
Abstract
Background
Many phytophagous insects, whose diet is generally nitrogen-poor, rely on gut bacteria to compensate for nutritional deficits. Accordingly, we hypothesized that insects in desert environments may evolve associations with gut bacteria to adapt to the extremely low nutrient availability. For this, we conducted a systematic survey of bacterial communities in the guts of weevils developing inside mud chambers affixed to plant roots in the Negev Desert of Israel, based on 16S rRNA gene amplicon sequencing.
Results
Our analyses revealed that gut bacterial communities in weevil larvae were similar across a wide geographical range, but differed significantly from those of the mud chambers and of the surrounding soils. Nevertheless, a high proportion of bacteria (including all of the core bacteria) found in the weevils were also detected in the mud chambers and soils at low relative abundances. The genus Citrobacter (of the Enterobacteriaceae family) was the predominant group in the guts of all individual weevils. The relative abundance of Citrobacter significantly decreased at the pupal and adult stages, while bacterial diversity increased. A mini literature survey revealed that members of the genus Citrobacter are associated with nitrogen fixation, recycling of uric acid nitrogen, and cellulose degradation in different insects.
Conclusions
The results suggest that although weevils could potentially acquire their gut bacteria from the soil, weevil host internal factors, rather than external environmental factors, were more important in shaping their gut bacterial communities, and suggest a major role for Citrobacter in weevil nutrition in this challenging environment. This study highlights the potential involvement of gut bacteria in the adaptation of insects to nutritional deficiencies under extreme desert conditions.
Similar content being viewed by others

Background
Many phytophagous insects, whose diet is often nutritionally suboptimal and nitrogen-poor, have been shown to rely on gut bacteria to complement their nutritional requirements [1,2,3]. For example, mutualistic interactions with diazotrophic bacteria (i.e., that fix atmospheric nitrogen) were reported in several wood-eating termite species [4, 5], a wood-eating cockroach [6], several bark and stag beetle species [7,8,9] [10], and fruit flies [11]. Associations with uricolytic bacteria that recycle uric acid nitrogen, which is later re-incorporated into insect tissues, has been reported in a wood-eating termite [12], a drupe-eating bug [13], fruit flies [11], and several herbivorous ants [14]. Some phytophagous insects also rely on their gut bacteria for the synthesis of essential molecules such as particular amino acids and vitamins. For example, plant sap-feeders (e.g., many hymenopterans), are often associated with obligate gut bacteria, such as Ishikawaella that supply them with essential amino acids [15, 16]. In addition, a large group of phytophagous insects feeding on bark or wood, which are rich in fastidious polymers, harbor gut bacteria responsible for the breakdown of ingested polymers into simpler forms that can be directly assimilated by the host insect [17]. In accordance, wood-eating insects were shown to harbor gut bacteria that aid them with cellulose digestion, while these bacteria are conspicuously absent from foliage and detritus-feeders [18].
Knowledge of insect gut bacteria has been primarily developed from insect pests in forest and agricultural ecosystems in an effort to promote pest control strategies (e.g., see reviews [2, 19]), while it is less well characterized in other environments. In desert ecosystems, soil nutrient availability, and particularly nitrogen, is considered to be limited because of low soil moisture coupled with high temperatures and high soil salinity [20, 21], most likely reducing nutrient uptake and nitrogen concentrations in desert plants [22]. Therefore, phytophagous insects in desert environments may experience extreme nitrogen limitation, which may lead them to evolve associations with specific gut bacteria as an adaptation to the exceptionally low nutrient availability.
Here, we took a first step in addressing this hypothesis by focusing on the gut bacteria of weevils (Conorhynchus palumbus Olivier and Menecleonus virgatus Schoenherr; Coleoptera: Curculionidae: Lixinae) that develop singly in a mud chamber affixed to the roots of two summer annual plants of the genus Salsola (Salsola inermis Forssk and S. incanescens Mey; Chenopodiaceae) [23] (Fig. 1). These weevils are widely distributed in the Negev Desert of Israel [24]. The weevil completes most of its life cycle during the summer within a mud chamber underground, where the larva presumably feeds on the plant fluids [25]. The adults emerge during the spring and are active leaf chewers. S. inermis and S. incanescens plants grow in salty desert loessial soils, which are low in nutrient availability (e.g., with a relative nitrogen content of ~ 0.02% in the soil [25]); thus, these plants are also poor in nutrient content (e.g., with a relative nitrogen content of ~ 0.66% in S. inermis [25], versus a global average of 1.58% in plant leaves [26]). Hence, it is unclear how these weevils obtain sufficient nutrients for their development.

Mud chamber affixed to the root of Salsola inermis, and larva, pupa, and adult (Menecleonus virgatus) in the mud chamber
Many weevils have been shown to harbor maternally transmitted intracellular bacteria (e.g., bacteriome-localized Nardonella, Sodalis and Curculioniphilus) that synthesize and provide essential amino acids to promote weevil development [27,28,29,30]. In addition, gut bacteria of various weevils have been reported to be involved in polymer degradative activities [8, 31,32,33,34,35], e.g., diterpene-degrading bacteria in conifer-feeding weevils [36, 37]. Nitrogen-fixing and nitrogen-recycling bacteria have also been suggested to supplement their host’s diet in phloem-feeding weevils [8, 9].
In this study, to investigate the potential role of gut bacteria of desert weevils, we conducted 16S rRNA gene amplicon sequencing of bacterial communities in the guts of these weevils throughout their life cycle and at different geographical locations. We also investigated bacterial communities in surrounding soils and in the mud chamber itself to examine to what extent the external soil environment vs. the internal physiological environment inside the weevil guts shapes the composition of their gut bacterial communities. Finally, we tested whether weevil gut communities are dominated by certain bacteria, as the high dominancy and stability of certain bacteria are often linked with their functional importance to their insect hosts [36, 38]. To this end, we conducted a literature survey to learn about the potential biological functions of the most dominant bacteria within the weevils.
Results
Bacterial community composition in guts of C. palumbus weevils, mud chambers, and the surrounding soils
Permutational multivariate ANOVA (PERMANOVA) and principal coordinates analysis (PCoA) based on unweighted UniFrac distance showed that weevils, mud chambers, and soils significantly differed in their bacterial communities (PERMANOVA: F2,74 = 21.73, R2 = 0.38, P = 0.001; Fig. 2a). Post hoc pairwise tests showed that bacterial communities from the weevil guts were significantly different from those of the mud chambers (P = 0.003) and of the surrounding soils (P = 0.003); and that those of the mud chambers were also significantly different from those of the surrounding soils (P = 0.009), although PCoA plots did not indicate a clear segregation between them. Results of tests based on binary Jaccard index, abundance Jaccard index and weighted UniFrac distance were qualitatively similar (see Additional file 1: Appendix A).

