
Abstract
Background
Phytophthora blight caused by Phytophthora cajani is an emerging disease of pigeonpea (Cajanus cajan L.) affecting the crop irrespective of cropping system, cultivar grown and soil types. Current detection and identification methods for Phytophthora species rely primarily on cultural and morphological characteristics, the assessment of which is time-consuming and not always suitable. Sensitive and reliable methods for isolation, identification, zoospore production and estimating infection severity are therefore desirable in case of Phytophthora blight of pigeonpea.
Results
In this study, protocols for isolation and identification of Phytophthora blight of pigeonpea were standardized. Also the method for zoospore production and in planta infection of P. cajani was developed. Quantification of fungal colonization by P. cajani using real-time PCR was further standardized. Phytophthora species infecting pigeonpea was identified based on mycological characters such as growth pattern, mycelium structure and sporangial morphology of the isolates and confirmed through molecular characterization (sequence deposited in GenBank). For Phytophthora disease development, zoospore suspension of 1 × 105 zoospores per ml was found optimum. Phytophthora specific real-time PCR assay was developed using specific primers based on internal transcribed spacer (ITS) 1 and 2. Use of real-time PCR allowed the quantitative estimation of fungal biomass in plant tissues. Detection sensitivities were within the range of 0.001 pg fungal DNA. A study to see the effect of elevated CO2 on Phytophthora blight incidence was also conducted which indicated no significant difference in disease incidence, but incubation period delayed under elevated CO2 as compared to ambient level.
Conclusion
The zoospore infection method for Phytophthora blight of pigeonpea will facilitate the small and large scale inoculation experiments and thus devise a platform for rapid and reliable screening against Phytophthora blight disease of pigeonpea. qPCR allowed a reliable detection and quantification of P. cajani in samples with low pathogen densities. This can be useful in early warning systems prior to potential devastating outbreak of the disease.
Similar content being viewed by others


Background
Pigeonpea (Cajanus cajan (L.) Millsp.) is one of the important legume crops of rainfed agriculture in the semi-arid tropics. The Indian subcontinent, Eastern Africa and Central America, three main pigeonpea producing regions of the world cultivates pigeonpea either as a sole crop or intermixed. As pigeonpea contains high level of protein and some essential free amino acids like methionine, lycine and tryptophan, the importance of the pigeonpea to world vegetarian population is very significant. In India, pigeonpea is second most important legume after chickpea and alone contributes 72.5% of world cultivated area with 62.5% of world production [1]. The accelerated susceptibility of pigeonpea to diseases in Indian subcontinent is one of the main roots of its deteriorating productivity.
Phytophthora blight (PB), caused by Phytophthora cajani is an emerging disease in pigeonpea [2,3]. Information on total economic losses in India caused by PB is not available, but the disease is of rising importance since last a few years and has the potential to cause 100% yield losses in field conditions under favourable environment. Occurrence and widespread distribution of PB has been reported in areas especially when excessive rains fall within a short span of time and hot and humid weather persists during the crop season [2]. There is a lack of comprehensive knowledge on available resistant genotypes to P. cajani [3].
Identification of Phytophthora species by conventional diagnostic tests abased on morphology, and growth on selective media is time consuming, laborious and takes considerable skill. In addition, taxonomic expertise is required for correct identification within the closely related species. Again the prospective for quantification of biomass of pathogen is limited. In this regard, the aim of the study was also to develop a rapid, highly specific and very sensitive method for the potential quantification of P. cajani. The polymerase chain reaction (PCR) has long been used to detect the pathogens and is a highly sensitive and relatively fast method that allows detecting specific target DNA molecules in a complex mixture, offering an alternative to microbiological conventional procedures in fungal diagnostic. One of the most important factors in the development of such molecular methods is the reliability of the primer set and the targeted DNA sequence of interest organism [4]. Nuclear rDNA including the small and large subunits, 5.8S and the internal transcribed spacer (ITS) region, proved to be an ideal target for specific PCR primers, as each sequences is variable at the family, genus or species level [5]. The ITS region has been shown to be largely conserved within Phytophthora spp. but differ across species [6,7]. Most importantly, sequence information is available in this region for nearly all known species of Phytophthora [6]. Consequently, we designed P. cajani specific primers within the ITS region. The current technique has the further advantage of being able to be performed as real-time PCR, visualized using an intercalating dye such as SYBR green. Real-time PCR allows products to be distinguished based not only size but also on sequence, because melt temperatures will differ for same size but distinct products [8,9].
In order to establish a successful method for in vitro infection of P. cajani, a standardised protocol is needed to culture at its pathogenic state and to isolate the zoospores. However, knowledge about the conditions which govern infection by zoospores of P. cajani is yet unknown. Several workers have described inoculation techniques using mycelium [10,11], sporangium [12] and zoospores [13,14] for obtaining infection with Phytophthora spp.; however P. cajani being a putatively novel species, very limited information about laboratory protocols are available. Further, Amin et al. [15] recognized that P. cajani resembled P. drechsleri but noted that P. cajani produced larger sporangia and undifferentiated sporangiophores. They also considered that the homothallic nature of P. cajani differentiated it from P. drechsleri, which is considered to be heterothallic according to Savage et al. [16]. However, Kannaiyan et al. [17] expressed the opinion that PB caused by P. drechsleri var. cajani and P. cajani are probably the same. Detailed descriptions for both these pathogens have been provided in Phytophthora Diseases Worldwide by Erwin and Ribeiro [18]. In current study, attempts have been made to establish an in planta infection system of P. cajani in pigeonpea.
