
Abstract
Background
The equine faecal microbiota is very complex and remains largely unknown, while interspecies interactions have an important contribution to animal health. Clostridium difficile has been identified as an important cause of diarrhoea in horses. This study provides further information on the nature of the bacterial communities present in horses developing an episode of diarrhoea. The prevalence of C. difficile in hospitalised horses at the time of admission is also reported.
Results
Bacterial diversity of the gut microbiota in diarrhoea is lower than that in non-diarrhoeic horses in terms of species richness (p-value <0.002) and in population evenness (p-value: 0.02). Statistical differences for Actinobacillus, Porphyromonas, RC9 group, Roseburia and Ruminococcaceae were revealed. Fusobacteria was found in horses with diarrhoea but not in any of the horses with non-diarrheic faeces. In contrast, Akkermansia was among the three predominant taxa in all of the horses studied. The overall prevalence of C. difficile in the total samples of hospitalised horses at admission was 3.7 % (5/134), with five different PCR-ribotypes identified, including PCR-ribotype 014. Two colonised horses displayed a decreased bacterial species richness compared to the remaining subjects studied, which shared the same Bacteroides genus. However, none of the positive animals had diarrhoea at the moment of sampling.
Conclusions
The abundance of some taxa in the faecal microbiota of diarrhoeic horses can be a result of microbiome dysbiosis, and therefore a cause of intestinal disease, or some of these taxa may act as equine enteric pathogens. Clostridium difficile colonisation seems to be transient in all of the horses studied, without overgrowth to trigger infection. A large proportion of the sequences were unclassified, showing the complexity of horses’ faecal microbiota.
Similar content being viewed by others


Background
Equine gut microbiota is poorly characterised and studies are currently underway to increase understanding of how defined microbial communities are able to interfere with different bacterial aetiologies of diarrhoea in horses. Recently, high-throughput amplicon sequencing analysis has been introduced to investigate the intestinal microbiota of healthy horses and horses with colitis [1–3]. However, the equine faecal microbiome is still largely unknown, as are interspecies interactions and their contribution to animal health [1, 3].
In horses, diarrhoea and colitis have been associated with a number of different pathogenic agents, including Clostridium difficile, Clostridium perfringens, Salmonella spp and Escherichia coli [4–7]. Clostridium difficile is amongst the most important agents of diarrhoea and serious colitis in horses [8]. While both adult horses and foals can suffer C. difficile enteric disease, it seems that foals are more likely to be colonised by the bacterium [9]. A diagnosis of C. difficile infection (CDI) requires clinical suspicion as well as detection of pre-formed C. difficile toxins TcdA and/or TcdB (or transcripts) in non-enriched specimens. Other possible causes of acute colitis must also be ruled out [8]. However, in most cases of diarrhoea, the aetiology remains unclear and the prevalence of C. difficile colonisation in hospitalised horses has rarely been addressed [10, 11].
As in humans, the major risk factors for the development of CDI are antibiotic treatment and hospitalisation [8]. However, some cases of infection have been also reported in horses without previous exposure to these risk factors, including in foals at 2 to 5 days of age [12]. Furthermore, it has been reported that up to 7 % of horses carry spores of C. difficile without showing any signs of diarrhoea [10, 13].
The first objective of this study was to provide further information on the nature of the bacterial community present in horses developing diarrhoea, including possible alterations in the microbiota profiles as a result of antibiotic treatment, through comparison with faeces from horses without diarrhoea. This study also aimed to examine, by culture of horse faeces at admission to an Equine Clinic, the carriage rate of C. difficile. Isolates obtained were characterised in terms of PCR-ribotype, toxigenic activity and antibiotic resistance. Further metagenetic analyses were performed to compare the faecal microbiota of C. difficile in colonised and non-colonised horses.
Results
Bacterial community present in horses with and without diarrhoea
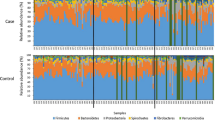
A group of 10 horses with diarrhoea at the moment of sampling were compared with 10 non-diarrhoeic horses via metagenetic analysis. All of the animals (n = 20) tested negative for C. difficile by faeces culture (Table 1). Pyrosequencing yielded between 4,000 and 5,000 reads per sample (Additional file 1). The microbiota composition for each horse is presented at phylum level (Fig. 1), genus level (Fig. 2) and species level (Additional file 2). The more abundant bacterial families found for both groups were Lachnospiraceae (between 7 % and 39 %), Ruminococcaceae (between 2.7 % and 28.7 %), Verrucomicrobiaceae (between 0 % and 43.1 %), and Prevotellaceae (between 0.6 % and 17.5 %). Bacterial diversity of the gut microbiota in diarrhoea was lower than in non-diarrhoeic horses (p-value: 0.0105). This effect was observed both in terms of species richness and in the population evenness (Fig. 3). Principal coordinate analysis (PCoA) of the diarrhoeic and non-diarrhoeic horses did show the sample distribution along the 3 main axes (PC1-PC2-PC3) (Additional file 3). Analysis of Molecular Variance (AMOVA) revealed a significant difference between the variance of both groups taken as a single group and the variance of each group (sum of squares (ss): 0.53 and 11.66 among and within groups respectively; AMOVA test statistic Fs: 1.57; p-value: 0.006). Unifrac weighted analysis further showed that both groups share different population structure (Wscore: 0.88 and p-value: <0.001). Thus, the microbiota structure of each group was showed statistically different from each other. The relative abundance of each genus in both groups were compared in order to identify the populations responsible for this difference (White t-test), leading to statistical differences for Actinobacillus, Porphyromonas, RC9 gut group, Roseburia and a taxonomically undefined population belonging to the Ruminococcaceae (Ruminococcaceae unclassified) (Fig. 4).

