiments: WGW, RL, QLZ, XYT. Contributed reagents/materials/analysis tools: WGW, RL, QLZ. Wrote the paper: WGW, RL, XYT, QZ. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Primers used in this paper. (DOCX 17 kb)
Additional file 2: Figure S1.
Sequencing saturation analysis (A) and distribution of gene coverage (B) in each library. (DOCX 78 kb)
Additional file 3: Figure S2.
Unigenes matching the 15 top species using BLASTx in the nr database. (DOCX 415 kb)
Additional file 4: Figure S3.
COG functional classification of all unigenes sequences. 14,172 (20.04 %) unigenes showed significant similarity to sequences in the COG databases and were clustered into 25 categories. (DOCX 124 kb)
Additional file 5: Figure S4.
Profiles order based on the P-value significance of number assigned versus expected. (DOCX 32 kb)
Additional file 6:
Differentially expressed genes. (XLSX 818 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, W., Li, R., Zhu, Q. et al. Transcriptomic and physiological analysis of common duckweed Lemna minor responses to NH4 + toxicity. BMC Plant Biol 16, 92 (2016). https://doi.org/10.1186/s12870-016-0774-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-016-0774-8