Host and pathogens are influenced by the interactive effects of multiple climatic factors. If any one of the factors of disease triangle (host, pathogen and environment) is altered, changes in the progression of a disease epidemic can occur [19]. High humidity, rising temperature, elevated CO2 and depletion of O3, all is events of imperative deviations in atmospheric components. Changes in these important climatic factors play a direct or/and indirect role in changes of pathosystem and in disease expression [20]. Anthropogenic emissions are drastically increasing the concentration of atmospheric CO2, as in 1750 atmospheric CO2 was 280 ppm has increased to 400 ppm in 2013 and the projected concentration to be1250 ppm by 2095 [21]. Elevated CO2 directly alters growth, development, metabolism and plant physiology which, in turn, has an impact on pathogen invasion and disease progress [22]. Elevated CO2 can also modify plant–pathogen interactions primarily through changes in host plants [23].
Till now it is not clear in the literature, whether the disease severity is enhanced or diminished by a higher CO2. The study of Thompson and Drake [24] showed that elevated CO2 reduced powdery mildew (Erysiphe graminis) on wheat and the severity of rust (Puccinia sparganioides Ellis & Barth) on C3 sedge (Scirpus olneyi) Grey. On the other side, higher temperature and increased CO2 concentration are also posing higher threat by increasing fungal disease perception namely late blight of potato, rice blast and sheath blight. In this context, there is a need to study effect of elevated CO2 on Phytophthora blight of pigeonpea, as in the last decade the disease dynamics of pigeonpea has changed drastically in India.
The overall objective of the study was to (i) standardize protocol for isolation and infection of P. cajani, (ii) assess the effect of elevated atmospheric concentration of CO2 on PB development and (iii) quantify P. cajani during in planta infection.
Results
Isolation of Phytophthora
Pigeonpea plants with typical symptoms of Phytophthora blight were sampled from the infected fields of multiple locations of two different states of India during 2012-2013 and 2013–2014. Blight symptoms included brown to dark brown lesions distinctly different from healthy green portions on main stem, branches and petioles. These lesions sometimes elongate and cause girdle and cracks on the stems. Varying levels of PB incidence was recorded in surveyed locations (5.25-69.9%) irrespective of years. There was no effect of soil type on PB incidence (Table 1). A total of 14 isolates of Phytophthora (nine from 2012–2013 and five from 2013–2014), one isolate from Varanasi-Uttar Pradesh, one isolate from each district of Mahbubnagar and Adilabad of Telangana State and rest 11 isolates from different experimental fields (BP5, BP09, BP14, BP15, DHF4, RL17 and RM1) of ICRISAT were obtained (Table 1). The pure culture of the isolates was maintained on 20% tomato extract agar media at 15 ± 1°C in dark condition.
Morphological identification
The growth pattern, mycelium structure and sporangial morphology of the individual isolates of Phytophthora were examined under microscope for identification of the species. Pathogen formed distinct colonies in rosaceous, stellate or cottony pattern irrespective of isolates. The mycelium of the isolates was coenocytic and branched. Some isolates formed hyphal swellings on V8 agar. Karyotyping of the isolates showed single or pair of nuclei within the hyphae. Sporangium produced by different isolates varied in structure from broadly ovoid, obpyriform to elongate and non papilate. Based on morphological characters, the pathogen was identified as Phytophthora cajani (Figure 1a-f). Amphigynous anthredia were also observed and resembled to Phytophthora cajani as given in the manual of Phytophthora: Identifying species by morphology and DNA fingerprints [25].

Morphology of Phytophthora cajani isolate ICPC 1: (a) growth in Petriplate; (b) coenocytic mycelium; (c) hyphal swellings; (d) non-papillate sporangia; (e) zoospores; (f) oospore.
Molecular characterization
To confirm the P. cajani at molecular level, the ITS sequence of 5.8S rDNA of the 14 isolates was amplified and sequenced. The sequences were purified and were used in a BLASTn against the Phytophthora database (http://www.phytophthoradb.org/). The BLAST result confirmed the identity of the isolates as P. cajani. The sequences were deposited under the GenBank (Table 1). The pair-wise nucleotide sequence identity matrix showed 99-100% sequence identity among the isolates. The phylogenetic relationship among the isolates and as well as with other Phytophthora spp. was made based on nucleotide sequences of ITS region. In phylogenetic tree, it was found that all the isolates of P. cajani were grouped together in the same clade with the nearest relative P. sojae isolate. There was no phylogenetic discordance within the P. cajani isolates (Figure 2).

The maximum likelihood phylogenetic tree showing the relationship among the P. cajani isolates of this study and other Phytopthora spp. based on ITS sequences of 5.8S rDNA. Scale bar represent the genetic distance, proportional to the number of nucleotide differences between branch nodes. The significance of the nodes was estimated with 1000 bootstrap repetitions. The isolates of the current study is marked by Italics font.
Zoospores production and in planta infection
Pathogenicity of all the 14 isolates was tested individually on the susceptible cultivar of pigeonpea ICP 7119. The pathogenicity of the isolates was tested on 50 seedlings. The complete mortality of seedlings was recorded two days after the inoculation. Lesions were seen on the stem and whole seedlings collapsed within 6–7 days after inoculation. All the 14 isolates were found to be virtually equal pathogenic to host ICP 7119 (data not shown). The P. cajani isolate ICPC 1 was taken to develop a method for zoospores production and in planta infection as it had shown consistent susceptible reaction. We developed the reproducible method for zoospore production from P. cajani and the method of inoculation for rapid screening for resistance to PB in pigeonpea genotype. The zoospores produced by P. cajani isolate ICPC 1 were measured under haemocytometer and inoculum concentration of 1.5 × 105 zoospores per ml was found to be optimum for PB development. The symptoms started after 2 days of inoculation and 100% seedling mortality was recorded at 3–4 days post inoculation. Un-inoculated plants did not show any disease symptom throughout the tests.