Microbiota faecal composition at phylum level for horses with and without diarrhoea. Bar chart detailing the relative abundance of the 17 core phylotypes common to the two groups of horses (with and without diarrhea)

Microbiota faecal composition at genus level (cumulated mean relative abundance >4 %) for horses with and without diarrhoea. Samples 13, 14, 15, 17, 23, 25, 26, 27, 28 and 29: horses with diarrhoea. Samples 11, 12, 16, 18, 19, 20, 21, 22, 24 and 30: horses without diarrhoea. The unclassified populations correspond to defined groups of the genus level for which a taxonomical classification assignation to the genus cannot be attributed. These populations are therefore labelled with the first defined superior hierarchical taxonomic level followed by “_unclassified” to prevent confusion

Bacterial biodiversity, bacterial richness and bacterial evenness in C. difficile negative horses with and without diarrhoea. Box plot of richness, evenness and diversity values showed that the microbiota structure of each group (diarrhoeic and non diarrhoeic horses) is statistically different from each other. Whiskers represent minimum and maximum value. Bottom and top of the box are the first and the third quartile. The median is shown as a band inside the box

Bacterial genus whose relative abundance was statistically different between the 2 groups. Result of a White test (p value <0.05). Box plot showing mean relative sequence abundance of Actinobacillus, Porphyromonas, RC9, Roseburia and Ruminococcaceae_unclassified in horses with and without diarrhea. The error bar indicates the diversity between samples in terms of proportions of sequences
Clostridium difficile prevalence in horses at admission and strain characterisation
During the three-month study period, the total number of horses admitted at the clinic for either emergencies or consultation was 302, with 141 hospitalisations. A total of 136 samples were collected from the 134 hospitalised horses enrolled in the study. Two horses were sampled on two different times because they suffered a diarrhoeal episode during hospitalisation. However, these horses did not test positive for C. difficile on any of the sample days. The overall prevalence of C. difficile in the faecal microbiota of hospitalised horses at admission was 3.7 % (5/134). All horses testing positive were adult animals aged between four and 16 years old.
Altogether, 52 of the total of 134 horses studied (38.8 %) presented gastrointestinal problems at admission, but C. difficile was isolated from only three of these animals (two with colic and one with proximal enteritis). In these three horses a nasogastric tube had been passed before the faecal collection. However, none of them had previously received an antibiotic treatment. Nineteen horses (14.2 %) had diarrhoea at admission but all tested negative for the bacterium (Additional File 4).
The remaining 82 horses sampled (61.2 %) were not affected by gastrointestinal disorders. In this group, C. difficile was detected in two horses. The clinical diagnoses in these two infected horses were multiple wounds and wound with tendon injury, respectively. Both had received a similar antibiotic treatment (gentamicin and penicillin with or without ceftiofur) and non-steroidal anti-inflammatory drugs (dypirone). Age (9 years) and the gender (female) of the two horses were also the same (Table 2). In terms of antimicrobial therapy, 54 out of 134 horses studied (40.3 %) had previously received antimicrobial therapy (prior to the study period), but most (n = 52) tested negative for C. difficile. Among them, the most common drug used was ceftiofur, which was administered to 16 (11.9 %) horses.
Four of the equine isolates were positive for tcdA, tcdB and binary toxin CDT genes while only one was non-toxinogenic. The presence of TcdB was confirmed by cytotoxicity assay using confluent monolayer MRC-5 cells. None of the isolates presented an 18, 39-base pair deletion or a deletion at 117 of the tcdC gene. Five different PCR-ribotypes were detected. Only one strain had a ribotype profile associated with the reference Cardiff collection number (014). The remaining isolates were identified as UCL237, UCL49, UCL23f and UCL36 (non-toxigenic PCR-ribotype). PCR-ribotypes UCL49, 014 and UCL23f were isolated from three animals with gastrointestinal problems while PCR-ribotypes UCL237 and UCL36 were recovered from the two horses with wounds (Table 2).
Only the non-toxigenic strain PCR-ribotype UCL36 showed resistance to metronidazole (minimum inhibitory concentration (MIC) = 40 μg/ml, average of two essays) and erythromycin. For clindamycin, only one isolate (PCR-ribotype 014) was susceptible, while all others were fully resistant. Intermediate resistance for penicillin was observed in all of the isolates tested. The isolate PCR-ribotype UCL237 also exhibited intermediate resistance to tetracycline, while all the rest were susceptible. There was no vancomycin, moxifloxacin or rifampicin resistance detected, but all the strains were resistant to ceftiofur (Table 2).
Microbiota composition for C. difficile positive and negative horses
Stool samples from all horses testing positive for C. difficile (n = 5) were studied in order to obtain further information about the microbiota composition of the colonised subjects. As during the entire study period (3 months) only five animals were positive for the bacterium, we could only use five C. difficile-negative horses as control group, but with similar clinical history (Additional file 5). In both C. difficile colonised and non-colonised horses, the dominant taxa were Lachnospiraceae (ranging from 3.2 to 20.8 %), Bacteroidales (ranging from 5 to 29.0 %) and Ruminococcaceae (ranging from 7 to 17.9 %). In the group of C. difficile positive horses, only one animal (10) presented a predominance of Bacteroides (36.8 %) and Akkermansia (19.5 %). The same Bacteroides genus was found in another C. difficile positive sample (01) at a level of 7.2 % (Fig. 1). Only 45 distinct OTUs (with a mean abundance greater than 4 %) were identified, representing 25-40 % of the relative abundance of the microbial taxa in the faecal samples (Fig. 5 and Additional file 6).