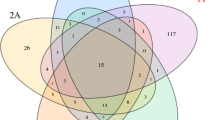
Bacterial community composition in the guts of Conorhynchus palumbus (N = 25), mud chamber (N = 25) and surrounding soil (N = 25). (a) PCoA plot displaying unweighted UniFrac distance. The percent variation explained by each principle coordinate is shown. (b) Venn diagram representing the number of OTUs that are unique to each of the sample type and shared between them. (c) Mean relative abundance of bacterial genera. For weevil, only those genera with > 1% mean relative abundance across all weevil samples are shown; for mud chamber and soil, only the top five genera are shown, whereas all remaining sequences are represented as others. S24–7, Actinomycetales, Rhodobacteraceae, and Frankineae are provided because the phylotypes were not classified to lower taxonomic levels. (d) Phylogenetic diversity. Columns with different letters are different at P < 0.05 based on Tukey’s post hoc tests
Consistently, as shown in the Venn diagram, the proportion of shared operational taxonomic unit (OTU) between soil and mud chamber (84%) was higher than the corresponding proportion of shared OTUs between weevil and mud chamber (61%) and between weevil and soil (62%) (Fig. 2b). The proportions of unique OTUs in weevils, mud chambers, and soils were 27, 17, and 29%, respectively, mainly corresponding to rare OTUs (relative abundance below 0.1%). All the 66 weevil core OTUs (defined as those that were present in at least 70% of all weevil samples) were observed in both mud chamber and surrounding soil but at a relatively low abundance and accounted for a total of 74 ± 3% (SE) of the relative abundances in weevil guts.
The differences in bacterial communities were evident also at the genus level (PERMANOVA: F2,74 = 43.03, R2 = 0.56, P = 0.001; Fig. 2c). The weevil gut bacteria were dominated by Citrobacter, followed by Clostridium, Streptococcus, and S24–7 (the Bacteroidetes family); while several members of Lachnospiraceae, Ruminococcaceae and Anaerolineaceae, were unique to it. The predominancy of the genus Citrobacter in weevils was mainly driven by the high relative abundance of a single OTU (though the analysis showed 28 OTUs binned to Citrobacter), which was also present but less abundant in the mud chambers and surrounding soils (mean relative abundances of 42.85 ± 3.13%, 2.87 ± 1.04%, and 0.53 ± 0.30%, respectively). The five most abundant bacteria in the mud chambers were Solirubrobacter, JG30-KF-CM45 (the Thermomicrobia order), Citrobacter, Rubrobacter, and Blastococcus; while Winogradskyella, Sulfitobacter and Actinokineospora were unique to it. The five most abundant bacteria in surrounding soils were WD2101 (the Phycisphaerae order), Sphingomonas, Solirubrobacter, Arthrobacter, and JG30-KF-CM45; while Halotalea, Crinalium, Niastella, Sediminibacter and several members of the Cyanobacteria phylum were unique to it. The soil bacterial community was qualitatively similar to what was found in a previous study in the same region of the Negev Desert [39] (Additional file 1: Appendix B).
A similarity percentage analysis (simper) revealed 12 bacteria contributing to 50% of the dissimilarity between the weevil gut and the mud chamber, including Citrobacter (contributing to 32% of the dissimilarity) being more abundant in the weevil guts, and the other 11 bacteria being more abundant in the mud chambers (Table 1). A similarity percentage analysis between the weevil gut and the surrounding soil showed that 13 bacteria contributed to 50% of the dissimilarity, including Citrobacter (contributing to 33% of the dissimilarity) and Clostridium being more abundant in the weevil gut, and the other 11 bacteria being more abundant in the surrounding soil (Table 1).
Weevil gut bacteria had the lowest alpha diversity based on the phylogenetic diversity, and there was no significant difference between the mud chamber and the surrounding soil (Generalized linear mixed model (GLMM): F2,74 = 6.45, P = 0.003; Fig. 2d). Similar results were obtained when comparing the Chao1 richness index, Shannon diversity and evenness index (Additional file 1: Appendix A).
Spatial variation: bacterial community composition in the guts of C. palumbus weevils at different sites
Plant species had no significant effect on C. palumbus weevil gut bacterial community composition (PERMANOVA: F1,52 = 1.05, R2 = 0.02, P = 0.329). In addition, there was no effect of the interaction between the sampling site and plant species (PERMANOVA: F3,52 = 1.10, R2 = 0.05, P = 0.299). Therefore, the data for the host plants S. inermis and S. incanescens were pooled for visualization and interpretation of the effects of geographical location on bacterial community composition.
PERMANOVA based on unweighted UniFrac distance (PERMANOVA: F10,52 = 6.22, R2 = 0.60, P = 0.740) showed that C. palumbus weevil gut bacterial communities were similar across locations (Fig. 3). Results based on binary Jaccard index, abundance Jaccard index and weighted UniFrac distance (Additional file 1: Table S1), as well as results based on relative abundance of genus (PERMANOVA: F10,52 = 2.24, R2 = 0.35, P = 0.154), showed a similar pattern. The communities were predominantly (at all sites) composed of Citrobacter, followed by Streptococcus, S24–7, and Clostridium. Again, the predominancy of Citrobacter in C. palumbus was mainly driven by the high relative abundance of a single OTU that was present in all individual C. palumbus weevils with a mean relative abundance of 71.52 ± 2.53%. The relative abundance of Citrobacter did not differ across locations (one-way ANOVA: F10,52 = 0.79, P = 0.636).

Mean relative abundance of bacterial genera in the guts of Conorhynchus palumbus larvae across 11 different sites (Ashalim: N = 6; Abu Haduba, Dimona2, Dimona1, Mamshit, Havat MaShash, Tlalim, Yeruham, and Neot Hovav: N = 5; Mitzpe Ramon: N = 4; Revivim: N = 3). S24–7 was provided because the phylotype was not classified to lower taxonomic level. Only those genera with > 1% mean relative abundance across all weevil samples are shown, whereas all remaining sequences are represented as others
Temporal variation: bacterial community composition in the guts of C. palumbus weevils at different developmental stages
PERMANOVA and PCoA based on unweighted UniFrac distance showed that the bacterial communities significantly differed among weevil life stages (PERMANOVA: F2,59 = 2.51, R2 = 0.08, P = 0.017; Fig. 4a,b; see Additional file 1: Appendix A for results of equivalent tests). Post hoc pairwise tests indicated no significant difference in bacterial communities between guts of larvae and those of pupae (P = 0.222) and adults (P = 1.00), while there was a significant difference in the gut bacterial communities of pupae and adults (P = 0.015).

Bacterial community composition in the guts of Conorhynchus palumbus at different developmental stages (larva: N = 44, pupa: N = 9, adult: N = 7). (a) PCoA plot displaying unweighted UniFrac distance. The percent variation explained by each principle coordinate is shown. (b) Venn diagram representing number of OTUs that are unique to each of developmental stage and shared between them. (c) Mean relative abundance of bacterial genera. Only those genera with > 1% mean relative abundance across all samples are shown, whereas all remaining sequences are represented as others. S24–7 was provided because the phylotype was not classified to lower taxonomic level. (d) Phylogenetic diversity. Columns with different letters are different at P < 0.05 based on Tukey’s post hoc tests
The PCoA plot further indicated two separate clusters of larvae. To understand what accounts for these differences we compared larval mass and date of collection between larvae of the two clusters. We found that larvae that clustered alone were those collected earlier in the season (before mid-August), and were smaller in size (fresh biomass: 155.1 ± 31.2 mg, N = 15), while larvae that clustered more closely with pupae and adults were those collected later in the season (after mid-August), and were larger in size (fresh biomass: 277.9 ± 23.5 mg, N = 29; t-test comparing larval mass: t43 = 42, P = 0.004), suggesting they were closer to pupation.
