
Abstract
Background
Anaplasma translocated substrate 1 (Ats-1) is an effector of type 4 secretory systems (T4SS) and the main virulence factor of Anaplasma phagocytophilum. Ats-1 is involved in the regulation of host cell biological processes, but the specific molecular mechanism of its action is unclear.
Results
In this study, we identified Ats-1 as involved in mitochondrial respiratory regulation of HEK293T cells by multi-omics analysis. After intracellular expression of Ats-1, adenosine triphosphate levels and the proliferation of HEK293T cells were both up-regulated, while HEK293T cells apoptosis was inhibited. Ats-1 targeted translocation to the mitochondria where it up-regulated the expression of NDUFB5, NDUFB3, NDUFS7, COX6C, and SLC25A5, thereby enhancing energy production and inhibiting HEK293T cells apoptosis while enhancing HEK293T cells proliferation, and ultimately facilitating Anaplasma phagocytophilum replication in HEK293T cells.
Conclusions
This study demonstrated that Anaplasma phagocytophilum Ats-1 induces anti-apoptosis and energy metabolism by upregulating the respiratory chain-mPTP axis in eukaryotic mitochondria. These results provide a better understanding of the pathogenic mechanism of Anaplasma phagocytophilum within host cells.
Similar content being viewed by others


Introduction
Human granulocytic anaplasmosis is a tick-borne zoonotic disease caused by Anaplasma phagocytophilum. A. phagocytophilum is an intracellular parasite that can survive long periods within host cells including neutrophils, monocytes, macrophages, epithelial cells, and others[1, 2]. After A. phagocytophilum infection occurs in animals and humans, symptoms such as leukopenia, thrombocytopenia, and increased serum aminotransferase (liver enzyme) activity are often present[1,2,3,4,5]. The disease is commonly present in the northeastern USA, northern Europe, and southeastern Asia [6,7,8,9]. The specific molecular mechanism of A. phagocytophilum infection of host cells is unclear, seriously hindering research into new treatment and preventive measures.T4SS transfers proteins or nucleoprotein complexes from bacteria to eukaryotic cells through eukaryotic cell membranes [10]. Three A. phagocytophilum T4SS effectors have been experimentally identified including AnkA, APH-0455, and Ats-1 [11,12,13]. Among these, Ats-1 is the first bacterial protein known to pass through five layers of membranes. When Ats-1 is in the mitochondria, it can block the loss of mitochondrial membrane potential and inhibit host cell apoptosis that is induced by etoposide [12]. Further, Ats-1 targeting A. phagocytophilum inclusion bodies binds to Beclin-1 and induces autophagy [14]. However, the regulatory mechanisms of Ats-1 remain unclear and require further investigation.
Proteomics and metabonomics have been widely used in recent years to study the mechanisms underlying various diseases like tuberculosis [15], liver fluke infection [16, 17], and diabetes [18]. In addition, isobaric tags used in the relative and absolute quantification (iTRAQ) [19,20,21]of proteins can be used to evaluate differentially expressed proteins under different biological conditions and has attracted widespread attention. However, iTRAQ analysis cannot achieve the identification of specific core proteins [22]. Parallel reaction monitoring (PRM) is an ion monitoring technique that is based on high resolution and high precision mass spectrometry that are used in the relative or absolute quantification of target proteins and peptides [23]. PRM can consequently be used to validate iTRAQ results. In addition, targeted metabolomics analyzes several target compounds or metabolites involved in certain pathways, while using standards to construct detection methods with high specificity, high sensitivity, and good repeatability.
In this study, proteomics (iTRAQ) was used to investigate the proteome changes in HEK293T cells after Ats-1 expression, and PRM, targeted metabolomics, and other experiments were used to verify the regulation of Ats-1 in HEK293T cells, which laid a foundation for the in-depth understanding of the molecular pathogenesis of A. phagocytophilum.
Materials and methods
Bioinformatics analyses
The Signalp 5.0 (http://www.cbs.dtu.dk/services/SignalP/) and TargetP 2.0 (http://www.cbs.dtu.dk/services/TargetP/) servers were used to predict signal peptides and mitochondrial transfer peptides of the Ats-1 protein, respectively. The secondary structure of the Ats-1 protein was predicted and analyzed with the SOPM software program (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). The tertiary structure of the Ats-1 protein was predicted and analyzed with the Swiss-Model software program (http://swissmodel.expasy.org/). The CNLS Mapper software program (http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi) was used to predict the nuclear localization signal (NLS) and the NetNES 1.1 server (http://www.cbs.dtu.dk/services/NetNES/) was used to analyze the nuclear export signal (NES).