Microbiota faecal composition at genus level (cumulated mean relative abundance >4 %) of C. difficile culture-positive and -negative horses. Samples HF_01, 03, 04, 09 and 10: C. difficile positive horses detected by faeces culture. Samples HF_02, 05, 06, 07 and 08: C. difficile negative horses detected by faeces culture
However, bacterial biodiversity, bacterial richness and bacterial evenness were not statistically different between C. difficile colonised and non-colonised horses (p-value > 0.05). Ordination analysis with PCoA supported the grouping of most of the individuals into one group (Additional file 7), which confirmed by non significant results from AMOVA (sum of squares (ss) 0.41 and 3.18 among and within groups respectively; test statistic for AMOVA (Fs) 1.03; p-value: 0.389) and Unifrac analysis of sample clustering (Wscore: 0.97 and p-value: 0.92). Indeed, White test abundance population comparison between groups performed at the different taxonomical levels identified two taxa (RF16 group and Clostridiales) for which the relative abundance was statistically different (Additional file 8).
Microbiota composition relation with gastro-intestinal disorder diagnostics
We grouped the microbiota profiling data from the 20 samples from diarrhoeic and non diarrhoeic samples with the 10 C. difficile positive and negative samples together and assigned them to diagnosis categories: Colic (n = 12), Enteritis (n = 1), Enterocolitis (n = 1), diarrhoea (n = 3) and others (13). We used this clustering in order to identify genus whose abundance could be related to one particular category. Statistical abundance comparison with ANOVA2 between colic, diarrhoea and other categories highlight a higher abundance of Escherichia and Streptococcus genera in colic group compared to the others and higher abundance of Akkermansia, Fusobacterium, Porphyromonas and Xylanibacter genera in the Diarrhoea group (Additional file 9). Enteritis and Enterocolitis group, having only one sample could not be included in statistical abundance comparison but each sample was dominated by 2 defined genus: Bacteroides and Parabacteroides in the Enteritis sample and Porphyromonas and Fusobacterium Enterocolitis sample (Additional file 10).
Discussion
High-throughput amplicon sequencing analysis is one of the methods of choice in the study of complex gut microbiota ecosystems [14]. However, most of the studies reported bacteria populations at the phylum and class level while the genus and species level were explored only in a few recent studies [15, 16]. Higher taxonomic resolutions (genus or species level) may reveal more differences in population structure than phylum or class level [17] and provide the degree of precision necessary for clinical diagnosis [18]. As previously demonstrated, selecting the accurate region of 16S ribosomal DNA (rDNA) gene to sequence is essential in determining the utility of microbial genomics for species-level assignments [19]. In the present study we report genus and species labelling based on V1-V3 region.
To determine whether the presence of diarrhoea was related to changes in the composition of the faecal microbiota of horses, a strict screening process was carried out among all the samples obtained over three months, in order that the two groups (with and without diarrhoea) were as similar as possible and therefore comparable. As observed in a previous study investigating the microbiota in the equine large intestine via 16S ribosomal DNA sequencing [1], a great part of the sequences (60 %) were not identical (less than 1 % mismatch) to sequence entries present in SILVA database (v1.15). Even among the sequences identical to known entries, the species name was seldom taxonomically defined. These findings underline the lack of knowledge regarding a good part of the horse gut microbiota stressing the need for further research on fundamental microbiology either on taxonomic as well on the functional level.
Interestingly, Akkermansia was found in 90 % of horses studied, with a relative abundance ranging between 0.03 % and 43.1 %. This bacterium is an appealing candidate to become a human probiotic, selected based on established mechanisms of preventative treatment of obesity and diabetes [20–22]. Only one previous study [23] describes the genus Akkermansia in the equine intestinal microbiota In this study we reported the presence of Akkermansia muciniphila and Akkermansia EU779370 in the faecal microbiota of horses with and without diarrhoea. Akkermansia EU779370 population identified in this study is 100 % identical to the Akkermansia EU779370 GenBank entry (for the sequenced V1-V3 section). It represents a potential new Akkermansia species as it shares only 90 % of nucleotide identity with Akkermansia muciniphila. The relevance of this finding deserves further investigation.
Overall, the composition of the microbiota of all horses studied was dominated by the same taxa as previously described [4]. However, the differences observed in the cumulative mean relative abundance among individuals in these dominant bacterial groups may be linked to recent dietary history [17] or to antimicrobial therapy [23]. Diarrhoea has been associated with changes in the faecal microbiota composition of humans but alterations in the equine gut microbiota has been rarely addressed [24]. In the present study, Actinobacillus (0.3 % mean abundance in diarrhoeic group versus 0.004 in non-diarrhoeic group) and Porphyromonas taxa (5.6 % mean abundance in diarrheic group versus 0.002 in non-diarrhoeic group) were detected more abundantly in horses with diarrhoea. These results contrast with a previous study of commensal bacteria in acute diarrhoea in children, where Porphyromonas species were in the lowest proportions during acute diarrhoea compared with levels during periods of normal gastrointestinal health [25]. Fusobacteria were found in horses with diarrhoea but not in any of the horses with normal faeces. In a previous study, Fusobacterium spp. was also found in higher percentages in horses with colitis, which could be a consequence of overgrowth due to bacterial dysbiosis or an aetiological agent of disease [4]. In human beings, Fusobacteria have been associated with colorectal carcinomas and adenomas [26]. However, there are no previous studies describing this bacterium as an equine enteropathogen [4].