The bacterial communities significantly differed among weevil life stages also at the genus level (PERMANOVA: F2,59 = 8.13, R2 = 0.22, P = 0.001; Fig. 4c). In this case, bacterial communities from the guts of larvae significantly differed from those of pupae (P = 0.018) and adults (P = 0.033), while there was no significant difference in the gut bacterial communities of pupae and adults (P = 0.414).
A similarity percentage analysis revealed four bacteria contributing to 50% of the dissimilarity between the adults and the larvae, including Citrobacter (contributing to 41% of the dissimilarity) being more abundant in the larvae, and Acinetobacter, Massilia, and Clostridium being more abundant in the adults (Table 2). A similarity percentage analysis between the guts of larvae and pupae showed that two genera contributed to 50% of the dissimilarity, including Citrobacter being more abundant in the larvae (contributing to 46% of the dissimilarity), and Clostridium being more abundant in the pupae (Table 2). Consistently, a significant decrease in the relative abundance of Citrobacter at pupal and adult stages (GLMM: F2,59 = 10.70, P < 0.001) was detected. This was mainly driven by the high abundance of a single OTU that was present in all individual C. palumbus weevils with a mean relative abundance of 71.34 ± 3.91%, 31.64 ± 10.7%, and 32.56 ± 9.86% in the larvae, pupae, and adults, respectively.
The weevil at the larval stage had the lowest alpha diversity based on phylogenetic diversity, while there was no significant difference between weevil pupal and adult stages (GLMM: F2,59 = 8.20, P < 0.001; Fig. 4d). Similar results were obtained when comparing the Chao1 richness index, Shannon diversity and evenness index (Additional file 1: Appendix A).
Bacterial community composition in the guts of the different weevil species
PERMANOVA based on unweighted UniFrac distance (PERMANOVA: F1,95 = 2.9, R2 = 0.02, P = 0.093; see Additional file 1: Table S1 for results of equivalent tests) and on relative abundance of genus (PERMANOVA: F1,95 = 0.49, R2 = 0.01, P = 0.601) indicated no significant difference in the gut bacterial community compositions of C. palumbus and M. virgatus (Fig. 5a). The gut bacteria of the two weevil species were dominated by Citrobacter, followed by Streptococcus, S24–7, and Clostridium. However, C. palumbus and M. virgatus differed in their dominant OTUs (both of which were also present but were less abundant in mud chambers and surrounding soils). Specifically, the mean relative abundance of the dominant OTU (classified as Citrobacter) present in C. palumbus was 71.73 ± 1.91%, while its relative abundance was 0.15 ± 0.87% in M. virgatus. Similarly, the mean relative abundance of the dominant OTU (also classified as Citrobacter) present in M. virgatus was 66.63 ± 3.84%, while its relative abundance was 0.67 ± 0.20% in C. palumbus.

(a) Mean relative abundance of bacterial genera in the guts of weevil larvae Conorhynchus palumbus (N = 88) and Menecleonus virgatus (N = 8). (b) Neighbour-Joining phylogenetic tree constructed from MUSCLE alignment of the V4 region of the 16S rRNA gene of two dominant representative Citrobacter sequences obtained in this study, different Citrobacter sequences obtained from a range of insects, and representative Enterobacter and Klebsiella sequences (of the Enterobacteriaceae family) obtained from the other weevil species in MEGA v10.0. Pseudomonas sp. (of the Pseudomonadaceae family) was used as the out group. Genbank accession No. is shown for each sequence obtained from Genbank, while the OTU No. (the same as in the deposited datasets in Genbank) is shown for the sequence obtained in this study. Numbers on the branches are bootstrap values. Only bootstrap values greater than 70 are shown. Sequences in bold were obtained in this study
The phylogenetic tree constructed by Neighbour-Joining analyses of the V4 region of the 16S rRNA gene showed that the representative dominant Citrobacter sequences isolated from C. palumbus and M. virgatus were phylogenetically close to each other and were the closest to C. koseri isolated from the palm weevil (Rhynchophorus ferrugineus) (Fig. 5b).
Discussion
The objective of this study was to characterize the gut bacteria of weevils associated with Salsola plants, as a first step to address the hypothesis that these bacteria are involved in the adaptation of insects to extreme desert environments. Our results revealed high stability of bacterial community composition among weevils of the different species, geographical locations, and host plants.
Several gut bacteria have been found to be associated with weevils in this study, including the predominant Citrobacter (of the Enterobacteriaceae family), as well as the less abundant Clostridium, Streptococcus, and the Bacteroidales family S24–7. Our phylogenetic analysis indicated that Citrobacter in our weevils was most closely related to strains isolated from the palm weevil. Citrobacter have been previously reported to dominate in several species of weevil beetles, e.g., the palm weevil, R. ferrugineus, and the bark beetle, Dendroctonus armandi [32, 35, 40,41,42], whereas it was less abundant in others, e.g., the bark beetles, D. valens and D. mexicanus [43]. Other than in weevils, Citrobacter has been commonly reported to form associations with a variety of insects, such as termites, flies and moths (see literature survey, Additional file 1: Appendix C). The occurrence of Citrobacter in the gut flora of insects of different orders may suggest its general role in supplementing their diets. Indeed, previous studies have reported that different members of Citrobacter are associated with nitrogen fixation, uric acid recycling, and cellulose degradation in the guts of different insects (Additional file 1: Appendix C) [12, 44,45,46,47,48]. In addition, different members of Citrobacter from soils are commonly associated with other processes of nitrogen metabolism, e.g., denitrification and nitrification [49, 50]. Nitrogen fixation has been previously demonstrated to occur in C. palumbus larvae [23]. Although Klebsiella spp. was suggested as the main nitrogen-fixing bacteria, Citrobacter could also play a role in this function [24]. Uric acid recycling by Citrobacter may also potentially be important for the nitrogen budgets of weevils in our study system. Cellulose degradation, however, is less likely to occur as the weevil larvae do not feed on plant tissues [25].
Although in lower relative abundance than Citrobacter, other bacterial groups may have potential impacts on the weevils. For example, the genus Clostridium that is widely distributed in soils, and in the guts of humans and animals, and have also been reported at a low relative abundance in few species of weevil beetles [41, 42], has been shown to be able to ferment complex molecules including cellulose, hemicellulose, and pectin [51]. In addition, the Bacteroidales family S24–7 has been commonly found within murine gut, where it contributes to carbohydrate degradation [52], as well as within insect gut where its function has not yet been characterized [53]. Different members of Streptococcus occur mostly in human mouth and respiratory tracts, and less commonly in soils [54], but have been occasionally found in insects as potential pathogens [55], as well as in other weevil species where its function has not yet been characterized [42, 56,57,58]. As such, these less common bacterial genera could also have an impact on the diet and the health of the weevils.