Cells
HEK293T cells were provided by the Cell Resource Center at the Institute of Basic Medical Sciences of the Chinese Academy of Medical Sciences/Peking Union Medical College (Beijing, China), and were cultured in DMEM (Solarbio, Beijing, China) medium containing 10% fetal bovine serum (FBS) (Gibco, Brooklyn, NY, USA) in an incubator with a 5% CO2 atmosphere.
Construction of a eukaryotic expression vector and establishment of a transfection model
The Ats-1 gene was synthesized according to the sequence deposited in Genbank (Accession: FJ210653) and was cloned into the pcDNA3.1 vector with a His tag by Sangon Biotech (Shanghai, China).The pcDNA3.1-Ats-1 plasmid was extracted with a large-scale plasmid extraction kit (Tiangen, Beijing, China) according to the manufacturer’s instructions. HEK293T cells were cultured until reaching cell densities of 80–90%. The cells were divided into three groups including group A: pcDNA3.1-Ats-1 plasmid transfection into cells, group P: transfection with the pcDNA3.1 empty vector, and group H: the no transfection control group. The Lipofectamine plasmid 3000 Transfection Reagent (Invitrogen, Carlsbad, USA) was used for transfection. After 48 h, the supernatant was discarded and native lysis buffer (Solarbio, Beijing, China) along with protease inhibitors (Solarbio, Beijing, China) were added, followed by total protein extraction and storage at − 80 °C.
Verification of transfection by Western blot analysis
For western blotting (WB), the proteins were resolved on SDS–polyacrylamide gel electrophoresis (SDS–PAGE) gels followed by standard WB. Rabbit anti-6×His tag antibody (Abcam, Cambridge, MA, USA) (1:2,000) as the primary antibody. Goat anti-rabbit IgG H&L (Abcam, Cambridge, MA, USA) (1:4,000) was used as the secondary antibody and a SuperSignal West Femto Trial Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used for color development. β-Actin protein was used as the control, using mouse anti-β-actin monoclonal antibody (Sino Biological, Beijing, China) (1:2,000) and goat anti-mouse IgG H&L (Abcam, Cambridge, MA, USA) (1:4,000).
iTRAQ, PRM, and targeted metabolomics
The total cellular proteins from the three groups of cells were extracted, quantified, detected, digested, desalted, and labeled for iTRAQ according to methods described by Ross et al. [24]. An EASY-nLCTM 1,200 na upgrade UHPLC system with a Q ExactiveTM HF-X mass spectrometer were used to generate mass spectra for detecting the original data. The Proteome Discoverer 2.2 software program was used to search the database (homo_sapiens_uniprot_2019.01.18.fasta), followed by quantification of peptides and proteins. iTRAQ LC-MS/MS proteome analysis was performed by Novogene Co., Ltd. (Beijing, China). Additional quantification (PRM) of eight differentially expressed proteins was also conducted by Novogene Co., Ltd. (Beijing, China), along with targeted metabolomics to quantify and analyze carbon-related compounds that were present in all three sample groups. Three samples in each group were used for iTRAQ, PRM, and targeted metabolomic analysis.
Quantitative real-time PCR (qRT-PCR) detection
The total RNAs from the three groups of transfected cells were extracted with an Ultrapure RNA Kit (CWBIO, Beijing, China). RNA purity and concentration were determined with a nucleic acid detector (Nanodrop ND-2000, Wilmington, DE, USA). RNA was then reverse transcribed into cDNA using a HiFiScript cDNA Synthesis Kit (CWBIO, Beijing, China). The UltraSYBR mixture (High ROX) (CWBIO, Beijing, China) was used for quantitative detection. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal reference (primer sequence shown in Supplementary Table 1). qRT-PCR was conducted using a QuantStudio™ 5 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA).
Adenosine triphosphate (ATP) level detection
Cell lysate protein concentration was detected using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA), followed by measurement of absorbance at 570 nm with a multi-function enzyme labeling instrument (SUNRISE, Tecan Austria Gmbh, Grodig, Austria). Protein concentrations were then calculated according to a standard curve. The ATP concentrations were also measured for the three groups of cells after transfection using an Enhanced ATP Assay Kit (Beyotime, Shanghai, China) following the manufacturer’s instructions. Chemiluminescence was detected using a multi-functional detector (Synergy 2 Biotek USA).