After clustering of the different feces samples into more defined diagnosis categories, genus Escherichia and Streptococcus were more abundant in horses with diarrhoea without any other symptoms. A more refined analysis revealed that the species involved were Escherichia coli and Streptococcus equinus. If the first is long known to be associated with diarrhoea, the second is the most common Streptococcus found in horse feces. In the colic group, Fusobacterium and Porphyromonas genera were found in higher amount compared to other groups but were composed of yet unknown species. As mentioned above, little is known regarding the involvement of these bacteria in gastro-intestinal disorders. Finally, two horses suffered from enteritis and enterocolitis and their 16S profiling revealed a domination (above 30 % of the sample sequences) of genus Bacteroides in the enteritis case and genus Porphyromonas in the enterocolitis case. The Bacteroides population was mainly represented by Bacteroides heparinolyticus. This species, originally characterised as an agent of periodontitis in human [27], is phylogenetically related to Bacteroides fragilis which is well-known enterotoxinogenic bacteria involved in human infections [28]. However, there is still no evidence of B. heparinolyticus involvement in gut disorder in horse or in human and it is thus unclear whether its abundance is related to the symptomatology.
We are aware that the limited size of the analysed cohort reduces the strength and the scope of our results. Larger cohort studies will be needed to improve our knowledge on diarrhoea impact on horse microbiota.
In the present study, the carriage of C. difficile at the time of admission was examined. The overall prevalence found was 3.7 % (5/134). None of the positive animals had diarrhoea at the moment of sampling which may suggest that C. difficile colonisation in these horses was transient in most cases. In the literature, there is only one previous study that investigated the presence of C. difficile in horses at admission to a veterinary teaching hospital [11]. Our results correlate with the findings of this study, which reported a prevalence of 4.8 % (4/82). In another previous study conducted at the same Belgian Equine Clinic, we observed a C. difficile colonisation rate of 13.7 % (10/73). However, in that study, animals were not only sampled at the time of admission but also tracked during their hospitalisation, which could explain the higher prevalence found [10].
From the five C. difficile positive horses found, three of them presented gastrointestinal problems (colic and proximal enteritis) with a nasogastric tube passed before sampling. Nasogastric tube placement has been previously identified as a risk factor for C. difficile infection [11]. Two other horses without enteric perturbations were also colonised by C. difficile. Both had suffered wounds and were treated with antibiotics. Previous studies reported intestinal flora perturbations and antibiotic exposure as the most significant risk factors for C. difficile proliferation in horses [5].
There were five different PCR-ribotypes detected among the five C. difficile positive animals, which suggests that a wide variety of C. difficile strains circulate in horses, as previously reported [11, 29], including PCR-ribotype 014. All the isolates were resistant to ceftiofur and four out of five were also resistant to clindamycin, which agrees with the findings of previous studies [10, 30]. Only one isolate was resistant to metronidazole and erythromycin. Surprisingly, this strain (PCR-ribotype UCL36) was the only non-toxigenic isolate. A high degree of resistance to antimicrobials (including erythromycin and clindamycin) in non-toxigenic strains has been reported previously [30] but the role in disease development or prevention is still unknown [31].
We were unable to identify C. difficile by pyrosequencing analysis in the stool samples with positive cultures. In humans, it is considered that feces harbour up to 1012 bacteria per gram [32]. Thus, our sampling of thousands of sequences limits our detection ability to populations above 109 bacteria per gram. The horses testing positive by culture did not have any clinical signs of C. difficile disease and the isolate was obtained only after three days of enrichment. While the results of high-throughput amplicon sequencing analysis are limited by the small number of animals positive for C. difficile, and by the fact that none of the animals suffered CDI, the findings for each colonised horse should not be dismissed as they provide a first insight, albeit limited, about the impact of C. difficile colonisation in the horses’ gut microbiota, which merits further investigation.
Conclusions
Metagenomic analysis is a promising tool to identify correlations between changes in the gut microbiota and intestinal diseases. The abundance of Actinobacillus, Porphyromonas and Fusobacteria in the faecal microbiota of diarrhoeic horses deserves special attention, as it can be a result of microbiome dysbiosis, and therefore a cause of intestinal disease, or in the case of Fusobacteria, may act as equine enteric pathogen. Furthermore, the high proportion of Akkermansia in all of the horses studied and its role in the intestine merits further investigation. Clostridium difficile colonisation seems to be transient in all of the horses studied without overgrowth to trigger infection. For a great variety of bacterial species the currently available systems are not able to confidently assign taxonomy, which shows how complex and still unknown the equine microbiome is.
Methods
Inclusion criteria and sampling
Samples were collected over a three month period (October to December 2013) at the Equine Clinic, Department of Companion Animals and Equids, Faculty of Veterinary Medicine, University of Liege.
All hospitalised horses during this period with a clinic stay of at least one day were eligible. Subjects were all selected without regard to their diagnosis or the possible duration of hospitalisation. The exclusion criteria were horses exhibiting dysphoric mood, intolerable stress, or any other disease for which sampling by rectal manipulations was not recommended. Samples were collected between day one and day two following admission. In addition, all horses developing an episode of diarrhoea during their hospital stay were sampled for a second time. Horses were documented for data relating to clinical history, diagnostic findings and treatment received, including the prescription of antimicrobial agents. Faecal sampling was performed directly via rectal. Samples obtained were scored as normal faeces, diarrhoea or bloody diarrhoea faeces. All samples were processed on the same day immediately after transport (at room temperature) to the laboratory (approximately 15 min after sampling). In cases of emergency admission (i.e., during the night or on a non-working day), two samples per horse were collected. The first sample was collected in an individual identified sterile 50 ml tube for further culture to detect C. difficile; the second was collected using the Stool DNA stabiliser (PSPR Spin Stool DNA Plus Kit 00310, Invitek) and stored at 4 °C in the hospital for a maximum of three days before processing. After culture of faeces, all samples were frozen immediately at −80 °C before DNA extraction.