We found no evidence of the Nardonella—the most ancient and widespread bacterial endosymbiont in weevils of the superfamily Curculionoidea)—nor of its alternate clades (e.g., Sodalis and Curculioniphilus) [59], although the location of these bacteriome-localized bacteria is often closely associated with the larval gut [59, 60]. The absence of Nardonella from our samples concurs with previous evidence that Nardonella is absent from weevils of the sub-family Lixinae [61], as well as from weevils of other groups (e.g., [30]). Notably, similar to our results, in the weevil Irenimus aequalis, Nardonella was absent, and the dominant bacterium was a member of the Enterobacteriaceae family [30].
Bacterial composition in the weevil guts was associated with the weevil developmental stage. Similarly, previous studies show changes in the bacterial community composition throughout the insect host’s life cycle in response to diet shifts or to changes in internal morphology and physio-chemical conditions inside the insect [62, 63]. In fact, changes (either an increase or decrease) in bacterial diversity and shifts in the dominant gut bacteria across insect life stages seem to be the norm [3, 62,63,64,65]. For example, shifts in the dominant bacteria from Proteobacteria in the larvae (leaf chewer) to Firmicutes in the pupae and the adults (nectar feeder), accompanied with changes in gene expression, were demonstrated in the cotton leafworm [63]. In other cases, a change in bacterial diversity occurs while the dominant bacteria remain consistent, as shown in a forest cockchafer, in which the gut bacteria of the diapausing adults represented a subset of those of the larvae [66]. In our study system, the higher community diversity in the pupae and adults, coupled with the decrease in the relative abundance of the predominant group Citrobacter, may be due to the fact that the gut is renewed during metamorphosis [67]. More specifically, these changes could be associated with the absence of feeding activity at these stages, as the weevil’s gut is emptied in the pupal stage (pers. obs. by F. Meng), and possibly already at late larval stages, and the adults sampled in this study were collected while diapausing.
The stable gut bacterial community of the weevils across a wide geographic range and host plants implies the functional importance of the dominant taxa. This notion is supported by a number of studies that have demonstrated high stability of functionally relevant microbial communities in different insects, including other species of weevil beetles [38, 41, 68]. For example, in the wood-feeding pine and palm weevils, gut bacteria exhibited a highly stable microbial community with high prevalence of the Enterobacteriaceae across different locations, and the Enterobacteriaceae members were demonstrated to detoxify toxins and degrade cellulose in the weevils’ diets [32, 36, 37]. In contrast, other studies demonstrated large spatial variation in bacterial communities, possibly resulting from geographic isolation [69] and variation in external environmental conditions. For example, in the black chafer beetle, the variation in bacterial communities across geographic locations was related to climatic factors and soil properties [65].
A high proportion of bacteria OTUs (including all weevil core OTUs) observed in the weevil guts were also observed in the environment, suggesting that the weevils may potentially acquire most of their gut bacteria from the soil. However, the pronounced differences in bacterial composition between the weevils and the soil (as apparent in the PCoA plots), combined with the high stability of the gut bacterial community (especially of the core bacteria) of the weevils, suggests that internal factors in the weevil guts are more important than external environmental factors, in shaping it. In particular, the reduction in the diversity of bacterial species in weevil guts, relative to their surroundings, could be attributed to the selection of specific bacterial populations, mediated via weevil gut morphology and/or physiology, as demonstrated in other insects, including termites [70] and the bean bug [71]. Consistently, the high relative abundance and persistence of Citrobacter and, particularly, of a specific OTU may suggest that Citrobacter could be selected by the weevil internal environment to support weevil nutrition. The fact that Citrobacter was common in C. palumbus and M. virgatus, but was represented by a different OTU in each species may suggest that the weevils acquire it vertically, as suggested for fruit flies [72]. However, this may also be explained by horizontal transmission [73], combined with differences in the internal morphology or physiological environment of the two weevil species, potentially selecting for different Citrobacter strains [70], especially given that both OTUs were also found in the surrounding environments in this study and that Citrobacter members are widely distributed in soils [49, 74]. Further investigations are needed to clarify the function and mode of the transmission of Citrobacter associated with these weevils.
In view of the extremely low nitrogen content of Salsola plants, we suggest that gut bacteria could contribute to the fitness of weevils in confronting their challenging nutritional environment. Similarly, the desert locust, which is widely distributed in the desert regions of northern Africa, the Middle East, and southwest Asia, contained a relatively simple but abundant gut bacteria including different members of Enterobacteriaceae [75,76,77,78]. The persistence of these bacteria in the guts of the desert locust was suggested as an adaptation to overcome the limited nitrogen in this environment. Additional studies on insect gut bacteria in desert ecosystems, combining metagenomic, metatranscriptomic, biochemical analyses and experimentation, are crucial to determine the generality of such interactions and their adaptive role under extreme climatic conditions.
Conclusions
The study shows highly stable gut bacterial community and predominancy of the genus Citrobacter in weevils across a wide geographical range in the Negev Desert, suggesting a major role for Citrobacter in weevil nutrition. This is one of very few studies that explicitly examine insect gut bacteria in the desert, highlighting the potential involvement of such bacteria in the adaptation of insects to extreme environmental conditions.
Methods
Study area
The study was conducted in the Negev Plateau in Israel’s Negev Desert (30°38′49.3″~ 31°18′50.5″N, 34°40′22.2″~ 35°03′00.2″E). The average maximum daily temperature is ~ 33 °C in July–August, and the annual rainfall ranges from 34 to 187 mm with a mean of ~ 100 mm, with all rainfall occurring during the winter months from November to April; hence, the summers in this region are characterized as hot and dry (the Israel Meteorological Service; http://www.ims.gov.il/IMSEng/CLIMATE).
Sample collection
To characterize the spatial variation of gut bacteria, we collected weevil, soil, and mud chamber samples in 11 sites (Additional file 1: Appendix D) during August 2017, when the beetles were at their larval stage. To characterize changes in gut bacterial community composition throughout the weevil life cycle, samples were collected monthly from two sites: Ashalim (dominated by S. incanescens) and Neot Hovav (dominated by S. inermis), starting from July 2017 (early larval developmental stage) till November 2017 (weevil diapausing adult stage when the adult is still inside the mud chamber underground), for a total of five sampling events for each site.
In each site, we excavated Salsola plant roots to collect 3–10 weevils (according to availability) together with their mud chamber. In addition, we took a soil sample from a depth of ~ 10 cm (typical depth of the weevil mud chamber) from the rhizosphere of each individual Salsola plant. We cleaned the digging tools, tweezers, and gloves with 75% ethanol between each sample collection. All the samples were transported to the laboratory in sterile vials in the icebox within 6 h and immediately stored at − 80 °C for subsequent DNA extraction.
A total of 72 weevils (including 49 larvae, 10 pupae, and 13 adults) were sampled to characterize temporal variation in bacterial composition throughout the weevil life cycle, and a total of 56 weevil larvae (including 11 larvae which were also used for temporal variation) were sampled to characterize spatial variation in bacterial composition. According to a previous survey, three weevil species develop affixed to the roots of Salsola plants in the Negev Desert [24], and hence, we used genetic markers to identify the weevil larvae and pupae to species (see below), while all the sampled weevil adults were morphologically identified as C. palumbus. Since many mud chambers broke during the excavation process, we only used intact ones (given that we were specifically interested in the bacteria of the mud chambers’ inner layers) to avoid potential contamination with associated surrounding soils. This resulted in a total of 25 intact mud chamber samples and 25 surrounding soil samples used for subsequent analyses.