PB incidence under elevated CO2
Incubation period, measured as the time in days between inoculation and disease symptoms was significantly delayed under elevated CO2 as compared to ambient (p < 0.001). PB symptoms appeared after 30 hours of inoculation under ambient condition, after 36 hours under elevated CO2 levels of 550 ppm and 40 hours under 700 ppm but, it was observed that disease progressed faster under elevated CO2 as compared to ambient. No significant effect of ambient and elevated CO2 concentrations on PB disease incidence was observed after a certain period of inoculation (48 hours and 72 hours after inoculation) (Figure 3).

Comparison on progression of PB incidence under ambient condition and elevated CO 2 .
Development of qPCR for the quantification of Phytophthora cajani
Specificity of the primers
The specificity of the qPCR primers designed from ITS sequences of ribosomal DNA was assessed using DNA from targeted Phytophthora and non-targeted other fungal species such as Fusarium udum, Macrophomina sp, Alternaria alternata. The assessment showed that the specific primers, qPCR_F2 and qPCR_R2 used in this study only amplified 146 bp PCR products from targeted species, P. cajani (Table 2). The PCR product was sequenced and sequence obtained from the amplicon was aligned with targeted sequence. Specificity of the primers was confirmed by its 100% similarity with P. cajani. No signal was generated from any non-targeted fungal pathogen indicating excellent specificity and sensitivity of the primers.
The standard curve
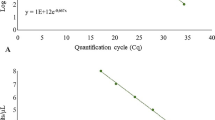
A standard curve was generated by plotting the cycle threshold value (Ct) versus the logarithm of the concentration of each serial dilution of DNA in a 10 fold over a 7-log range from 10 to 1 × 10−4 ng/μL. A good correlation was observed between Ct values and DNA concentration of standard. The slope of linear regression curve was −3.325 with the correlation coefficient R2 = 0.9822 demonstrating the PCR efficiency of 99.87% (Figure 4a) Therefore, the standard curve obtained in this study indicated that the nominated primer was highly specific over a linear series of magnitude.

Calibration of qPCR for quantification of P. cajani : (a) Standard curve analysis: Standard curve showing the correlation between the log10 DNA amounts (ng) vs. the Ct values for 10 fold dilution of P. cajani pure genomic DNA. (b) Melting curve analysis: The melting curve (SYBR Green florescence versus temperature) of specific amplifications from the ITS sequences of 5.8S rDNA at different concentrations. The single pick of targeted amplicon at melting temperature (Tm) 79.8°C indicates the specificity of the qPCR primers to P. cajani. No contaminating product was detected in PCR reaction.
The dissociation analysis showed that the SYBR® Green consistently generated a single peak at 79.8°C in the PCR reactions, demonstrating the presence of only one specific product in the reaction (Figure 4b). The developed qPCR system was used to analyse the amount of P. cajani DNA present in an unknown sample by interpolating its Ct value against the standard curve. The qPCR sensitivity result showed at least 0.001 pg fungal DNA was needed for a positive signal in reaction for SYBR-Green detection (results not shown).
Phytophthora cajani colonization under elevated CO2
Fungal colonization was delayed with the increase of CO2 concentration. In ambient condition, the fungal DNA reached to the minimum detection limit 0.001 pg in 1.0 ng of host plant DNA after 20 h of the inoculation with zoospores. In case of elevated CO2 concentration of 550 ppm and 700 ppm, the fungal colonization was detected at 27 h post inoculation. The amount of fungal DNA was found to be increased in the host tissues with the time period. The maximum fungal colonization was recorded at 72 h of post inoculation. In ambient, the maximum 0.25 ng of P. cajani DNA was detected within the 1.0 ng root DNA of pigeonpea, whereas 0.18 ng and 0.16 ng fungal DNA was measured within the 1.0 ng root DNA of the plants grown in 550 ppm and 750 ppm CO2, respectively (Figure 5). No cross reactivity was found with non-inoculated plant samples.

Chronological colonization profile of P. cajani in root tissues of inoculated pigeonpea (cv. ICP 7119) seedlings grown in three different CO 2 conditions, ambient, 550 ppm and 700 ppm respectively. Absolute quantification of fungal DNA was determined in real time PCR assay using ITS sequences of 5.8S rDNA. Error bar represents the standard error of three biological replicationsat the 95% confidence interval.
Discussion
We studied the effect of elevated CO2 on PB of pigeonpea. The study included isolation and molecular characterisation of Phytophthora cajani and simultaneously developing a standard protocol for zoospore production and reliable inoculation method for in planta infection process. Our current study documented first time the molecular identification of P. cajani on pigeonpea and further quantified the P. cajani in pigeonpea seedlings grown under ambient and elevated CO2.
