
Abstract
Background
Pseudomonas fluorescens strain MFE01 secretes in abundance two Hcp proteins (haemolysin co-regulated proteins) Hcp1 and Hcp2, characteristic of a functional type 6 secretion system. Phenotypic studies have shown that MFE01 has antibacterial activity against a wide range of competitor bacteria, including rhizobacteria and clinically relevant bacteria. Mutagenesis of the hcp2 gene abolishes or reduces, depending on the target strain, MFE01 antibacterial activity. Hcp1, encoded by hcp1, may also be involved in bacterial competition. We therefore assessed the contribution of Hcp1 to competition of P. fluorescens MFE01 with other bacteria, by studying MFE01 mutants in various competitive conditions.
Results
Mutation of hcp1 had pleiotropic effects on the MFE01 phenotype. It affected mucoidy of the strain and its motility and was associated with the loss of flagella, which were restored by introduction of plasmid expressing hcp1. The hcp1 mutation had no effect on bacterial competition during incubation in solid medium. MFE01 was able to sequester another P. fluorescens strain, MFN1032, under swimming conditions. The hcp2 mutant but not the hcp1 mutant conserved this ability. In competition assays on swarming medium, MFE01 impaired MFN1032 swarming and displayed killing activity. The hcp2 mutant, but not the hcp1 mutant, was able to reduce MFN1032 swarming. The hcp1 and hcp2 mutations each abolished killing activity in these conditions.
Conclusion
Our findings implicate type 6 secretion of Hcp1 in mucoidy and motility of MFE01. Our study is the first to establish a link between a type 6 secretion system and flagellin and mucoidy. Hcp1 also appears to contribute to limiting the motility of prey cells to facilitate killing mediated by Hcp2. Inhibition of motility associated with an Hcp protein has never been described. With this work, we illustrate the importance and versatility of type 6 secretion systems in bacterial adaptation and fitness.
Similar content being viewed by others


Background
Environmental bacteria are in perpetual war against several competitors, and thus require weapons to conquer new territory. The type 6 secretion system (T6SS) of Gram-negative bacteria is an effector translocation apparatus resembling an inverted bacteriophage puncturing device [1-3]. It is involved in a broad variety of functions, including antibacterial activity [4,5] and bacterial communication [6]. For example, Proteus mirabilis uses the killing activity of T6SS for self-recognition: this T6SS seems to be activated when opposing P. mirabilis swarms meet. The result of its action is a visible boundary called the Dienes line [7,8].
The T6SS machinery comprises at least 13 proteins, the core components, and sometimes, additional proteins [9,10]. Some of the core component proteins can affect systems other than T6SS. Hcp proteins, extracellular components of this secretion machinery, are released into the medium, and therefore may serve as markers of a functional T6SS apparatus [11]. Silverman and colleagues demonstrated that P. aeruginosa Hcp are not only structural proteins but also play a crucial role as chaperone and receptor for T6SS effectors. Various Hcp proteins transport their own effectors and this effector selection by Hcp seems to be specific [12].
The diversity of T6SS regulation reflects the vast array of its functions. The P. aeruginosa H1-T6SS gene cluster exhibits posttranscriptional regulation involving two sensor kinases, RetS and LadS [13-15]. High concentrations of synthetic c-di-GMP have negative effects on sensor kinase RetS, leading to P. aeruginosa H1-T6SS being turned on [16]. It is likely that a similar regulatory pathway controls the expression of P. protegens Pf-5 and P. syringae pv. syringae T6SS gene clusters [17,18]. However, T6SS regulation involves systems that regulate other genes suggesting regulatory cross-talk between T6SS and other virulence factors.