Weevil dissection and DNA extraction
Prior to the dissection, the samples were washed in three different solutions: 1% dish soap, 100% ethanol, and phosphate buffer pH 7.0 (Caisson Labs, Smithfield, UT, USA), and this procedure was repeated three times [47]. The weevils were dissected with the aid of insect pins to excise the whole gut. The gut was transferred to a 2-mL sterilized microcentrifuge tube with 3-mm and 40-μm radius glass beads, and 100 μl of EDTA pH 8.0 (50 mM, bioWORLD, Dublin, OH, USA) was added. Tweezers and pins were cleaned with 70% ethanol and fire between each dissection. All the solutions were sterilized, and all the procedures were performed in a sterile environment.
For larval lysis, samples were vortexed at maximum speed for 30 s, followed by 30 s of freezing in liquid N2; this procedure was repeated three times. The homogenized samples were incubated for 1 h at 37 °C with 120 μl of lysozyme (10 mg/ml, Amresco, Solon, OH, USA). Samples were then centrifuged at 13,000 g in 4 °C for 10 min, and the supernatant was removed to allow DNA purification. DNA was then extracted using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s protocols. This Purification Kit was used after comparing the DNA extraction efficiency of two widely used commercial kits (i.e., Wizard Genomic DNA Purification Kit and Qiagen DNeasy Blood & Tissue Kit).
Soil and mud chamber DNA extraction
Mud chamber samples consisted of 1 g of soil taken from the internal portion of the mud chamber. The soil sample was homogenized in a mortar and pestle with liquid N2, and DNA was extracted from 1 g of subsamples of each soil sample. DNA was extracted using the PowerSoil DNA Isolation Kit (MO BIO, Carlsbad, CA, USA) according to the manufacturer’s protocols. This DNA Isolation Kit is widely used and highly recommended for DNA extraction from environmental samples (including soil) [79].
To ensure sample quality, PCR amplification of the entire 16S rRNA gene (with a positive control and water as a negative control) was conducted for each sample after DNA extraction, following a general PCR protocol [80]. No PCR products were detected for any of the negative controls while there were clear bands for positive controls on a 2% agarose gel.
Weevil species identification
We amplified the gene cytochrome c oxidase subunit I to identify weevil larvae and pupae to species, as previously described [24]. Briefly, PCR was performed in a 20-μl reaction volume, containing 10 μl Taq Bio-ReadyMix (Bio-Lab, Jerusalem, Israel), 1 μl template, 6.6 μl DNase-free water, 0.7 μl of each primer for C. palumbus (forward:5′- TTAGTCCCTCTCATACTAGGAGCC − 3′, reverse: 5′- GAAGAGAAAGAAGGAGTAAAATAGCGG − 3′), and 0.5 μl of each primer for M. virgatus (forward:5′- ACTTCCGCCATCTTTAACCTTGT − 3′, reverse: 5′- GGTAGTTCGGTCAGGTGT − 3′). The cycling parameters were: initial denaturation at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 30 s, and a final extension at 72 °C for 5 min. We identified 108 C. palumbus (a PCR product size of 354 bp) including 74 collected from S. inermis and 34 collected from S. incanescens, and eight M. virgatus (a PCR product size of 235 bp) including two collected from S. inermis and six collected from S. incanescens from all samples.
Identification and taxonomic classification of 16S rDNA fragments
The Research Laboratory Hylab (Rehovot, Israel) conducted amplicon sequencing of the DNA samples. Briefly, 20 ng of metagenomic DNA was amplified in a 25 μl PCR reaction by PrimeStar Max DNA Polymerase (TAKARA Bio Inc., Otsu, Japan) for 20 cycles. The PCR products were purified using Ampure XP beads. Then the 2 μl of the first PCR was amplified to add the adaptor and index sequences in a 10 μl reaction for 10 cycles with the Fluidigm Access Array primers for Illumina. The PCR product was then purified and was sequenced on the Illumina MiSeq platform. The V4 region of the 16S rRNA gene was amplified using the universal bacterial forward primer 515f (5′-GTGYCAGCMGCCGCGGTAA-3′) and reverse primer 806r (5′-GGACTACNVGGGTWTCTAAT-3′), and 250 bp paired-end reads were generated.
Sequence processing was done by using the bioinformatics platform QIIME v1.9.1 [81]. We removed sequencing reads if they contained ambiguities or homopolymers (> six nucleotides in length), or if the average Phred quality score was less than 25 using the default setting in QIIME. Primer sequences were trimmed, and chimeric sequences were eliminated using USEARCH (version 6.1) and the “gold” reference database [82]. Sequences that passed these quality filters were classified into OTUs using the de novo clustering method at 97% similarity with USEARCH and nontarget reads (i.e., chloroplasts and unclassified) were removed. Representative OTUs were then aligned against the SILVA bacterial database (https://mothur.org/wiki/Silva_reference_files) with a threshold confidence level of 50%, and taxonomic classification was carried out with Mothur (version 1.36.1) using the Wang approach [83].
Our samples resulted in 4,384,135 quality sequences averaging 26,410 sequences per sample (ranging from 66 to 72,207). Of the initial 167 samples (including 117 weevil, 25 mud chamber, and 25 surrounding soil samples), we failed to detect bacterial DNA in one weevil pupa sample, and an additional six adult weevil samples yielded < 3700 sequences (ranging from 66 to 1275 sequences). This led to a total of 160 samples that were used for downstream analyses, corresponding to 10,220 OTUs: ranging from 158 to 2904 OTUs, with an average of 987 per sample for weevils, 1986 per sample for mud chambers, and 2557 per sample for the surrounding soil (see Additional file 1: Appendix E for full details on sample sizes used for analyses and sequencing results).
Data analysis
We tested whether the different sample types (i.e., weevil, mud chamber and surrounding soil), weevil species (i.e., C. palumbus and M. virgatus), host plant species (i.e., S. inermis and S. incanescens), weevil developmental stages (i.e., larva, pupa and adult), and sampling sites, differed in their presence/absence of OTUs using binary Jaccard index (taxonomic) and unweighted UniFrac distance (phylogenetic), as well as relative abundances of OTUs using abundance Jaccard index (taxonomic) and weighted UniFrac distance (phylogenetic) with QIIME v1.9.1. The abundance Jaccard index and weighted UniFrac distance were generated from a CSS normalized OTU table with QIIME v1.9.1. The differences in microbial community composition at higher level were also assessed based on the relative abundances of genera. The statistical significance in these comparisons were assessed by using PERMANOVA as implemented in the “adonis” function in the R package vegan [84]. Pairwise comparisons for significant PERMANOVA results were conducted and corrected for multiple testing using a Bonferroni correction as implemented in the “pairwise.adonis” function in the R package pairwiseAdonis [85]. The observed dissimilarity for significant pairwise comparison results was assessed using similarity percentage analyses as implemented in the “simper” function in the R package vegan. PCoA were carried out in QIIME to visualize the significantly different dissimilarity matrices. The individual plant ID was regarded as a random effect (i.e., specified “strata = individual plant ID” in the adonis test) when differences in microbial community composition among different sample types (weevil, mud chamber, and soil) were assessed, while sampling site was regarded as a random factor (i.e., specified “strata = sampling site” in the adonis test) when differences across weevil life cycle were assessed. Results of binary Jaccard index, abundance Jaccard index, unweighted and weighted UniFrac distance were similar, and hence we present only the result of unweighted distance and the results of others are presented in the supplementary material.