The ITS sequence of rDNA is the most proposed DNA region in molecular fungal ecology and has been recommended as the sole universal fungal barcode for fungi [26]. For Phytophthora spp., ITS have been shown to be useful for species identification, although some related species share identical ITS sequences [5,27]. In addition, universal ITS1 and ITS4 primers that has been widely used in most of the labs, was successfully used in this study to identify the P. cajani from the isolated fungal samples. In the current study, fourteen P. cajani isolates were identified and characterised at molecular level using ITS1 and ITS4 primers from different locations of India. Although P. cajani is a novel species, phylogenetic relationship of P. cajani with the other taxa of Phytophthora based on nucleotide sequence is now well recognized. In our study, it was found that all 14 P. cajani isolates grouped into same clade with nearest relative P. sojae. Based on DNA distance analysis of the combined ITS1, 5.8S subunit, and ITS2 regions of the genomic ribosomal RNA tandem gene repeat, Cooke et al. [28] showed that P. cajani placed into Clade 7b where the nearest relative P. sojae does belong. There was no discordance phylogenetic relationship noted within the isolates.
Phytophthora species are notorious oomycete pathogen that causes diseases through wide range of inoculum, e.g. mycelium, sporangia, oospore and zoospore. A suitable protocol is required in order to establish a successful infection in planta. Several researchers have worked on different Phytophthora species to enable an efficacious and repeatable method for in planta infection, but for Phytophthora sp. on pigeonpea, none of the methods tested in the past have been found consistently reliable (unpublished data). Various methods reported for screening Phytophthora in different crops included; in vitro leaf disk infection of brussels sprout plants (Brassica oleracea var. gemmifera) by infectious zoospores of P. brassicae [14]; root dip of pea in zoospore suspension of P. pisi [29]; leaf detached method in Nicotiana tabacum for P. parasitica var. nicotianae [11]; germination of soybean seeds in soil supplemented with zoospores suspension of P. megasperma var. sojae [30] and rapid radicle assay in pepper (Capsicum annuum L.) for P. capsici [31] etc. But no effective screening technique was reported for resistant selection of pigeonpea to P. cajani. This is the first time we have established a successful method for obtaining high concentrations of zoospores followed by its use for standardization of inoculation technique in pigeonpea. Use of zoospores to establish in planta infection is convenient and suitable to carry out small and large scale inoculation experiments. Testing of previously published techniques [11,14,29-31] with various alterations was carried out, including those found useful for other Phytophthora species to develop a suitable method for obtaining high concentrations of P. cajani zoospores. Finally, we were successful in developing a repeatable, reliable and economical screening technique using zoospore suspension for PB development.
Visual estimation of infection in the field is time-consuming. Furthermore, clear differentiation of the Phytophthora species based on morphology requires expert knowledge. However, with the advent of real-time PCR, plant pathologist possesses the unprecedented ability to accurately quantify the specific pathogen within a host plant [32]. Since last one decade efforts have been made in detection and quantification of various plant pathogens e.g. Macrophomina phaseolina [33], Phytoplasma [34], different Fusarium species [35], Phytophthora species [29]. In this study, real-time PCR assay based on SYBR green chemistry was developed for P. cajani quantification.
To determine the effect of elevated CO2 on colonization and disease establishment by P. cajani in pigeonpea, we targeted 5.8S rDNA to quantify the fungal DNA in host tissue. The sensitivity of the assay was determined using dilution series of pure DNA of known concentration from the P. cajani. In general, the detection limit was 0.001 pg per 1.0 ng of host DNA. The reproducibility of the real time assay was evaluated for initial DNA content, presence of PCR inhibitors etc. The low variation between assays and single pick in melting curve indicated high reproducibility of the real-time PCR. However, the variation was eliminated by normalizing samples with the control.
We observed delayed colonization of P. cajani and disease establishment under elevated CO2. However, pigeonpea seedlings showed no significant difference in disease incidence under elevated and ambient CO2 levels. The amount of fungal DNA was found to be increased in the host tissues with the time period and was more in ambient as compared to elevated CO2. However, it is not clear that reduction of fungal growth performance is because of negative effects of elevated CO2 on fungal performance. Tomato plants showed increased tolerance to infection by Phytophthora parasitica at elevated 700 μMmol−1 of CO2. The results suggested that more vegetative growth in aerial parts and as well as in root systems under higher CO2 might have reimbursed the loss of growth caused by the root pathogen [36]. Reduction of powdery mildew (E. graminis) on wheat and barley and the severity of rust (Puccinia sparganioides Ellis & Barth) are also reported [24]. On the other side, higher threats of Phytophthora infestans in potato and diseases of rice, like blast (Pyricularia oryzae) and sheath blight (Rhizoctonia solani) is increased day to day due to elevated CO2 concentration and higher temperature [37]. For instances, it was found that in vitro exponential growth rates of Phyllosticta minima, a maple fungal pathogen were enhanced by 17% under elevated CO2, discounting the possibility that disease reductions by elevated CO2 [38].
Increased CO2 levels can influence both the host and the pathogen in various means. Under elevated CO2, sometimes internal physiological adjustment occurs in plant systems that alter sugar concentration, C:N ratio by producing phenolics compounds in the cells and nutritive quality may lead to unfavourable condition for growth of pathogenic organism and reduced the disease severity. In few study, it was found that elevated CO2 increased the thickening of the roots in plants by more production of root border cells [39] and lead to the increased amounts of root exudates into the rhizodeposition [40,41]. The fungal penetration also inhibit in root cells due to thickening of cell walls by deposition in root due to elevated CO2. These changes make the environment more favourable for plant defence. As elevated CO2 directly involve in change of the vegetative growth of the plants, it is assumed that the amendment of the auxin, cytokinin ratio in plant cells also occurs simultaneously. It is reported that the auxin content modulates the infectivity of Phytophthora parasitica in Arabidopsis [42]. Therefore, our further study is aimed to investigate the effect of elevated CO2 coupled with other climate variables such as temperature on PB of pigeonpea and investigate the expression of resistance at biochemical level.