Environmental bacteria are often surrounded by an extracellular matrix, forming a protective capsule called the glycocalyx [19]. This extracellular matrix is generally composed of bacterial exopolysaccharides (EPS). EPS are involved in a variety of functions, including microcolony formation, protection against bacteriophages and mucoid phenotypes. The mucoid phenotype of Pseudomonas sp. is believed to be a global adaptive stress response to adverse environmental conditions [20,21]. It is characterized by overproduction of EPS alginate leading to shiny, raised and opaque colonies. The mucoid phenotype is a major factor contributing to P. aeruginosa infection in cystic fibrosis (CF) patients [22,23] but is unstable in vitro [24]. Some P. aeruginosa CF isolates acquire a mucoid phenotype through mutation of the anti-sigma factor MucA, a negative regulatory factor that sequesters AlgU, a positive regulator of alginate production [22,25]. A proteomic study by Rao and coworkers found that mucoid P. aeruginosa strains do not express T6SS genes [26]. Unlike P. aeruginosa, some Vibrio cholerae strains are mucoid and have an active T6SS [27]. Some environmental P. fluorescens strains produce alginate or neutral and amino sugars which give a mucoid phenotype [28,29]. The P. fluorescens mucoid phenotype, like that in P. aeruginosa, was reported to be unstable [29]. The mucoid phenotype can occur following mutation of the negative regulator of the alginate biosynthetic operon, muc [30]. We previously characterized the strain MFE01, a mucoid environmental P. fluorescens isolate. It constitutively secretes two characteristic T6SS proteins at 28°C: Hcp1 and Hcp2. It also exerts antibacterial activity during contact on a solid surface with competitive bacteria; this activity is associated with Hcp2 [31]. The aim of this work was to study the role of Hcp1 and the link between T6SS and the mucoid phenotype of MFE01.
Results and discussion
Some mucoid P. fluorescens strains secrete Hcp abundantly
The phenotypes of four P. fluorescens strains were observed after growth on LB agar plates at 28°C. Environmental strains MFE01 [31] and MFE07 (this study) had a stable shiny aspect, characteristic of mucoid phenotypes at 28°C. Skin strain MFP05 [32] and clinical strain MFN1032 [33] did not have a mucoid phenotype at this temperature (Figure 1A and B).

Hcp secretion and mucoid phenotype in P. fluorescens strains. A: Mucoidy was assessed on LB agar at 28°C. +: mucoid, −: non mucoid. B: Mucoidy at 28°C or 37°C of MFE01. +: mucoid, −: non mucoid. The images shown are representative of three assays. C: Concentrated supernatants of cultures in late exponential growth phase, grown at 28°C or 37°C, were analysed by SDS-PAGE (15% separation gel) and Coomassie staining. Bands (indicated by arrows) with an approximate molecular mass of 21 kDa were observed in MFE01 and MFE07 supernatants at 28°C. Mass spectrometry identified these major supernatant proteins as Hcp proteins.
MucA is a negative regulator of the mucoid phenotype [22] in Pseudomonas species. We introduced the mucA gene of the Pseudomonas protegens strain Pf-5 (previously described as a P. fluorescens strain) [34] into MFE01. The expression of mucA did not switch-off mucoidy at 28°C, suggesting that the mucoid phenotype of MFE01 is not due to a mutation of the mucA gene or is MucA independent (Figure 1A). Scanlan and Buckling showed that the environmental P. fluorescens strain SBW25 has an unstable mucoid phenotype; although the genetic mechanisms of this phenotype remain unknown, they do not include mutations at many of the loci commonly reported to be involved in mucoid conversion, including mucA and algU. They also demonstrated that lytic phage exerts a selection pressure by selecting the mucoid phenotype [29].
We prepared supernatants of cultures of mucoid P. fluorescens strains MFE01 and MFE07 and found that these strains secreted large amounts of proteins identified as Hcp by Mass Spectroscopy (MS) (Figure 1C). The spot for the protein secreted by MFE01 matched significantly with gi: 398989489 (T6SS effector, Hcp1 family from Pseudomonas sp GM24) with a score of 120 for a score threshold of 84. The spot for the protein secreted by MFE07 spot matched significantly with gi:77458272 (hypothetical protein Pfl01_2045 from Pseudomonas fluorescens Pf0-1, Hcp1 family) with a score of 65 for a score threshold of 54. The non-mucoid P. fluorescens strains MFP05 and MFN1032 did not secrete detectable amounts of Hcp proteins into the extracellular medium.
The optimal growth temperature of MFE01 is 28°C but it can grow at 37°C. Clearly visible at 28°C, the mucoid phenotype and Hcp secretion were both switched off at 37°C, suggesting a common regulation of these two phenotypes (Figure 1B,C).