16S rDNA pyrosequencing and data analysis
Among all the faecal samples collected, clinical history of each subject was investigated in order to select two homogenous groups of horses (with and without diarrhoea) with the same number of individuals in each group. Both groups were matched for age, pathologies, previous hospital stay and medical treatment. A total of 20 faecal samples (ten with diarrhoea and ten without diarrhoea) were further studied. A second selection was done on the pool of 140 horses to gather 2 homogenous groups of horses, either or not positive for C. difficile by classical microbiology (n = 5), which were matched for age, pathologies, previous hospital stay and medical treatment. Finally, the 30 samples were grouped together in a third analysis and clustered into defined diagnostic category based upon diagnostic (Table 1; Table 2; Additional file 5). The resulting diagnostic categories are: colic—horses suffering from abdominal pain (n = 12); diarrhoea—horses with 3 or more loose or liquid stools per day, without other symptom (n = 3); enteritis—horse with ileon inflammation (n = 1); enterocolitis—horse with ileon and colon inflammation (n = 1) and others—horses with non Gastro-intestinal disorders (n = 13).
Total DNA was extracted from the stool samples with the PSPR Spin Stool DNA Plus Kit 00310 (Invitek), following the manufacturer’s recommendations. The DNA was eluted into DNase/RNase-free water and its concentration and purity were evaluated by absorbance measurement using the NanoDrop ND-1000 spectrophotometer (NanoDrop ND-1000, Isogen). PCR-amplification of the V1-V3 region of the 16S rDNA was performed as previously described [33]. Primers E9-29 and E514-530 [33–35] were selected for their theoretical ability to generate the lowest possible amplification capability bias among the various bacterial phyla [36]. The oligonucleotide design included 454 Life Sciences’ A or B sequencing titanium adapters (Roche Diagnostics) and multiplex identifiers (MIDs) fused to the 5′ end of each primer. The master mix composition consisted of 5 units of FastStart high fidelity polymerase (Roche Diagnostics), 1x enzyme reaction buffer, 200 μM dNTPs (Eurogentec), 0.2 μM of each primer and 100 ng of genomic DNA in a volume of 100 μl. The amplification was carried out in a gradient thermocycler (Eppendorf) as follows: denaturation at 94 °C for 15 min followed by 25 cycles of 94 °C for 40 s, 56 °C for 40 s; 72 °C for 1 min; and a final elongation step at 72 °C for 7 min. PCR products were run on 1 % agarose gel electrophoresis and purified using the SV PCR purification kit (Promega Benelux). Picogreen dsDNA quantitation assay (Isogen) was performed in order to assess the quality and quantity of the products. All libraries were run in the same titanium pyrosequencing reaction using Roche multiplex identifiers, and amplicons were sequenced using the Roche GS-Junior Genome Sequencer instrument (Roche).
Sequence reads were processed using MOTHUR software package v1.32 [37] and denoised using the Pyronoise algorithm [38]. Trimming criteria of the reads was applied as follows: read lengths no shorter than 425 bp, an exact match to the barcode, and 1 mismatch allowed to the proximal primer [33]. Sequences were checked for the presence of chimeric amplifications using the UCHIME algorithm [39].
The read sets obtained were compared with a reference data set of aligned sequences of the corresponding region derived from the SILVA database (v1.15) of full-length rDNA sequences [40] implemented in MOTHUR [41]. The final reads were clustered into operational taxonomic units (OTUs) using the nearest MOTHUR neighbour algorithm with a 0.03 distance unit cut-off. Taxonomic identity was attributed to each OTU by comparison with the SILVA v1.15 database [37] (80 % homogeneity cut-off) [33]. When taxonomic identification fell below the 80 % treshold, the taxonomic level was labelled with the first defined level from higher level followed by the term “_unclassified”.
All unique sequences for each OTU were further compared with the SILVA data set version v1.15 using the BLASTN algorithm [42, 43], as MOTHUR is not suitable to taxonomic assignment beyond the genus level. For each OTU, a consensus detailed taxonomic identification was given based upon the identity (less than 1 % of mismatch with the aligned sequence) and the metadata associated with the most frequent hits leading to 3 kind of labelling : (i) the population is identical to a taxonomically defined species and is labelled “genus_species”; (ii) the population is identical to a reference sequence belonging to a still undefined species and is labelled “genus_NCBI Accession Number”; (iii) the sequence is not identical to any known sequence and is arbitrarily labelled with its OTU number [33].
Subsample datasets were obtained and used to evaluate ecological indicators (the richness and microbial diversity of the samples) using MOTHUR. Population structure and community membership were assessed with MOTHUR using distance matrices based on the Jaccard index (a measure of community membership; which considers the number of shared OTUs but not their abundance) and the Yue and Clayton measure of dissimilarity (a measure of community structure which considers shared OTUs and their relative abundances) [44]. Richness estimation (Chao1 estimator) [45], microbial biodiversity (non-parametric (NP) Shannon diversity index) [46], and the population evenness (Shannon evenness) [47] were calculated using MOTHUR. Chao 1 estimator was used to estimate the richness of the detected species (OTUs) in a sample [33].
Ordination and statistical analysis and biosample accession numbers
Ordination analysis were performed with Vegan package in R [48].