Conclusions
Little is known concerning the isolation method and the infection by zoospores of Phytophthora cajani, being a novel species on pigeonpea. The study included isolation and molecular characterisation of P. cajani and simultaneously established a standard protocol for zoospore production and in planta inoculation technique via isolated zoospores. The zoospore inoculation will facilitate the epidemiological studies and thus will be useful for developing a rapid and reliable screening technique against Phytophthora blight of pigeonpea. qPCR had allowed detection and quantification of P. cajani in samples with low pathogen densities. No significant effect of elevated CO2 on PB of pigeonpea was observed. To the best of our knowledge, this is the first report of molecular identification of P. cajani, zoospore inoculation method and real-time PCR application used to assess the extent of colonization by P. cajani during disease development in pigeonpea.
Methods
Plant material
The seeds of Pigeonpea cv. ICP 7119 (commonly known as HY-3C) used for this study were procured from the breeding unit of the International Crop Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru, Telangana State, India. The variety is reported highly susceptible to Phytophthora blight [43].
Fungal isolates and disease incidence
During a disease survey 2012–13 and 2013–14 Kharif seasons, pigeonpea plants exhibiting symptoms of PB were sampled from epidemic block in the field from different locations in the districts of Telangana State and Uttar Pradesh in India (Table 1). Symptomatic plant materials were placed in labelled plastic bags, which were transported in cooler box and stored in 4°C until fungal culture were purified from the samples in the laboratory (Legumes Pathology, ICRISAT). Stem tissues with typical PB lesions were selected for direct isolation of the Phytophthora. Symptomatic tissues were cut and surface sterilised by sodium hypochlorite (2% v/v) for 2–3 min followed by washing in sterile distilled water. The tissues were then placed onto V8 juice agar media (Himedia, Mumbai, India) amended with PARP antibiotics (pimarcin 400 μL; ampicillin 250 mg; rifampicin 1000 μL; and pentachloronitrobenzine 5 mlL−1 media). Plates were incubated at 25°C in the 12 h/12 h day-night photoperiod for 5 days. Putative Phytophthora colonies were transferred to 20% tomato extract agar slants and maintained under in vitro at 15 ± 1°C in dark condition by regular sub culturing after 15–20 days. The disease incidence of the individual fields from where the plants sampled were recorded and their details were provided in Table 1. Disease incidence (DI) was calculated by following formula:
The P. cajani isolates used in the present study are available with ICRISAT for further use by any scientific community. These isolates are provided based on standard material transfer agreement.
Morphological identification
Individual isolates were grown on V8 juice agar plate at 25-28°C under continuous fluorescent light. After 5 to 7 days, 5 mm disc of an actively grown hyphal culture was inoculated on 5% tomato juice extract in Petri plates and incubated at 25°C. After 24 hr, the extract was replaced with sterile distilled water and incubated for another 24 hr in the same conditions. After this, the colony and sporangial morphology was observed under the microscope. Other structures, oogonia, antheridia and oospores were also observed on V8 juice agar and tomato agar 2–3 weeks after incubation at 30°C.
Identification with ITS sequencing
The internal transcribed spacer region (ITS1 and ITS2) including 5.8 s gene of the ribosomal DNA (rDNA) of each isolate was sequenced for DNA-based identification. An agar disk about 5 mm of each isolate grown in V8 juice agar were transferred into 50 mL of tomato juice broth in a 250 mL flask for 5 to 7 days at 25°C. Mycelial mats were harvested by discarding the media and dried on autoclaved blotting paper. Dried mycelial mat was stored at −80°C. Genomic DNA (gDNA) was extracted from mycelia of Phytophthora with the PureLink Plant Total DNA Purification kit (Invitrogen, USA) as per manufacturer’s protocol using 100 mg of fungal tissue. The purified DNA was evaluated in 0.8% (w/v) agarose gel stained with 0.2 μg/mL ethidium bromide and visualized under UV light. The quantity and quality of total DNA was determined by the OD260/280 using Nanodrop spectrophotometer (Thermo Scientific, USA). The extracted DNA was stored at −20°C.
PCR amplification of the ITS regions (ITS1 and ITS2) and 5.8S ribosomal DNA was performed with the ITS1 (F) and ITS4 (R) (Table 2) [44]. Reactions were performed in 50 μL volumes composed of 5 μL of 10X PCR buffer, 3.0 μL of 50 mM MgCl2, 1.0 μL of 10 mM dNTP mix (2.5 mM of each dNTP), 0.5 μL of each 10 mM primer, 0.5 μL of 5 U/μLTaq DNA polymerase Brazilian origin (Invitrogen, USA), 1 μL of extracted DNA and nuclease-free water for volume make up to 50 μL. Thermal cycling conditions consisted of an initial denaturation of 94°C for 5 min; 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and a final extension step of 72°C for 10 min. Amplified products were confirmed and concentrations estimated visually in with 1% agarose gel electrophoresis. The PCR products were then sequenced in an automated sequencer using ITS1(F) and ITS4 (R) by a commercial service.