CCK-8 detection of cell proliferation
After 48 h post-transfection, the three groups of cells were inoculated in a 96 well culture plate at 2 × 103 cells per well and then cultured at 37 °C for 2 h in an incubator with a 5% CO2 atmosphere. Cell proliferation was detected with a Cell Counting Kit-8 (Dojindo Laboratories, Shanghai, China). The absorbance value (A450) of each well was then determined with a multi-function enzyme labeling instrument (SUNRISE, Tecan Austria Gmbh, Grodig, Austria).
Detection of HEK293T cells apoptosis by flow cytometry
HEK293T cells in logarithmic growth phase were inoculated into six-well cell culture plates at 2 × 104 cells/well. Cells were divided into three groups for transfection, and three duplicate wells were set up in each group and cultured for 48 h at 37 °C in a 5% CO2 incubator. Cell apoptosis was evaluated with the Annexin V-FITC/PI Apoptosis Detection Kit (YEASEN, Shanghai, China) and flow cytometry (BD FACSAria™ III, BD Biosciences, Franklin Lakes, NJ, USA).
Data analysis
The data reported here represent the results from three independent experiments, with values as means ± SD. The GraphPad Prism software program was used to construct figures and the SPSS software program was used for statistical analyses. NSK tests and one-way ANOVA tests were used to compare means among different groups of data, wherein *: p < 0.05, *: p < 0.01, ***: p < 0.001, and ****: p < 0.0001 were considered statistically significant.
Results
Ats-1 Bioinformatics analyses
To more comprehensively understand the function of the Ats-1 protein, it was analyzed using several bioinformatics software tools. The results showed that Ats-1 had Sec and Tat signal peptides (Supplementary Fig. 1 A) in addition to mitochondrial transfer signals (Supplementary Fig. 1 B). Protein secondary structures indicated that α-helices of Ats-1 were the most abundant structures, followed by β-folding and β-turning angles, with each accounting for 39.89%, 8.78%, and 6.38%, respectively (Supplementary Fig. 1 C). The tertiary structure prediction of Ats-1 (Supplementary Fig. 1 D) revealed a protein coverage rate of 26.98%. The CNLS Mapper software program and the NetNES 1.1 server prediction results showed that Ats-1 had no NLS or NES (Supplementary Fig. 1 E, 1 F). Predictive analysis further revealed that Ats-1 may transfer to the endoplasmic reticulum and across the bilayer lipid membrane of mitochondria, but not into nuclei.
Successful expression of Ats-1 in HEK293T cells
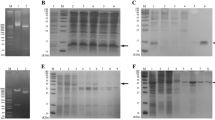
After transfection of HEK293T cells with pcDNA3.1-Ats-1 for 24 and 48 h, total cell proteins were collected and analyzed by Western blot. Two Ats-1 protein forms have been identified in cells including the full-length 48 kDa protein and the 35 kDa truncated Ats-1 protein [12]. Clear target bands were observed at 48 kDa and 35 kDa, indicating obvious expression of Ats-1 proteins (Fig. 1 A). Semi-quantitative analysis with the ImageJ software program revealed that target protein expression at 48 h after transfection was significantly higher than at 24 h (p < 0.05) (Fig. 1 B). Thus, Ats-1 proteins were successfully expressed in HEK293T cells.

(A) Ats-1 was expressed in HEK293T cells for 24 and 48 h, then transfected with empty vector and untransfected HEK293T cells for Western blot analysis. Each band in the figure represents the results of three independent experiments. Arrows 1 and 2 indicate full-length Ats-1 and cleaved Ats-1 proteins, respectively [14]. Molecular mass markers are shown on the left. The original diagram is shown in Supplementary Fig. 2. (B) Western blot results that were semi-quantitatively analyzed using the ImageJ software program. All data are shown as means ± SD from three independent tests. *: p < 0.05.
Proteomic analysis of HEK293T cells after Ats-1 expression
To fully understand proteome changes in host cells after Ats-1 expression, HEK293T cells were transfected with empty pcDNA3.1 vector (group P) and pcDNA3.1-Ats-1 recombinant vector (group A). Cells were then collected 48 h later for LC-MS based proteomics analyses. The mass spectrometry results were compared against a human database (homo_sapiens_uniprot_2019.01.18.fasta) to identify proteins. The expression levels of 862 proteins were identified as significantly different between the two groups (p < 0.05, fold change > 2). A total of 407 proteins were up-regulated in group A, while 455 proteins were down-regulated (Fig. 2).

Volcano plots showing differentially expressed proteins detected in Ats-1-transfected HEK293T cells compared to control HEK293T cells (A. vs. P). The x‑axis represents log2 (fold change) values while the y‑axis represents − log10(p) values.