Principal coordinate analysis (PCoA) was applied to visualise the biodiversity between the two groups [49]. Statistical analysis regarding community structure and composition were performed with AMOVA and UNIFRAC implemented in MOTHUR v1.32. Analysis of molecular variance (AMOVA) was used for estimating population differentiation [50]. Unifrac unweighted analysis, which accounts for the relative abundance of each of the taxa within communities, was used to evaluate differences in population structure between pairs of sample categories [51]. The differences were considered significant for a p-value of less than 0.05; all the results given are the means ± the standard deviations of the results between the samples of each category [33]. Statistical differences between bacterial biodiversity, richness and evenness were assessed using two-sided unpaired t-test using PRISM 6 (Graphpad Software). In order to highlight statistical differences in the bacterial population abundance between categories, a White tests (paired comparisons) and ANOVA with Tukey post-hoc test were performed using STAMP software [52]. All the biosample sequences have been deposited at the National Center for Biotechnology Information (NCBI) [53] and are available under de Bioproject ID PRJNA279335.
Clostridium difficile culture, identification and characterisation
For isolation of C. difficile, one gram of faeces was inoculated into 9 ml of CCFT (cycloserine cefoxitin fructose taurocholate) broth as previously described [54] and incubated anaerobically for 72 h at 37 °C. A 10 μl aliquot of the enriched broth was spread on CCFT plates and incubated anaerobically at 37 °C for three days. One presumptive colony per plate was subcultured onto blood agar 5 % Sheep Blood (Biorad) and checked using a C. difficile latex agglutination rapid test Kit DR 1107A (Oxoid). Identification, toxin gene profile, deletions in the regulator gene tcdC, and gyrA mutation (gene associated with moxifloxacin resistance) were determined using the Genotype Cdiff system (Hain Lifescience) according to the manufacturer’s instructions. The supernatant from each pure culture was tested for cytotoxicity assay (TcdB) using confluent monolayer MRC-5 cells, as previously described [54]. A PCR ribotyping method based on capillary gel was performed using the primers recommended by Bidet et al. [55]. The isolates with a PCR-ribotype profile from which reference strains were available in our laboratory were designated with the relevant Cardiff international number. Otherwise, isolates were identified with an internal nomenclature beginning with UCL.
Antibiotic resistance
Clostridium difficile isolates (n = 5) were tested for susceptibilities to a panel of nine antimicrobial agents by disc diffusion (n = 7) and E-test (n = 2). The antimicrobials studied were chosen because they have been associated with C. difficile infection or its treatment, or because they were widely used in the equine teaching hospital used in the study.
Resistance to rifampin (25 μg), erythromycin (15 μg), oxitetracycline (30 μg), vancomycin (30 μg), penicillin (10 μg), clindamycin (2 μg), and ceftiofur (30 μg) (Becton-Dickinson) was tested by disc diffusion assay on Brucella Blood Agar with hemin and vitamin K1 (Becton-Dickinson) according to the French Society of Microbiology [56] protocols. Zone diameters were read after 24 h of anaerobic incubation at 37 °C and interpreted as previously described [10].
Susceptibility to metronidazole and moxifloxacin was determined using the Etest method (Lucron ELITechGroup) on Schaedler with Vit K1 and 5 % sheep blood (Becton-Dickinson) according to the manufacturer’s instructions. Plates were incubated anaerobically at 37 °C for 48 h. The susceptibility and resistance breakpoints for metronidazole (s ≤8 μg/ml; r ≥32 μg/ml) and moxifloxacin (s ≤ 2 μg/ml; r ≥8 μg/ml) used for interpretation were those recommended by the Clinical and Laboratory Standard Institute [57]. Bacteroides fragilis ATCC 25285 was tested as a quality control.
Metagenetic analysis
Rectal faecal samples of all animals positive for C. difficile (n = 5) were analysed by 16S rDNA pyrosequencing (as described above) in order to recover further information about the microbiota composition of horses colonised by the bacterium. A further group of non-colonised horses (n = 5) was used as a control. Control subjects were selected on basis of their similarity to colonised horses, including same clinical history (pathologies and antimicrobial treatment), age and previous hospital stay, to obtain two groups as similar as possible.
Ethics
This study required no experimental research on animals, only the use of collected feces. Therefore, this study did not require approval from Animal Ethics Committee following Belgian (royal decree M.B.10.07.2013) and European legislation (2010/63/UE).
References
Daly K, Stewart CS, Flint HJ, Shirazi-Beechey SP. Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes. FEMS Microbiology Ecology. 2001;38:141–51.
Dougal K, de la Fuente G, Harris PA, Girdwood SE, Pinloche E, Newbold CJ. Identification of a core bacterial community within the large intestine of the horse. PloS one. 2013;8:e77660.
O’ Donnell MM, Harris HMB, Jeffery IB, Claesson MJ, Younge B, O’ Toole PW, et al. The core faecal bacterial microbiome of Irish Thoroughbred racehorses. Letters in applied microbiology. 2013;57:492–501.
Niwa C, Kato H, Hobo S, Kinoshita Y, Veno T, Katayama Y, et al. Postoperative Clostridium difficile infection with PCR ribotype 078 strain identified at necropsy in five throughbred racehorses. Veterinary Research. 2013;173:607.
Donaldson MT, Palmer JE. Prevalence of Clostridium perfringens enterotoxin and Clostridium difficile toxin A in feces of horses with diarrhea and colic. Journal of the American Veterinary Medical Association. 1999;215:358–61.
Hartnack AK, Van Metre DC, Morley PS. Salmonella enterica shedding in hospitalized horses and associations with diarrhea occurrence among their stablemates and gastrointestinal-related illness or death following discharge. Journal of the American Veterinary Medical Association. 2012;240:726–33.
Van Duijkeren E, van Asten AJ, Gaastra W. Characterization of Escherichia coli isolated from adult horses with and without enteritis. The Veterinary quarterly. 2000;22:162–6.