Phylogenetic analysis
The DNA sequences of all P. cajani isolates were aligned in BioEdit v. 7.2.5 and edited manually for generating consensus sequences. The sequence of each isolate was subjected to BLAST analysis using the database of Phytophthora (www.phytophthoradb.org) and GenBank (NCBI, Bethesda, MD). The sequences were then submitted to NCBI data base. Multiple sequence alignment was performed with CLUSTAL X version 1.81 and pairwise sequence identities among the P. cajani isolates were compared using Gene Doc version 2.6.002. Phylogenetic tree was constructed with 35 other Phytophthora spp. as out groups using the program MEGA6.06. Model was predicted by JModeltest 2.1.7 v20141120 software. The best suited model for the data was NrT + G + I on the basis of BIC, AIC and DT scores [45]. The tree was constructed with Maximum Likelihood statistical mode using the NT93 with gamma (G) plus invariable parameter (I) and taking 1000 replications for bootstrap value.
Standardization of zoospore production
For in planta infection of pigeonpea seedlings, method for preparation of zoospores suspension culture from P. cajani was standardised. Isolate ICPC 1 was chosen as it was consistently virulent on susceptible genotype. One piece (5 mm) of mycelial mat from pure culture of isolate ICPC 1 was inoculated in 100 ml conical flasks containing 25 mL 5% tomato extract broth and incubated in darkness at 25°C for 72 h. After post inoculation period, the tomato extract broth was decanted and replaced with 25 mL sterile pond water, which was immediately decanted again and replaced with 25 mL fresh pond water, in which the mycelium was further incubated at 25°C in dark for 4 h. The procedure was repeated soon after the completion of incubation period and incubated for 20 h, in these cases with single changes of water. Finally, mycelial growth of isolate was removed from the flask and zoospores were harvested in water suspension. The zoospores were checked under microscope and the concentration of zoospores was measured in a haemocytometer. The suspension was used for in planta infection with further dilution.
In planta infection system under elevated CO2
The apparently healthy seeds of ICP 7119 were surface sterilized in sodium hypochlorite (1% v/v) for 2–3 min and then washed in sterile double distilled water. The seeds were sown (1 seed/well) in seed trays filled with autoclaved sand:vermiculite (9:1) mixture at 2 cm depth and well watered. The trays were then divided into three different sets, each treatment with three replications with 50 seedlings per tray and kept in three separate plant growth chambers adjusted with three different experimental conditions. Plant growth chambers were tuned to adjust different CO2 concentration and termed as E1 for ambient (~380 ppm), E2 for elevated CO2 to 550 ppm and E3 for elevated CO2 to 700 ppm, and maintained at 28°C with >75% humidity. After seven days, seedlings were inoculated with P. cajani zoospore suspension (1.5 × 105 zoospores/mL) so that the roots were submerged up to collar region in each well. Similar number of seedlings inoculated with only sterilized water served as un-inoculated control. The experiment was designed in completely randomized design (CRD) manner and disease incidence was recorded everyday up to the complete mortality. The critical differences (CD) value of disease incidence was calculated at 1% level at 36 h, 48 h and 72 h.
For quantification of P. cajani infection in planta, seedlings were harvested 0 h (immediate after infection), 2 h, 20 h, 27 h (30), 48 h and 72 h of post inoculation, washed and preserved in −80°C for further experiments.
Total genomic DNA (gDNA) extraction from plant samples
Total gDNA from Phytophthora infected plant samples was isolated using PureLink Plant Total DNA Purification kit (Invitrogen, USA) as per manufacturer’s protocol. About 100 mg of root tissue was ground in liquid N2 and resuspended in 250 μL Resuspension Buffer (supplied in the kit). The tissues were homogenized with vigorous vortexing until sample was completely resuspended. About 15 μL 20% SDS and 15 μL RNase A (20 mg/mL) were added to the tissue resuspension and incubated at 55°C for 15 minutes to complete lysis of tissues. Total gDNA was eluted in 50 μL of Elution Buffer and stored at −20°C for further downstream application. The purified DNA was evaluated in 0.8% agarose gel as well as by UV spectrophotometry. The extracted DNA was stored at −20°C.
Development and evaluation of qPCR primers
To quantify the growth of P. cajani within the host tissue under different experimental conditions (E1, E2 and E3), the real-time PCR was carried out. Three pairs of sequence specific primers were designed from internal transcribed spacer (ITS) sequences of P. cajani using IDT Primer Quest software (eu.idtdna.com/Primerquest/Home/Index). ITS sequences (Acc nos. KJ010534-KJ010538) were aligned using BioEdit v. 7.2.5. and primers were designed from conserved region of the ITS sequences. To test the specificity of the primers pairs, PCR was carried out separately with the individual primers sets using pure gDNA of P. cajani in 50 μL reaction mixture containing: 5 μL of 10 X Taq polymerase buffer, 1.5 μL 50 mM MgCl2, 1 μL of 10 mM each dNTP, 1 μM of each primer, 1 μL of 5u/1 μL Taq polymerase (Invitrogen, USA), about 100 ng of gDNA and H2O up to 50 μL. Reaction conditions were: 94°C for 4 min, (94°C for 45 s, 60°C for 45 s and 72°C for 30 s) × 35 cycles followed by incubation at 72°C for 10 min. The amplified products were separated by 1.5% (w/v) gel electrophoresis and sequenced. The expected size of PCR amplicons was constantly generated from the real-time PCR analysis.