Unterweger and colleagues observed a correlation between the mucoid phenotype and Hcp secretion in V. cholerae smooth strains [27]. Sigma-54 controls T6SS genes transcription, so they introduced vasH, encoding a sigma-54 activator protein, into rough (non mucoid) V. cholera strains impaired in Hcp secretion: the expression of vasH restored the mucoid phenotype, but not the Hcp secretion. However, vasH can activate transcription of genes other than T6SS genes via sigma-54 [35,36] suggesting that the correlation between mucoidy and T6SS in V. cholerae may be due to a common regulation system.
Hcp1 of P. fluorescens MFE01 is involved in mucoid phenotype
We constructed hcp mutants, and found that the hcp1 mutation (MFE01∆hcp1 strain) lead to the loss of mucoidy at 28°C whereas the hcp2 mutation (MFE01∆hcp2 strain) did not affect the mucoid phenotype (Figure 2). The introduction of a plasmid carrying the hcp1 gene into MFE01∆hcp1 (MFE01∆hcp1+hcp1 strain) restored the wild-type phenotype, suggesting a direct link between hcp1 expression and mucoidy. We analysed and compared the extracellular matrices (ECM) of MFE01 and MFE01∆hcp1. The mucoid phenotype is a consequence of exopolysaccharide (EPS) accumulation [37], so we determined ECM sugar composition by Gas–liquid Chromatography (GLC). No significant difference between MFE01 and MFE01∆hcp1 was observed concerning the proportion of characteristic sugars of the Pseudomonas EPS (rhamnose, mannose, galactose and glucose; Figure 3A). Thus, MFE01∆hcp1 is able to secrete an EPS similar to that produced by MFE01. Nevertheless, the MFE01 ECM contained twice as much EPS as the MFE01∆hcp1 ECM suggesting that the loss of the mucoid phenotype was a consequence of less EPS accumulation (Figure 3B). In Pseudomonas aeruginosa, biofilm formation and T6SS expression are negatively regulated by the sensor RetS and retS mutants produce a hyperbiofilm phenotype [16]. Biofilm formation by MFE01 and mutants was then assayed. The moderate biofilm biovolume was not significantly different between MFE01 and MFE01∆hcp1 or MFE01∆hcp2 mutants (Figure 3C). These findings indicate that mucoid phenotype and biofilm formation are not co-regulated by T6SS in MFE01.

Effects of hcp1 and h cp2 mutations on mucoid phenotype. Representative images of mucoidy of MFE01, MFE01Δhcp1, MFE01Δhcp2, MFE01Δhcp1+pPSV35 and MFE01Δhcp1+hcp1 at 28°C on LB agar (n = 3). +: mucoid, −: non mucoid.

Analysis of the sugar composition of the extracellular matrix and effect of hcp1 and hcp2 mutations on biofilm biovolume. A: Relative quantity of major sugars (Rha = Rhamnose, Man = Mannose, Gal = Galactose and Glc = Glucose) determined by gas chromatography in extracellular matrix (ECM) of MFE01 and MFE01Δhcp1. Values are mean results of four independent experiments, and the error bars represent standard error of the mean. Strains were compared pairwise by non-parametric Mann–Whitney two-tailed tests. ns: no significant difference. B: Gas chromatographic determinations of the absolute quantity of exopolysaccharides (EPS) in the extracellular matrix (ECM) of MFE01 and MFE01Δhcp1. Values are mean results of four independent experiments and the error bars represent standard error of the mean. Strains were compared pairwise by non-parametric Mann–Whitney two-tailed tests. *p-value <0.05. C: Biovolume of biofilms. The biofilms were grown on glass surface at 28°C for 48 h under a flow of LB medium. Biovolumes were obtained by COMSTAT analyses. Values are mean results of at least five independent experiments and the error bars represent standard error of the mean. Statistical analyses were performed using Non-parametric Mann–Whitney Tests (two tailed). ns means no significant difference in biovolume (p-value >0.05) relative to the MFE01 biofilm biovolume assay.
ECM protein fractions from supernatants of MFE01, MFE01∆hcp1 and MFE01∆hcp1+hcp1 were studied by SDS-PAGE (Figure 4A). Proteins of approximate molecular mass of 38 kDa were present in ECM extracts from MFE01 and MFE01∆hcp1+hcp1 but not in MFE01∆hcp1. MS identified these 38 kDa proteins as flagellin proteins: they matched significantly with flagellin from Pseudomonas moraviensis (gi: 515142380) with a score of 94 for a score threshold of 86. Thus, the mucoid phenotype appears to involve the accumulation of both EPS and flagellin in ECM, which are perturbed by hcp1 deletion. This was unexpected because the literature generally reports an inverse cross-talk between the mucoid phenotype and flagellar assembly. For example, the alternative sigma factor, σ22 (synonym AlgT or AlgU), is a positive regulator of alginate biosynthesis and a negative regulator of flagellum biosynthesis [38].