To determine the biological processes and pathways involved in Ats-1 expression, significantly differentially expressed proteins were compared against functional databases including the Gene Ontology (GO) database and the Kyoto Encyclopedia of Genetics and Genomes (KEGG) [25, 26]database. GO analysis is divided into three functional categories: biological processes (BP), cellular components (CC), and analytical functions (MF). Among the annotations of differentially expressed proteins into the three functional categories, reactive oxygen species metabolic processes were identified in BP, while oxidoreductase activity via acting on the aldehyde or oxo groups of donors and oxoglutarate dehydrogenases (succinyl-transferring) were identified in the MF category (Fig. 3). These three BP are all related to mitochondrial function. In both the GO and KEGG analysis, oxidative phosphorylation and pyruvate metabolism were both highly enriched (Fig. 4), which are also related to mitochondrial function. Thus, differentially expressed proteins after Ats-1 expression in HEK293T cells were particularly associated with mitochondrial-related pathways.

Gene ontology (GO) analysis of total proteins based on the GO categories of cellular components (CC), biological processes (BP), and molecular functions (MF).

KEGG enrichment analyses of differentially abundant proteins (Fisher’s exact test, p < 0.05).
PRM validation of mitochondrial respiratory chain-related protein expression
Niu [12] also observed that Ats-1 contained a signal peptide targeting mitochondria, and that after the recombinant vector PGADT7-Ats-1 was transfected into yeast, the Ats-1 protein was observed in mitochondria. The differential proteins identified in the experiments of this study were also enriched with mitochondrial functions based on GO and KEGG analyses. Seven of 862 differentially expressed proteins related to mitochondrial biological function (Table 1) were selected for PRM analysis alongside iTRAQ analysis (Fig. 5). After Ats-1 expression, the expression levels of five proteins (NDUFB3, NDUFB5, NDUFS7, COX6C, and SLC25A5) based on iTRAQ were the same as identified by PRM (i.e., they were all up-regulated).

Proteins identified by iTRAQ analysis (A. vs. P groups) and verified by PRM analysis. The iTRAQ data were validated using seven proteins.
Ats-1 Up-regulates the expression of 80% of central carbon pathway-related metabolites
Upon Ats-1 expression, five proteins were up-expressed in the mitochondrial respiratory chain suggesting a potential influence on cellular energy metabolism. The central carbon pathway is the main source of energy for host cells. Consequently, we detected the expression levels of metabolites related to central carbon pathways after Ats-1 expression using targeted metabonomics. A total of 40 differential metabolites were subsequently detected (Figs. 6 and 7), with most differential metabolites involved in the pentose phosphate pathway. Thirteen of the metabolites (32.5%) exhibited altered expression, with nine up-regulated and four down-regulated (Fig. 7). Nine of these (22.5%) were involved in the TCA cycle pathway, of which eight were up-regulated and one was down-regulated. The metabolic pathway with the third largest number of differentially expressed metabolites was glycolysis, wherein the expression of six metabolites was altered (15%, five up-regulated and one down-regulated). Although the pentose phosphate pathway exhibited the largest number of differentially expressed metabolites, the pathway does not lead to the production or consumption of ATP. In contrast, the TCA cycle is an effective pathway to oxidize polysaccharides and other substances to conserve energy. Glycolysis is the pathway by which most organisms oxidize glucose, contribute to energy conservations, and involves many different metabolites. These data reveal that Ats-1 expression leads to the up-expression of metabolites within various energy conservation pathways and a general enhancement of cellular energy conservation.

Targeted metabolic analysis showing coverage of the detected substances relative to the central carbon metabolite.

Relative quantitative analysis of central carbon-related substances (A. vs. P groups). The x‑axis represents central carbon-related substances and the y‑axis represents log2(fold change) values.
Verification of mitochondrial mRNA levels related to differential proteins based on qRT-PCR
To evaluate the effects of Ats-1 expression on the mRNA expression of seven mitochondrial-related differentially expressed proteins, qRT-PCR assays were conducted. Compared with group P, Ats-1 expression significantly increased the mRNA expression of NDUFB3, NDUFB5, and COX6C and significantly inhibited the mRNA expression of SDHC and NDUFS7, but did not significantly affect the mRNA expression of SLC25A4 and SLC25A5 (Fig. 8 A). Comparative analysis of the qRT-PCR (Fig. 8 A) and PRM (Fig. 5) results demonstrated that the mRNA levels of NDUFB3, NDUFB5, COX6C, and SLC25A4 were consistent with protein levels (i.e., they were up-regulated), while the other three proteins exhibited differing trends. The combined results of iTRAQ, PRM, and qRT-PCR analyses revealed that the expression of Ats-1 affects the expression of NDUFB3, NDUFB5, NDUFS7, COX6C, and SLC25A5 within the respiratory chain of mitochondria (Table 2).