Real-time PCR reaction
Real-time PCR was carried out in a total volume of 20 μL consisting of 10 μL 2x SYBR Green PCR Master Mix (Applied Biosciences, USA), 500 nM of each primer (qPCR_F2 and qPCR_R2) (Table 2), and 1 ng of each template DNA. Sterile bi-distilled water was added up to a final volume of 20 μL. The PCR thermal cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 10 s, and 62°C for 30 s (during which the fluorescence was measured). Following the final amplification cycle, a melting curve was constructed by measuring the fluorescence continuously when heating from 60 to 95°C at the rate of 0.5°C per second. qPCR reactions were performed in a PikoReal 24 Real-Time PCR Detection System (Thermo Scientific, USA). To generate the standard curve, DNA from pure culture of P. cajani was subjected to qPCR with a 10-fold dilutions ranging from 10 ng to 0.01 pg under the same conditions described above. Quantification values were automatically determined by the PikoReal software version 2.0 (Thermo Scientific, USA) and the threshold cycle (Ct) values were then obtained. The standard curve is a plot of the Ct versus log DNA concentration. In all the experiments, appropriate negative controls containing no template were subjected to the same procedure to eliminate or to detect whether any possible DNA contamination present. Each sample was amplified in triplicate replication in every experiment.
Abbreviations
- ITS:
-
Internal transcribed spacer
- qPCR:
-
Quantitative polymerase chain reaction
- PB:
-
Phytophthora blight
- rDNA:
-
Ribosomal DNA
- ICRISAT:
-
International Crops Research Institute for the Semi-Arid Tropics
- PARP:
-
Pimarcin ampicillin, rifampicin and pentachloronitrobenzine
- DI:
-
Disease incidence
- gDNA:
-
Genomic DNA
- UV:
-
Ultra violet
- BLAST:
-
Basic local alignment search tool
- NCBI:
-
National Center for Biotechnology Information
- MEGA6.06:
-
Molecular evolutionary genetics analysis version. 6.06
- CRD:
-
Completely randomized design
- CD:
-
Critical differences
- IDT:
-
Integrated DNA technologies
- Ct:
-
Cycle threshold
References
FAO. FAO Year book (Production). Food and Agriculture Organization of the United Nations. 2012.
Pande S, Sharma M, Mangla UN, Ghosh R, Sundaresan G. Phytophthora blight of Pigeonpea [Cajanus cajan (L.) Millsp.]: an updating review of biology, pathogenicity and disease management. Crop Prot. 2011;30:951–7.
Sharma M, Pande S, Rao JN, Kumar PA, Reddy DM, Benagi VI, et al. Prevalence of phytophthora blight of pigeonpea in the Deccan Plateau of India. Plant Pathol J. 2006;22:309–13.
Grünwald NJ, Martin FN, Larsen MM, Sullivan CM, Press CM, Coffey MD, et al. Phytophthora-ID.org: a sequence-based Phytophthora identification tool. Plant Dis. 2011;95:337–42.
Cooke DEL, Duncan JM. Phylogenetic analysis of Phytophthora species based on ITS1 and ITS2 sequences of the ribosomal RNA gene repeat. Mycol Res. 1997;6:667–77.
Cooke DEL, Schena L, Cacciola SO. Tools to detect, identity and monitor Phytophthora species in natural ecosystem. J Plant Pathol. 2007;89:13–28.
Lee SB, Taylor JW. Phylogeny of five fungus-like Protoctistan Phytophthora species, inferred from the Internal Transcribed Spacer of Ribosomal DNA. Mol Biol Evol. 1992;9:636–53.
Ririe KM, Rusmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154–60.
Tajima K, Aminov RI, Nagamine T, Matsui H, Nakamura H, Bennoy Y. Diet-dependent shifts in bacterial population in the rumen revealed with real-time PCR. Appl Environ Microbiol. 2001;7:2766–74.
Stewart S, Robertson AE. A modified method to screen for partial resistance to Phytophthora sojae in soybean. Crop Sci. 2012;52:1181–6.
Tedford EC, Miller TL, Nielsen MT. A detached-leaf technique for detecting resistance to Phytophthora parasitica var. nicotianae in tobacco. Plant Dis. 1990;74:313–6.
Nowakowska M, Nowicki M, Kłosinska U, Maciorowski R, Kozik EU. Appraisal of artificial screening techniques of tomato to accurately reflect field performance of the late blight resistance. Plos One. 2014;9:1–12. doi: 10.1371/journal.pone.0109328.
Ariadna M, Bosland PW. A rapid technique for multiple-race disease screening of phytophthora foliar blight on single Capsicum annuum L. plants. Hort Sci. 2010;45:1563–6.
Bouwmeester K, Govers F. A novel method for efficient and abundant production of Phytophthora brassicae zoospores on Brussels sprout leaf discs. BMC Plant Biol. 2009;9:111.
Amin KS, Baldev B, Williams FJ. Phytophthora cajani, a new species causing stem blight on Cajanus cajan. Mycologia. 1978;70:171–6.
Savage EJ, Clayton CW, Hunter JH, Brenneman JA, Laviola C, Gallegly ME. Homothallism, heterothallism and interspecific hybridization in the genus Phytophthora. Phytopathology. 1968;58:1004–21.
Kannaiyan J, Ribeiro OK, Erwin DC, Nene YL. Phytophthora blight of pigeonpea in India. Mycologia. 1980;72:169–81.
Erwin DC, Ribeiro OK. Phytophthora diseases worldwide. St Paul, Minnesota, USA: APS Press; 2005.
Rogers DJ, Randolph SE. Climate change and vector-borne diseases. Adv Parasit. 2006;62:345–81.
Drake J. Population effects of increased climatic variation. Proc Royal Soc London Series B Biol Sci. 2005;272:1823–7.
Savary S, Ficke A, Aubertot JN, Hollier C. Crop losses due to diseases and their implications for global food production losses and food security. Food Sec. 2012;4:519–37.