In addition, we tested whether sampling sites and weevil developmental stages differed in relative abundances of the predominant genus Citrobacter. Plant species had no significant effect on the relative abundance of Citrobacter, and hence the data for the host plants S. inermis and S. incanescens were pooled for subsequent analyses. For spatial variation, one-way ANOVA was then conducted to assess the effect sampling sites on the relative abundance of Citrobacter, while, for temporal variation, GLMMs with sampling site as a random factor, followed by Tukey’s post hoc tests, were performed to examine the effects of weevil developmental stage on the relative abundance of Citrobacter.
Given that we only identified eight M. virgatus weevils, these were used for an analysis on the differences between weevil species, while all other analyses (including the differences between different sampling types, sampling sites, and weevil developmental stages) were based only on the common weevil species C. palumbus. In addition, only C. palumbus larvae were used for the comparison between weevil species, provided that only M. virgatus larvae were found.
We calculated alpha diversity by performing phylogenetic diversity (estimates using the PD Whole Tree), Chao1 richness (expressed as the number of observed unique OTUs) and Shannon diversity estimates in QIIME. Evenness index was also calculated as: Evenness index = Shannon diversity/ln (the number of observed OTUs derived for each sample). Prior to alpha diversity calculation, we rarefied OTU tables in QIIME such that all samples were randomly rarefied to 3700 sequences (the lowest number among all samples) per sample such that they had equal sampling effort, thus removing heterogeneity among samples. We used GLMMs with individual plant ID and sampling site as random factors, followed by Tukey’s post hoc tests, to examine the effects of sample type and weevil developmental stage, respectively, on the alpha diversity. The results of Chao1 richness index, Shannon diversity, evenness index and phylogenetic diversity were similar, and hence we present only the result of phylogenetic diversity and the results of others are presented in the supplementary material.
Availability of data and materials
The sequence data that support the findings of this study were deposited into Targeted Locus Study project at GenBank under the accession KDDD00000000 (https://www.ncbi.nlm.nih.gov/nuccore/KDDD00000000). The version described in this paper is the first version, KDDD01000000.
Abbreviations
- GLMM:
-
Generalized linear mixed model
- OTU:
-
Operational taxonomic unit
- PCoA:
-
Principal coordinates analysis
- PERMANOVA:
-
Permutational multivariate ANOVA
References
Douglas AE. The microbial dimension in insect nutritional ecology. Funct Ecol. 2009;23(1):38–47.
Hansen AK, Moran NA. The impact of microbial symbionts on host plant utilization by herbivorous insects. Mol Ecol. 2014;23(6):1473–96.
Yun J-H, Roh SW, Whon TW, Jung M-J, Kim M-S, Park D-S, et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microbiol. 2014;80(17):5254–64.
Breznak J, Brill W, Mertins J, Coppel HC. Nitrogen fixation in termites. Nat. 1973;244:577.
Benemann JR. Nitrogen fixation in termites. Sci. 1973;181:164–5.
Breznak JA, Mertins JW, Coppel HC. Nitrogen fixation and methane production in a wood-eating cockroach, Cryptocercus Punctulatus scudder (Orthoptera: Blattidae). Univ Wisc For Res Notes. 1974;184:1–2.
Bridges JR. Nitrogen-fixing bacteria associated with bark beetles. Microb Ecol. 1981;7(2):131–7.
Morales-Jiménez J, Zúñiga G, Villa-Tanaca L, Hernández-Rodríguez C. Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb Ecol. 2009;58(4):879–91.
Morales-Jiménez J, Vera-Ponce de León A, García-Domínguez A, Martínez-Romero E, Zúñiga G, Hernández-Rodríguez C. Nitrogen-fixing and Uricolytic Bacteria associated with the gut of Dendroctonus rhizophagus and Dendroctonus valens (Curculionidae: Scolytinae). Microb Ecol. 2013;66(1):200–10.
Kuranouchi T, Nakamura T, Shimamura S, Kojima H, Goka K, Okabe K, et al. Nitrogen fixation in the stag beetle, Dorcus (Macrodorcus) rectus (Motschulsky) (Col., Lucanidae). J Appl Entomol. 2006;130(9–10):471–2.
Behar A, Ben-Yosef M, Lauzon C, et al. Structure and function of the bacterial community associated with the Mediterranean fruit fly. In: Bourtzis K, Miller T, editors. Insect Symbiosis. Boca Raton: CRC Press; 2009. p. 251–71.
Potrikus CJ, Breznak JA. Gut bacteria recycle uric acid nitrogen in termites: a strategy for nutrient conservation. Proc Natl Acad Sci. 1981;78(7):4601–5.
Kashima T, Nakamura T, Tojo S. Uric acid recycling in the shield bug, Parastrachia japonensis (Hemiptera: Parastrachiidae), during diapause. J Insect Physiol. 2006;52(8):816–25.
Hu Y, Sanders JG, Łukasik P, D’Amelio CL, Millar JS, Vann DR, et al. Herbivorous turtle ants obtain essential nutrients from a conserved nitrogen-recycling gut microbiome. Nat Commun. 2018;9(1):964.
Nikoh N, Hosokawa T, Oshima K, Hattori M, Fukatsu T. Reductive evolution of bacterial genome in insect gut environment. Genome Biol Evol. 2011;3(1):702–14.
Kaiwa N, Hosokawa T, Nikoh N, Tanahashi M, Moriyama M, Meng XY, et al. Symbiont-supplemented maternal investment underpinning host’s ecological adaptation. Curr Biol. 2014;24(20):2465–70.
Breznak JA, Brune A. Role of microorganisms in the digestion of lignocellulose by termites. Annu Rev Entomol. 1994;39(1):453–87.
Martin MM. Cellulose digestion in insects. Comp Biochem Physiol Part A Physiol. 1983;75(3):313–24.
Dillon RJ, Dillon VM. The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol. 2004;49(1):71–92.
West NE, Skujins J. Nitrogen in desert ecosystems. New York: Hutchinson & Ross; 1978.
Hartley A, Barger N, Belnap J, Okin GS. Dryland ecosystems. In: Marschner P, Rengel Z, editors. Nutrient cycling in terrestrial ecosystems. Berlin, Heidelberg: Springer; 2007. p. 271–307.
Gutierrez JR, Whitford WG. Chihuahuan Desert annuals: importance of water and nitrogen. Ecol. 1987;68(6):2032–45.
Shelef O, Helman Y, Friedman A-L-L, Behar A, Rachmilevitch S. Tri-Party underground Symbiosis between a weevil, Bacteria and a desert plant. PLoS One. 2013;8(11):e76588.
Bar-Shmuel N, Rogovin E, Rachmilevitch S, Friedman ALL, Shelef O, Hoffmann I, et al. Tripartite symbiosis of plant-weevil-bacteria is a widespread phenomenon in the Negev Desert. Sci Rep. 2018;8(1):2420.