Ats-1 enhanced the respiratory activity and proliferation of HEK293T cells. (A) Analysis of qRT-PCR results. NDUFB5, COX6C, SLC25A4, and SLC25A5 were up-regulated after transfection with Ats-1 at the mRNA level, while SDHC and NDUFS7 were down-regulated. (B) ATP measurements in HEK293T cells. ATP levels significantly increased in cells after transfection with Ats-1 compared to transfection with an empty vector. (C) Cellular proliferation tests. Based on the CCK-8 method, the OD values were positively correlated with the number of living cells, thereby indirectly reflecting the proliferation of cells. Cellular proliferation was enhanced after transfection with Ats-1 compared to transfection with an empty vector. The results represent the results from three independent experiments. Data are shown as means ± SD from three independent tests. Ns: no significant difference, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
Ats-1 Increases ATP synthesis in HEK293T cells
The NDUFB3, NDUFB5, and COX6C proteins were up-regulated in the iTRAQ, PRM, and qRT-PCR analysis results, and they are all important components of the mitochondrial respiratory chain complex. Further, many metabolites in the TCA cycle were up-regulated, suggesting that the synthesis of ATP in host cells was altered after Ats-1 expression. Consequently, the levels of ATP synthesis in group A and group P were analyzed. The amount of ATP synthesis in group A was significantly higher than in group P (p < 0.01) (Fig. 8 B). Thus, Ats-1 expression affects the energy metabolism of host cells and increases ATP synthesis.
Ats-1 Promotes the proliferation of HEK293T cells
Given that host cell ATP synthesis increases after Ats-1 expression, we speculated that Ats-1 expression promotes cellular proliferation. Detection of CCK8 was evaluated, revealing that the OD value of group A was significantly higher than that of group P (p < 0.05) (Fig. 8 C). Thus, the expression of Ats-1 promoted host cell proliferation.
Ats-1 Inhibits apoptosis of HEK293T cells
The results begged the question of whether Ats-1 expression could alter apoptosis rates. The apoptosis rates of each treatment group were consequently evaluated by flow cytometry. The apoptosis rate was 35.2% in group H (Fig. 9 A), 17.57% in group P (Fig. 9 B), and 11.91% in group A (Fig. 9 C). The apoptosis rate in group A was significantly lower than in group P (p < 0.05) (Fig. 9 D). Thus, Ats-1 expression can inhibit the apoptosis of HEK293T cells.

HEK293T cells apoptosis rate measurements. (A) Flow cytometry analysis of HEK293T cells apoptosis after 48 h of untransfection. (B) Flow cytometry analysis of HEK293T cells apoptosis 48 h after transfection with an empty vector. (C) Flow cytometry analysis of HEK293T cells apoptosis 48 h after transfection with Ats-1. (D) Statistical analysis of HEK293T cells apoptosis rate. Data are shown as means ± SD for three independent tests. *: p < 0.05.
Discussion
Bioinformatics analysis helps us to better understand Ats-1. Online software predicted the presence of the Sec signal peptide and Tat signal peptide in Ats-1 (Supplementary Fig. 1 A), which are associated with protein secretion. Combined with GO functional annotation and KEGG enrichment analysis of the A .vs. P groups, the differential proteins were enriched in both the membrane and membrane components in the cellular component category (Fig. 3), and enriched in protein output in the KEGG pathway (Fig. 4). Ats-1 may regulate the process of protein secretion in host cells. Ats-1 also has a mitochondrial transfer signal, which is consistent with the results that Ats-1 targets translocated mitochondria [12]. In the prediction results, Ats-1 had no nuclear localization signal or nuclear output signal. There is currently no relevant literature on Ats-1 entry into the nucleus. Because the team has not yet isolated A. phagocytophilum and cannot directly infect host cells (HL-60) with pathogens, and because HEK293T cells are easily transfected with recombinant plasmids, this study observes Ats-1 transfection into HEK293T cells.