Ainsworth EA, Davey PA, Bernacchi CJ, Dermody OC, Heaton EA, Moore DJ, et al. A meta-analysis of elevated [CO2] effects on soybean (Glycine max) physiology, growth and yield. Glob Change Biol. 2002;8:695–709.
Chakraborty S, Luck J, Hollaway G. Impacts of global change on diseases of agricultural crops and forest trees. CAB Reviews: Perspectives in Agriculture, Veterinary Science, Nutrition and Natural Resources 2008, 3 No. 054.
Thompson GB, Drake BG. Insects and fungi on C3 sedge and a C4 grass exposed to elevated atmospheric CO2 concentrations in open-top chambers in the field. Plant Cell Environ. 1994;17:1161–7.
Gallegly ME, Hong CX. Phytophthora: identifying species with morphology and DNA fingerprints. St Paul, Minnesota, USA: APS Press; 2008.
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. In Proceedings of National Academy of Science: 2012; USA; 2012:6241–46.
Flier WG, Grünwald NJ, Kroon LPNM, Van Den Bosch TBM, Garay-Serrano E, Lozoya-Saldaña H, et al. Phytophthora ipomoeae sp. nov., a new homothallic species causing leaf blight on Ipomoea longipedunculata in the Toluca Valley of central Mexico. Mycol Res. 2002;106:848–56.
Cooke DEL, Drenth A, Duncan JM, Wagels G, Brasler CM. A molecular phylogeny of Phytophthora and related oomycetes. Fungal Genet Biol. 2000;30:17–32.
Hosseini S, Karlsson M, Jensen DF, Heyman F. Quantification of Phytophthora pisi DNA and RNA transcripts during in planta infection of pea. Eur J Plant Pathol. 2012;132:455–68.
Eye LL, Sneh B, Lockwood JL. Inoculation of soybean seedlings with zoospores of Phytophthora megasperma var sojae for pathogenicity and race determination. Phytopathology. 1978;68:1769–73.
Chang SH, Kwack MS, Kim YS, Lee JY, Kim KD. A rapid radicle assay for pre-screening antagonistic bacteria against Phytophthora capsici on pepper. Mycobiology. 2001;29:218–23.
Heid CA, Stevens J, Livak KJ, Williams PM. Real-time quantitative PCR. Genome Res. 1996;6:986–94.
Babu KB, Saxena AK, Srivastava AK, Arora DK. Identification and detection of Macrophomina phaseolina by using species-specific primers and probe. Mycologia. 2007;99:797–803.
Nair S, Roshna OM, Soumya VP, Hegde V, Kumar MS, Manimekalai R, et al. Real-time PCR technique for detection of areca nut yellow leaf disease phytoplasma. Australas Plant Path. 2014. doi 10.1007/s13313-014-0278-7.
Nicolaisen M, Suproniené S, Nielsen LK, Lazzaro I, Spliid NH, Justesen AF. Real-time PCR for quantification of eleven individual Fusarium species in cereals. J Microbiol Meth. 2009;76:234–40.
Jwa NS, Walling LL. Influence of elevated CO2 concentration on disease development in tomato. New Phytol. 2001;149:509–18.
Gautam HR, Bhardwaj ML, Kumar R. Climate change and its impact on plant diseases. Curr Sci India. 2013;105:25.
McElrone AJ, Reid CD, Hoye KA, Hart E, Jackson RB. Elevated CO2 reduces disease incidence and severity of a red maple fungal pathogen via changes in host physiology and leaf chemistry. Glob Change Biol. 2005;11:1828–36.
Madhu M, Hatfield JL. Dynamics of plant root growth under increased atmospheric carbon dioxide. Agron J. 2013;105:657–69.
Rouhier H, Read DJ. Plant and fungal responses to elevated atmospheric CO2 in mycorrhizal seedlings of Betula pendula. Environ Exp Bot. 1999;42:231–41.
Nguyen C. Rhizodeposition of organic C by plant: mechanisms and controls. Sustain Agric 2009, 97–123. doi: 10.1007/978-90-481-2666-8_9.
Evangelisti E, Govetto B, Minet-Kebdani N, Kuhn ML, Attard A, Ponchet M, et al. The Phytophthora parasitica RXLR effector penetration-specific effector 1 favours Arabidopsis thaliana infection by interfering with auxin physiology. New Phytol. 2013;199:476–89.
Reddy MV, Nene YL, Raju TN, Sheila VK, Sarkar N, Remanandan P, et al. Pigeonpea lines field-resistant to phytophthora blight. Int Pigeonpea Newslett. 1991;13:20–2.
White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc: Guide Methods Appl. 1990;18:315–22.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26.
Acknowledgements
The funding support from Department of Science and Technology-Climate Change Division and National Food Security Mission (NFSM), Department of Agriculture & Cooperation, Ministry of Agriculture, Govt. of India is gratefully acknowledged. The authors are thankful for technical assistance from Mr. Bal Krishna from Legumes Pathology group.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MS conceived the study, analysed the results and drafted the manuscript. RG participated in the design of the study, analysed the results and helped in writing the manuscript. AT and RT helped in conducting and analysing the experiments. All authors read and approved the final version.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Sharma, M., Ghosh, R., Tarafdar, A. et al. An efficient method for zoospore production, infection and real-time quantification of Phytophthora cajani causing Phytophthora blight disease in pigeonpea under elevated atmospheric CO2 . BMC Plant Biol 15, 90 (2015). https://doi.org/10.1186/s12870-015-0470-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-015-0470-0