Meng F, Rundel PW, Sharifi MR, Bar-Shmuel N, Segoli M. The unique interaction between the summer annual desert plant Salsola inermis Forssk and weevils residing on its roots: mutualism or parasitism? Ecol Entomol. 2019. https://doi.org/10.1111/een.12772.
Craine JM, Elmore AJ, Aidar MPM, Bustamante M, Dawson TE, Hobbie EA, et al. Global patterns of foliar nitrogen isotopes and their relationships with climate, mycorrhizal fungi, foliar nutrient concentrations, and nitrogen availability. New Phytol. 2009;183(4):980–92.
Conord C, Despres L, Vallier A, Balmand S, Miquel C, Zundel S, et al. Long-term evolutionary stability of bacterial endosymbiosis in Curculionoidea: additional evidence of symbiont replacement in the Dryophthoridae family. Mol Biol Evol. 2008;25(5):859–68.
Toju H, Tanabe AS, Notsu Y, Sota T, Fukatsu T. Diversification of endosymbiosis: replacements, co-speciation and promiscuity of bacteriocyte symbionts in weevils. ISME J. 2013;7(7):1378.
Kuriwada T, Hosokawa T, Kumano N, Shiromoto K, Haraguchi D, Fukatsu T. Biological role of Nardonella endosymbiont in its weevil host. PLoS One. 2010;5(10):e13101.
White JA, Richards NK, Laugraud A, Saeed A, Curry MM, McNeill MR. Endosymbiotic candidates for parasitoid defense in exotic and native New Zealand weevils. Microb Ecol. 2015;70(1):274–86.
Ben Guerrero E, Soria M, Salvador R, Ceja-Navarro JA, Campos E, Brodie EL, et al. Effect of different lignocellulosic diets on bacterial microbiota and hydrolytic enzyme activities in the gut of the cotton boll weevil (Anthonomus grandis). Front Microbiol. 2016;7:2093.
Muhammad A, Fang Y, Hou Y, Shi Z. The gut entomotype of red palm weevil Rhynchophorus ferrugineus Olivier (Coleoptera: Dryophthoridae) and their effect on host nutrition metabolism. Front Microbiol. 2017;8:2291.
Morales-Jiménez J, Zúñiga G, Ramírez-Saad HC, Hernández-Rodríguez C. Gut-associated bacteria throughout the life cycle of the bark beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microb Ecol. 2012;64(1):268–78.
Boone CK, Keefover-Ring K, Mapes AC, Adams AS, Bohlmann J, Raffa KF. Bacteria associated with a tree-killing insect reduce concentrations of plant defense compounds. J Chem Ecol. 2013;39(7):1003–6.
Butera G, Ferraro C, Colazza S, Alonzo G, Quatrini P. The culturable bacterial community of frass produced by larvae of Rhynchophorus ferrugineus Olivier (Coleoptera: Curculionidae) in the canary island date palm. Lett Appl Microbiol. 2012;54(6):530–6.
Berasategui A, Axelsson K, Nordlander G, Schmidt A, Borg-Karlson A-K, Gershenzon J, et al. The gut microbiota of the pine weevil is similar across Europe and resembles that of other conifer-feeding beetles. Mol Ecol. 2016;25(16):4014–31.
Berasategui A, Salem H, Paetz C, Santoro M, Gershenzon J, Kaltenpoth M, et al. Gut microbiota of the pine weevil degrades conifer diterpenes and increases insect fitness. Mol Ecol. 2017;26(15):4099–110.
Sudakaran S, Salem H, Kost C, Kaltenpoth M. Geographical and ecological stability of the symbiotic mid-gut microbiota in European firebugs, Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae). Mol Ecol. 2012;21(24):6134–51.
Baubin C, Farrell AM, Šťovíček A, Ghazaryan L, Giladi I, Gillor O. Seasonal and spatial variability in total and active bacterial communities from desert soil. Pedobiologia (Jena). 2019;74:7–14.
Hu X, Li M, Zhang F, Chen H. Influence of starvation on the structure of gut-associated bacterial communities in the Chinese white pine beetle (Dendroctonus armandi). Forests. 2016;7(6):126.
Tagliavia M, Messina E, Manachini B, Cappello S, Quatrini P. The gut microbiota of larvae of Rhynchophorus ferrugineus Oliver (Coleoptera: Curculionidae). BMC Microbiol. 2014;14(1):136.
Hernández-García JA, Briones-Roblero CI, Rivera-Orduña FN, Zúñiga G. Revealing the gut bacteriome of Dendroctonus bark beetles (Curculionidae: Scolytinae): diversity, core members and co-evolutionary patterns. Sci Rep. 2017;7(1):13864.
Hernández-García J, Gonzalez-Escobedo R, Briones-Roblero C, Cano-Ramírez C, Rivera-Orduña F, Zúñiga G. Gut Bacterial Communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): A Metagenomic Analysis across Different Geographical Locations in Mexico. Int J Mol Sci. 2018;19(9):2578.
Fontes-Perez H, Olvera-García M, Chávez-Martínez A, Rodriguez-Almeida FA, Arzola-Alvarez CA, Sanchez-Flores A, et al. Genome sequence of Citrobacter sp. CtB7.12, isolated from the gut of the desert subterranean termite Heterotermes aureus. Genome Announc. 2015;3(6):e01290–15.
Handique G, Phukan A, Bhattacharyya B, Baruah AALH, Rahman SW, Baruah R. Characterization of cellulose degrading bacteria from the larval gut of the white grub beetle Lepidiota mansueta (Coleoptera: Scarabaeidae). Arch Insect Biochem Physiol. 2017;94(2):e21370.
Anand AAP, Vennison SJ, Sankar SG, Prabhu DIG, Vasan PT, Raghuraman T, et al. Isolation and characterization of Bacteria from the gut of Bombyx mori that degrade cellulose, Xylan, pectin and starch and their impact on digestion. J Insect Sci. 2010;10(107):1–20.
Behar A, Yuval B, Jurkevitch E. Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol Ecol. 2005;14(9):2637–43.
French JRJ, Turner GL, Bradbury JF. Nitrogen fixation by Bacteria from the hindgut of termites. J Gen Microbiol. 1976;95(2):202–6.
Huang HK, Tseng SK. Nitrate reduction by Citrobacter diversus under aerobic environment. Appl Microbiol Biotechnol. 2001;55(1):90–4.
Kannan V, Raju P. Nitrification by some Diazotrophic Enterobacteria. Indian J Biotechnol. 2003;2:240–5.
Hipp H, Andreesen J, Gottschalk G. The genus Clostridium—nonmedical. In: Balows A, Truper HG, Dworkin M, et al., editors. The Prokaryotes. 2nd ed. New York: Springer; 1992. p. 1800–66.
Ormerod KL, Wood DLA, Lachner N, Gellatly SL, Daly JN, Parsons JD, et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome. 2016;4(1):36.
Hussin NA, Zarkasi KZ, Ab Majid AH. Characterization of gut bacterial community associated with worker and soldier castes of Globitermes sulphureus Haviland (Blattodea: Termitidae) using 16S rRNA metagenomic. J Asia Pac Entomol. 2018;21(4):1268–74.