The iTRAQ and PRM (Fig. 5) results did not perfectly coincide and this difference may be due to several factors. First, the instruments and quantitative principles behind the two technologies differ. Moreover, iTRAQ methods are not targeted and quantitative, but rather are a high-throughput identification method for proteins. Consequently, some false positive results can be achieved. Thus, PRM and qRT-PCR are needed to verify iTRAQ results. Five of the seven target proteins (NDUFB3, NDUFB5, NDUFS7, COX6C, and SLC25A5) exhibited the same expression trend (i.e., up-regulation) that nevertheless confirm the repeatability and reliability of iTRAQ data. NDUFS7 mRNA levels were down-regulated (Fig. 8 A), while protein levels were up-regulated (Fig. 5). Previous studies have shown that post-transcriptional regulation, translation regulation, and degradation regulation play important roles in determining protein concentrations relative to mRNA levels [27]. Therefore, corresponding correlations between protein levels and mRNA levels may not always coincide. Nevertheless, protein functionality can still be assessed and the PRM results at the protein level were the most significant aspects of this study.
Based on the results of this study, the regulation of Ats-1 in host cells was hypothesized (Fig. 10) to clarify the roles and functional mechanisms of Ats-1. After A. phagocytophilum enters the host cell, it secretes Ats-1 protein within the cytoplasm and a part of the Ats-1 protein enters the mitochondria, leading to direct up-regulation of complex I and complex IV components (NDUFB3, NDUFB5, NDUFS7, and COX6C), thereby enhancing the function of complexes I and IV. The expression of Ats-1 in cells can directly or indirectly enhance the expression intensity of the pentose phosphate, glycolysis, and TCA cycle pathways [25, 26] within mitochondria. Numerous redox reactions then take place in the glycolysis, tricarboxylic acid cycle, and pentose phosphate pathways [25, 26], resulting in increased production of hydrogen protons (H+) and electrons (e−). According to Mitchell’s [28] chemical permeation theory, hydrogen protons (H +) and electrons (e −) transfer in the mitochondrial electron transport chain and couple with ATP synthesis via the oxidative phosphorylation coupling reaction, resulting in increased ATP production. These processes inhibit the apoptosis of host cells, promote the proliferation of host cells, and thereby contribute to the survival of A. phagocytophilum in host cells. Under normal circumstances, mPTP allows some small ions to pass freely through the membrane and activate ATP synthetase through oxidative phosphorylation to maintain mitochondrial membrane potential and ion balance inside and outside the cell. The component protein (SLC25A5) of mPTP is up-regulated in this pathway and the biological function is enhanced, thereby leading to increased ATP synthetase activity and increased ATP production. Both of the two regulation routes can increase metabolic energy and inhibit host cell apoptosis, thereby providing more time for A. phagocytophilum replication in host cells.

Schematic showing the role of Ats-1 in host cells. The solid lines represent known results and the dashed lines represents hypothesized activities.
Numerous bacterial proteins have been shown to inhibit host cell apoptosis via mitochondrial action. For example, the outer membrane protein PorB of Neisseria meningitides [29] and the Chlamydia-secreted protease-like activity factor termed CPAF [30] can regulate host cell apoptosis through various mitochondrial mechanisms. Niu et al. [12]showed that Ats-1 blocks the loss of mitochondrial membrane potential and inhibits host cell apoptosis induced by etoposide. This study also shows that Ats-1 can inhibit host cell apoptosis through mitochondrial activities. In contrast to previous studies, Ats-1 expression inhibited host cell apoptosis by up-regulating five proteins (NDUFB3, NDUFB5, NDUFS7, COX6C, and SLC25A5) within the mitochondrial respiratory chain. It is unclear whether these five upregulated proteins (NDUFB3, NDUFB5, NDUFS7, COX6C, and SLC25A5) act on other signaling pathways to regulate cellular pathways. Studies in recent years have shown that NDUFB3 is a key protein involved in the oxidation of phosphate and is involved in Parkinson’s disease, Huntington’s disease, and Alzheimer’s disease pathways [31]. Further, some researchers have speculated that COX6C is an important component of the apoptotic pathway and may play a regulatory role in the early stages of influenza infection [32]. Whether the five up-regulated proteins identified here play an important role in A. phagocytophilum infection nevertheless requires further study.
A. phagocytophilum Ats-1 and E. chaffeensis Etf-1 are both T4SS effectors that share 21% homology at the amino acid level [33]. Etf-1 is located in mitochondria and blocks the apoptosis of host cells that is mediated by mitochondria, such that infected host cells can survive and E. chaffeensis can replicate in cells [33]. A. phagocytophilum Ats-1 also targets translocation to the host cell mitochondria to inhibit host cell apoptosis, thereby promoting the survival of A. phagocytophilum in host cells [12]. Thus, the functions of the two proteins are similar. Intracellular Etf-1 nano-antibody (Nbs) is a feasible treatment for blocking intracellular E. chaffeensis infections [33]. Consequently, the development of intracellular anti-Ats-1-Nbs would be an attractive area to research.