Diab SS, Songer G, Uzal FA. Clostridium difficile infection in horses: a review. Veterinary microbiology. 2013;167:42–9.
Rodriguez-Palacios A, Borgmann S, Kline TR, LeJeune JT. Clostridium difficile in foods and animals: history and measures to reduce exposure. Animal health research reviews / Conference of Research Workers in Animal Diseases. 2013;14:11–29.
Rodriguez C, Taminiau B, Brévers B, Avesani V, Van Broeck J, Leroux AA, et al. Carriage and acquisition rates of Clostridium difficile in hospitalized horses, including molecular characterization, multilocus sequence typing and antimicrobial susceptibility of bacterial isolates. Veterinary microbiology. 2014;172:309–17.
Medina-Torres CE, Weese JS, Staempfli HR. Prevalence of Clostridium difficile in horses. Veterinary microbiology. 2011;152:212–5.
Jones RL, Adney WS, Shideler RK. Isolation of Clostridium difficile and detection of cytotoxin in the feces of diarrheic foals in the absence of antimicrobial treatment. Journal of clinical microbiology. 1987;25:1225–7.
Arroyo LG, Stämpfli HR, Weese JS. Potential role of Clostridium difficile as a cause of duodenitis-proximal jejunitis in horses. Journal of medical microbiology. 2006;55(Pt 5):605–8.
Zhou Y, Burnham C-AD, Hink T, Chen L, Shaikh N, Wollam A, et al. Phenotypic and genotypic analysis of Clostridium difficile isolates: a single-center study. Journal of clinical microbiology. 2014;52:4260–6.
Shankar V, Hamilton MJ, Khoruts A, Kilburn A, Unno T, Paliy O, et al. Species and genus level resolution analysis of gut microbiota in Clostridium difficile patients following fecal microbiota transplantation. Microbiome. 2014;2:13.
Shahinas D, Silverman M, Sittler T, Chiu C, Kim P, Allen-Vercoe E, Weese S, Wong A, Low DE, Pillai DR: Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. mBio 2012. 3 doi: 10.1128/mBio.00338-12.
Fernandes KA, Kittelmann S, Rogers CW, Gee EK, Bolwell CF, Bermingham EN, et al. Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PloS one. 2014;9:e112846.
Salipante SJ, Sengupta DJ, Rosenthal C, Costa G, Spangler J, Sims EH, et al. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PloS one. 2013;8:e65226.
Conlan S, Kong HH, Segre JA. Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PloS one. 2012;7:e47075.
Cani PD, Everard A. [Akkermansia muciniphila: a novel target controlling obesity, type 2 diabetes and inflammation?]. Médecine Sciences. 2014;30:125–7.
Shin N-R, Lee J-C, Lee H-Y, Kim M-S, Whon TW, Lee M-S, et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63:727–35.
Underwood MA. Intestinal dysbiosis: novel mechanisms by which gut microbes trigger and prevent disease. Preventive medicine. 2014;65:133–7.
Costa MC, Stämpfli HR, Arroyo LG, Allen-Vercoe E, Gomes RG, Weese J. Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs. BMC veterinary research. 2015;11:19.
Costa MC, Arroyo LG, Allen-Vercoe E, Stämpfli HR, Kim PT, Sturgeon A, et al. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PloS one. 2012;7:e41484.
Balamurugan R, Janardhan HP, George S, Raghava MV, Muliyil J, Ramakrishna BS. Molecular studies of fecal anaerobic commensal bacteria in acute diarrhea in children. Journal of pediatric gastroenterology and nutrition. 2008;46:514–9.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome research. 2012;22:299–306.
Okuda K, Kato T, Shiozu J, Takazoe I, Nakamura T. Bacteroides heparinolyticus sp. nov. Isolated from Humans with Periodontitis. International Journal of Systematic Bacteriology. 1985;35:438–42.
Sears CL. The toxins of Bacteroides fragilis. Toxicon : official journal of the International Society on Toxinology. 2001;39:1737–46.
Thean S, Elliott B, Riley TV. Clostridium difficile in horses in Australia - a preliminary study. Journal of Medical Microbiology. 2011;60:1188–92.
Martirossian G, Sokół-Leszczyńska B, Mierzejewski J, Meisel-Mikołajczyk F. [Occurrence of Clostridium difficile in the digestive system of dogs]. Medycyna doświadczalna i mikrobiologia. 1992;44:49–54.
Natarajan M, Walk ST, Young VB, Aronoff DM. A clinical and epidemiological review of non-toxigenic Clostridium difficile. Anaerobe. 2013;22:1–5.
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Applied and environmental microbiology. 1999;65:4799–807.
Delcenserie V, Taminiau B, Delhalle L, Nezer C, Doyen P, Crevecoeur S, et al. Microbiota characterization of a Belgian protected designation of origin cheese, Herve cheese, using metagenomic analysis. Journal of dairy science. 2014;97:6046–56.
Bindels LB, Neyrinck AM, Salazar N, Taminiau B, Druart C, Muccioli GG, et al. Non Digestible Oligosaccharides Modulate the Gut Microbiota to Control the Development of Leukemia and Associated Cachexia in Mice. PloS one. 2015;10:e0131009.
De Maesschalck C, Eeckhaut V, Maertens L, De Lange L, Marchal L, Nezer C, et al. The effects of xylocpaxoligosaccharides on performance and microbiota in broiler chickens. Applied and environmental microbiology. 2015;81(17):5880–8.
Wang Y, Qian P-Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PloS one. 2009;4:e7401.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 2009;75:7537–41.
Quince C, Lanzén A, Curtis TP, Davenport RJ, Hall N, Head IM, et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nature methods. 2009;6:639–41.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (Oxford, England). 2011;27:2194–200.