Hardie J, Whiley RA. The Genus Streptococcus—Oral. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, et al., editors. The Prokaryotes. New York: Springer; 2006. p. 76–107.
Bulla LA Jr, Rhodes RA, St. Julian G. Bacteria as insect pathogens. Annu Rev Microbiol. 1975;29:163–90.
Briones-Roblero CI, Hernández-García JA, Gonzalez-Escobedo R, Soto-Robles LV, Rivera-Orduña FN, Zúñiga G. Structure and dynamics of the gut bacterial microbiota of the bark beetle, Dendroctonus rhizophagus (Curculionidae: Scolytinae) across their life stages. PLoS One. 2017;12(4):e0175470.
Delalibera I, Vasanthakumar A, Klepzig KD, Raffa KF. Composition of the bacterial community in the gut of the pine engraver, Ips pini (say)(Coleoptera) colonizing red pine. Symbiosis Symbiosis. 2007;43:97–104.
Rinke R, Costa AS, Fonseca FPP, Almeida LC, Júnior ID, Henrique-Silva F. Microbial diversity in the larval gut of field and laboratory populations of the sugarcane weevil Sphenophorus levis (Coleoptera, Curculionidae). Genet Mol Res. 2011;10(4):2679–91.
Buchner P. Endosymbiosis of animals with plant microorganisms. 1965; Interscience, N Y.
Toju H, Hosokawa T, Koga R, Nikoh N, Meng XY, Kimura N, et al. “Candidatus Curculioniphilus buchneri,” a novel clade of bacterial endocellular symbionts from weevils of the genus Curculio. Appl Environ Microbiol. 2010;76(1):275–82.
Zhang G, Browne P, Zhen G, Johnston A, Cadillo-Quiroz H, Franz N. Endosymbiont diversity and evolution across the weevil tree of life. bioRxiv. 2017:171181.
Hammer TJ, McMillan WO, Fierer N. Metamorphosis of a butterfly-associated bacterial community. PLoS One. 2014;9(1):e86995.
Chen B, Teh BS, Sun C, Hu S, Lu X, Boland W, et al. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci Rep. 2016;6:29505.
Engel P, Moran NA. The gut microbiota of insects - diversity in structure and function. FEMS Microbiol Rev. 2013;37:699–735.
Huang S, Zhang H. The impact of environmental heterogeneity and life stage on the hindgut microbiota of Holotrichia parallela larvae (Coleoptera: Scarabaeidae). PLoS One. 2013;8(2):e57169.
Arias-Cordero E, Ping L, Reichwald K, Delb H, Platzer M, Boland W. Comparative evaluation of the gut microbiota associated with the below- and above-ground life stages (larvae and beetles) of the Forest cockchafer, Melolontha hippocastani. PLoS One. 2012;7(12):e51557.
Johnston PR, Rolff J. Host and Symbiont jointly control gut microbiota during complete metamorphosis. PLoS Pathog. 2015;11(11):e1005246.
Adams AS, Aylward FO, Adams SM, Erbilgin N, Aukema BH, Currie CR, et al. Mountain pine beetles colonizing historical and naive host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl Environ Microbiol. 2013;79(11):3468–75.
Adams AS, Adams SM, Currie CR, Gillette NE, Raffa KF. Geographic variation in bacterial communities associated with the red turpentine beetle (Coleoptera: Curculionidae). Environ Entomol. 2010;39(2):406–14.
Brune A, Friedrich M. Microecology of the termite gut: structure and function on a microscale. Curr Opin Microbiol. 2000;3:263–9.
Kim JK, Kim NH, Jang HA, Kikuchi Y, Kim C-H, Fukatsu T, et al. Specific Midgut region controlling the Symbiont population in an insect-microbe gut symbiotic association. Appl Environ Microbiol. 2013;79(23):7229–33.
Behar A, Jurkevitch E, Yuval B. Bringing back the fruit into fruit fly-bacteria interactions. Mol Ecol. 2008;17(5):1375–86.
Guo Z, Lu Y, Yang F, Zeng L, Liang G, Xu Y. Transmission modes of a pesticide-degrading symbiont of the oriental fruit fly Bactrocera dorsalis (Hendel). Appl Microbiol Biotechnol. 2017;101(23–24):8543–56.
Borenshtein D, Schauer B. Borenshtein D, Schauer BD (2006) The genus Citrobacter. In: Dworkin M, Falkow S, Rosenberg E, et al (ed). The Prokaryotes, 3rd edn. New York:Springer;2006. p. 90–98.
Stevenson JP. The normal bacterial flora of the alimentary canal of laboratory stocks of the desert locust, Schistocerca gregaria Forskål. J Invertebr Pathol. 1966;8(2):205–11.
Hunt J, Charnley AK. Abundance and distribution of the gut flora of the desert locust, Schistocerca gregaria. J Invertebr Pathol. 1981;38:378–85.
Dillon RJ, Vennard CT, Charnley AK. A note: gut bacteria produce components of a locust cohesion pheromone. J Appl Microbiol. 2002;92(4):759–63.
Dillon R, Charnley K. Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res Microbiol. 2002;153:03–9.
Lear G, Dickie I, Banks J, Boyer S, Buckley HL, Buckley TR, et al. Methods for the extraction, storage, amplification and sequencing of DNA from environmental samples. N Z J Ecol. 2018;42(1):10–50A.
Shahinyan G, Margaryan A, Panosyan H, Trchounian A. Identification and sequence analyses of novel lipase encoding novel thermophillic bacilli isolated from Armenian geothermal springs. BMC Microbiol. 2017;17(1):103.
Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7.
Oksanen J, Blanchet FG, Kindt R, et al. Package ‘vegan’: community ecology package. 2013; version, 2.0.
Martinez AP. pairwiseAdonis: Pairwise multilevel comparison using adonis. 2017; R package version 0.0.1.
Acknowledgments
We thank Ishai Hoffmann, Tamir Rosenberg, Ofir Altstein, Enosh Tzin, Edo Bar-Zeev, Eyal Rahav, Yuval Gottlieb, Martin Kaltenpoth, and Efrat Sheffer for technical support and fruitful discussions. We thank Einat Zchori-Fein for comments on the manuscript. This is publication number 1056 of the Mitrani Department of Desert Ecology.
Funding
This research was supported by the Israel Science Foundation (grant No. 364/16). The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
MS, FM, and AB conceived the ideas; MS, FM, and NB designed the methodology; FM, NB, and MS performed the field experiments; FM and RS performed the molecular analyses; FM analyzed the data; and FM and MS led the writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Research procedures to insects were complied with the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Fauna and Flora.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Appendix A Bacterial communities of different sample types and in the guts of C. palumbus weevils at different developmental stages. Appendix B. Assessment of the representativeness of soil bacterial communities in Negev Desert. Appendix C. Suggested functions of different species of Citrobacter from a wide range of insects. Appendix D: Location of sampling sites. Appendix E. Sampling and sequencing details of all analyzed samples
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Meng, F., Bar-Shmuel, N., Shavit, R. et al. Gut bacteria of weevils developing on plant roots under extreme desert conditions. BMC Microbiol 19, 311 (2019). https://doi.org/10.1186/s12866-019-1690-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1690-5