Conclusion
In this study, A. phagocytophilum Ats-1 was observed to inhibit host cell apoptosis by up-regulating proteins involved in respiratory chains and mPTP within host cell mitochondria, providing new insights regarding the intracellular parasitism mechanism of A. phagocytophilum. A total of 407 up-regulated proteins and 455 down-regulated proteins were identified by iTRAQ analysis in HEK293T cells expressed by Ats-1. The function and classification of these differentially expressed proteins were annotated by GO and KEGG databases. In addition, seven differentially expressed proteins related to mitochondrial respiratory chain function were verified by PRM and qRT-PCR analyses. The expression of Ats-1 upregulated five proteins (NDUFB3, NDUFB5, NDUFS7, COX6C, and SLC25A5) involved in the mitochondrial respiratory chain and that led to increased ATP synthesis, enhanced cellular proliferation, and decreased apoptosis rates, ultimately providing more time for A. phagocytophilum to replicate and survive in host cells.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository [34] with the dataset identifier PXD029412. Other data generated or analyzed during this study are included in this published article.
References
Bakken JS, Dumler JS. Human Granulocytic Anaplasmosis. Infectious Disease Clinics of North America. 2015;29(2):341-55.
Schotthoefer AM, Hall MC, Satya V, Raza B, Frost HM. Clinical Presentation and Outcomes of Children With Human Granulocytic Anaplasmosis. Journal of the Pediatric Infectious Diseases Society. 2017;7(2):e9-e15.
Camacci ML, Paolo PR, Zachary P, Kamyar H, Alexander D, Cindy O, et al. Severe Human Granulocytic Anaplasmosis With Significantly Elevated Ferritin Levels in an Immunocompetent Host in Pennsylvania: A Case Report. Journal of Investigative Medicine High Impact Case Reports. 2018;6:2324709618758350.
Khan R, Ali A. Non-traumatic splenic rupture in a patient with human granulocytic anaplasmosis and focused review of the literature. Ticks and Tick-borne Diseases. 2018;9(3):735-7.
Welc-Falęciak R, Kowalec M, Zajkowska J, Pancewicz SA, Siński E. Clinical and molecular features of one case of human infection with Anaplasma phagocytophilum from Podlaskie Province in eastern Poland. Annals of Agricultural Environmental Medicine. 2015;22(3):414-7.
Wang F, Ma M, Luo S, Yan M, Tao L, Liu A, et al. Seroprevalence of tick-borne anaplasma phagocytophilum infection in healthy adult population and patients with acute undifferentiated fever from the yunnan province of China. Vector Borne Zoonotic Diseases. 2019;19(8):576-81.
Crosby FL, Lundgren AM, Hoffman C, Pascual DW, Barbet AF. VirB10 vaccination for protection against Anaplasma phagocytophilum. BMC Microbiology. 2018;18(1):217.
Lara Khoury E, Furie R. Inflammatory arthritis: a unique presentation of human anaplasmosis. Clin Rheumatol. 2018;38(1):257-9.
Uminski K, Kadkhoda K, Houston BL, Lopez A, Mackenzie LJ, Lindsay R, et al. Anaplasmosis: An emerging tick-borne disease of importance in Canada. IDCases. 2018;14:e00472.
Alvarez-Martinez CE, Christie PJ. Biological diversity of prokaryotic type IV secretion systems. Microbiology Molecular Biology Reviews. 2009;73(4):775-808.
Lin M, Rikihisa Y. Degradation of p22phox and inhibition of superoxide generation by Ehrlichia chaffeensis in human monocytes. Cellular Microbiology. 2007;9(4):861-74.
Niu H, Kozjak-Pavlovic V, Rudel T, Rikihisa Y, Valdivia RH. Anaplasma phagocytophilum Ats-1 Is Imported into Host Cell Mitochondria and Interferes with Apoptosis Induction. Plos Pathogens. 2010;6(2):e1000774.
Liu H, Bao W, Lin M, Niu H, Rikihisa Y. Ehrlichia type IV secretion effector ECH0825 is translocated to mitochondria and curbs ROS and apoptosis by upregulating host MnSOD. Cellular Microbiology. 2012;14(7):1037-50.
Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. PNAS. 2012;109(51):20800-7.