Silva. http://www.arb-silva.de/. Accessed 1 May 2015.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic acids research. 2007;35:7188–96.
Nucleotide Blast. https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome. Accessed 1 May 2015.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403–10.
Eshar D, Weese JS. Molecular analysis of the microbiota in hard feces from healthy rabbits (Oryctolagus cuniculus) medicated with long term oral meloxicam. BMC veterinary research. 2014;10:62.
Chao A, Bunge J. Estimating the number of species in a stochastic abundance model. Biometrics. 2002;58:531–9.
Chao A, Shen T-J: Nonparametric estimation of Shannon’s index of diversity when there are unseen species in sample. Environmental and Ecological Statistics, 10:429–443
Mulder CPH, Bazeley-White E, Dimitrakopoulos PG, Hector A, Scherer-Lorenzen M, Schmid B. Species evenness and productivity in experimental plant communities. Oikos. 2004;107:50–63.
Dixon P. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science. 2003;14:927–30.
Gower J. Wiley StatsRef: Statistics Reference Online. Chichester: John Wiley & Sons, Ltd; 2014.
Excoffier L, Smouse PE, Quattro JM. Analysis of Molecular Variance Inferred From Metric Distances Among DNA Haplotypes: Application to Human Mitochondrial DNA Restriction Data. Genetics. 1992;131:479–91.
Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology. 2005;71:8228–35.
Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics (Oxford, England). 2010;26:715–21.
National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/. Accessed 1 May 2015.
Rodriguez C, Taminiau B, Van Broeck J, Avesani V, Delmée M, Daube G. Clostridium difficile in young farm animals and slaughter animals in Belgium. Anaerobe. 2012;18:621–5.
Bidet P, Barbut F, Lalande V, Burghoffer B, Petit JC. Development of a new PCR-ribotyping method for Clostridium difficile based on ribosomal RNA gene sequencing. FEMS microbiology letters. 1999;175:261–6.
Société Française de Microbiologie. http://www.sfm-microbiologie.org/. Accessed 1 May 2015.
Standards - CLSI :CLSI. http://clsi.org/standards/. Accessed 1 May 2015.
Acknowledgements
The authors thank Cate Chapman for the support in English editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests, including non-financial competing interests.
Authors’ contributions
CR, BT, HA, MD and GD have designed the study. CR, BB, AL, MG and AB have preformed sampling and sample processing. VA and JvB have contributed toward typing. CR and BT have performed data analysis. All authors have participated at writing. All authors have read and approved the final manuscript.
Cristina Rodriguez and Bernard Taminiau contributed equally to this work.
Additional files
Additional file 1:
Quality analysis of the metagenetic libraries created for the horse faecal samples analysed. (DOCX 35 kb)
Additional file 2:
Microbiota faecal composition at species level (cumulative mean relative abundance >4 %) of horses with and without diarrhoea. Samples 13, 14, 15, 17, 23, 25, 26, 27, 28 and 29: horses with diarrhoea. Samples 11, 12, 16, 18, 19, 20, 21, 22, 24 and 30: horses without diarrhoea. (DOCX 146 kb)
Additional file 3:
Principal coordinate analysis of the diarrhoeic and non-diarrhoeic horses. The 3 graphs show the sample distribution along the 3 main axes (PC1-PC2-PC3). (DOCX 83 kb)
Additional file 4:
Clinical history comparison between C. difficile colonised and non-colonised horses. aAntimicrobial treatment of two or more antibiotics. bIncluding single or combined antimicrobial therapy (DOCX 63 kb)
Additional file 5:
Detailed information on five C. difficile negative horses studied via high-throughput amplicon sequencing analysis and compared with C. difficile colonised horses. NSAIDs: nonsteroidal anti-inflammatory drugs. Pen: penicillin; Gen: gentamicin; SXT: trimethoprim/sulfamethoxazole (DOCX 46 kb)
Additional file 6:
Microbiota faecal composition at species level (cumulative mean relative abundance >4 %) of C. difficile culture-positive and -negative horses. Samples 01, 03, 04, 09 and 10: C. difficile positive horses detected by faeces culture. Samples 02, 05, 06, 07 and 08: C. difficile negative horses detected by faeces culture. (DOCX 161 kb)
Additional file 7:
Principal coordinate analysis of C. difficile positive and negative horses. The 3 graphs show the sample distribution along the 3 main axes (PC1-PC2-PC3). (DOCX 73 kb)
Additional file 8:
Bacterial genus whose relative abundance was statistically different between C. difficile positive and negative horses. Result of a White test (p value <0.05) pairwise comparison. (DOCX 95 kb)
Additional file 9:
Bacterial genus whose relative abundance was statistically different between Colic, diarrhoea and Others diagnosis groups. The graph illustrates the mean relative abundance for the selected genera. Groups were compared by ANOVA 2 analysis with post-hoc Tukey-kramer multiple comparison. The inside table shows the pairwise difference (p-value < 0.05) represented by different letters. (DOCX 159 kb)
Additional file 10:
Abundance of the dominant genera found in the enteritis and enterocolitis samples. The graph shows the relative abundance of the 2 dominant genera found in enteritis and enterocolitis samples as well as their mean relative abundance in the other diagnosis categories. (DOCX 132 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rodriguez, C., Taminiau, B., Brévers, B. et al. Faecal microbiota characterisation of horses using 16 rdna barcoded pyrosequencing, and carriage rate of clostridium difficile at hospital admission. BMC Microbiol 15, 181 (2015). https://doi.org/10.1186/s12866-015-0514-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-015-0514-5