Dan A, Fernandez-Reyes D, Papadopoulos MC, Rojas SA, Krishna S. Identification of diagnostic markers for tuberculosis by proteomic fingerprinting of serum. Lancet. 2006;368(9540):1012-21.
Zhang F, Hu R, Elsheikha HM, Sheng Z, He J. Global serum proteomic changes in water buffaloes infected with Fasciola gigantica. Parasites Vectors. 2019;12(1):281.
Rioux MC, Carmona C, Acosta D, Ward B, Ndao M, Gibbs BF, et al. Discovery and validation of serum biomarkers expressed over the first twelve weeks of Fasciola hepatica infection in sheep. International Journal for Parasitology. 2008;38(1):123-36.
Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359(6380):1151-6.
Zhou D, Wang Z, Zhou C, He S, Elsheikha HM, Zhu X. Comparative proteomic analysis of virulent and avirulent strains of Toxoplasma gondii reveals strain-specific patterns. Oncotarget. 2017;8(46):80481.
Wang Z, Zhou C, Elsheikha HM, He S, Zhou D, Zhu X. Proteomic differences between developmental stages of Toxoplasma gondii revealed by iTRAQ-based quantitative proteomics. Frontiers in Microbiology. 2017;8:985.
Zhou C, Zhu X, Elsheikha HM, He S, Li Q, Zhou D, et al. Global iTRAQ-based proteomic profiling of Toxoplasma gondii oocysts during sporulation. Journal of proteomics. 2016;148:12-9.
Cehofski LJ, Honoré B, Vorum H. A review: proteomics in retinal artery occlusion, retinal vein occlusion, diabetic retinopathy and acquired macular disorders. International journal of molecular sciences. 2017;18(5):907.
Yu Q, Liu B, Ruan D, Niu C, Shen J, Ni M, et al. A novel targeted proteomics method for identification and relative quantitation of difference in nitration degree of OGDH between healthy and diabetic mouse. Proteomics. 2014;14(21-22):2417-26.
Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Molecular Cellular Proteomics. 2004;3(12):1154-69.
Minoru K, Susumu G, Shuichi K, Yasushi O, Masahiro H. The KEGG resource for deciphering the genome. Nucleic Acids Research. 2004;32(Database issue):D277-80.
Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Hirakawa M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Research. 2006;34(Database issue):D354-7.
Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature reviews genetics. 2012;13(4):227-32.
Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature. 1961;191(4784):144-8.
Massari P, Ho Y, Wetzler LM. Neisseria meningitidis porin PorB interacts with mitochondria and protects cells from apoptosis. Proceedings of the National Academy of Sciences. 2000;97(16):9070-5.
Pirbhai M, Dong F, Zhong Y, Pan K, Zhong G. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. Journal of Biological Chemistry. 2006;281(42):31495-501.
Zhang L, Guo X, Chu J, Zhang X, Yan Z, Li Y. Potential hippocampal genes and pathways involved in Alzheimer’s disease: a bioinformatic analysis. Genetics and Molecular Research. 2015;14(2):7218-32.
Othumpangat S, Noti JD, Beezhold DH. Lung epithelial cells resist influenza A infection by inducing the expression of cytochrome c oxidase VIc which is modulated by miRNA 4276. Virology. 2014;468:256-64.
Zhang W, Lin M, Yan Q, Budachetri K, Hou L, Sahni A, et al. An intracellular nanobody targeting T4SS effector inhibits Ehrlichia infection. Proceedings of the National Academy of Sciences. 2021;118(18):e2024102118.
Ma J, Chen T, Wu S, Yang C, Bai M, Shu K, et al. iProX: an integrated proteome resource. 2018.
Acknowledgements
We thank everyone who supported and worked hard on this research.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31760738).
Author information
Authors and Affiliations
Contributions
Ruirui Li, Zhongchen Ma, and Yong Wang participated in the conception and design of the study. Ruirui Li performed the laboratory work. Ruirui Li and Zhongchen Ma analyzed the data and wrote the manuscript. Wei Zheng and Yong Wang contributed to the analysis and helped in the manuscript discussion section. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ruirui Li, Zhongchen Ma, Wei Zheng authors have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, R., Ma, Z., Zheng, W. et al. Multiomics analyses reveals Anaplasma phagocytophilum Ats-1 induces anti-apoptosis and energy metabolism by upregulating the respiratory chain-mPTP axis in eukaryotic mitochondria. BMC Microbiol 22, 271 (2022). https://doi.org/10.1186/s12866-022-02668-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-022-02668-x