Effect of hcp1 and hcp2 mutations on flagellin secretion into the extracellular matrix and on motility. A: Protein fractions from the ECM of MFE01, MFE01Δhcp1 and MFE01Δhcp1+hcp1 were analysed by SDS-PAGE and Coomassie staining. Bands with approximate molecular mass ≈ 38 kDa, indicated by the arrows, were observed at the growth temperature of 28°C and were identified as flagellin proteins by MALDI-TOF (n = 3). Molecular weight markers are indicated. B: Swimming assay on LB in 0.3% agar at 28°C. MFE01, MFE01Δhcp2 and MFE01Δhcp1+hcp1 were motile but MFE01Δhcp1 and MFE01Δhcp1+ pPSV35 (empty plasmid used as control) were non motile (n = 3).
P. fluorescens MFE01 Hcp1 is involved in motility
We examined motility of our various strains to elucidate the relation between flagellin accumulation in the ECM and flagellum functionality. MFE01, MFE01∆hcp1, MFE01∆hcp2, MFE01Δhcp1+pPSV35 (empty vector control) and MFE01∆hcp1+hcp1 were assayed for swimming; this confirmed that MFE01∆hcp1 had lost motility that was restored by hcp1 introduction in trans. The restoration of wild-type phenotype in MFE01∆hcp1+hcp1 is inconsistent with a possible polar effect of the hcp1 deletion (Figure 4B).
The type 3 secretion system is related to the flagellum [39], but little is known about the relation between T6SS and the flagellar regulon. The IcmF of Vibrio cholerae, a T6SS protein, is involved in motility [40]. IcmF is an inner-membrane protein of the T6SS found in numerous pathogens, and has been implicated in intracellular multiplication inside host cells [41,42]. In an avian pathogenic E. coli, icmF mutation impaired motility [43]; the authors indicate that this contrasts with other mutants in T6SS genes, notably clpV and hcp, which had no motility defects. Their findings suggest that the motility defect of the icmF mutant was not due to a general defect of motility because of a non-functional T6SS. IcmF was somehow involved in flagellar regulation. The restoration of the motility of MFE01∆hcp1 by hcp1 introduction in trans implies that the function of T6SS associated to hcp1 expression is directly involved in motility in this strain.
The hcp1 mutant of P. fluorescens MFE01 retains virulence toward competitor bacteria
The Hcp concentration was slightly lower in the supernatant of MFE01∆hcp1 than that of the wild-type MFE01 at 28°C (Figure 5A). This is consistent with our previous observations [31]: most of the Hcp secreted by MFE01 is produced from the expression of the hcp2 gene. We have also demonstrated that MFE01 has a killing activity against various Gram-negative bacteria on solid medium. MFE01Δhcp2 reduced prey cell populations, but significantly less than MFE01 indicating that Hcp2 contributes to the killing activity, and that another factor is also involved. To determine whether Hcp1 could be this other factor, we co-cultured mutant MFE01Δhcp1 and the prey MFN1032 on a filter on solid media for 4h at 28°C. There was no significant difference of the MFN1032 population between the co-culture with wild-type MFE01 and the co-culture with MFE01∆hcp1 (Figure 5B). Thus, Hcp1 was not the key factor in this antibacterial activity even if it contributed to killing. Whereas, the MFE01∆hcp2 mutant had significantly less antibacterial activity than MFE01 and MFE01∆hcp1. The double mutant, MFE01∆hcp1∆hcp2, was even less bactericidal than MFE01∆hcp2 but nevertheless reduced the MFN1032 population. Another system independent of Hcp1 and Hcp2 seems to be involved in killing activity.

Hcp secretion and killing activity of P. fluorescens MFE01 and derivatives strains. A: Concentrated supernatants of MFE01 and MFE01Δhcp1 cultures were analysed by SDS-PAGE and Coomassie staining. Bands with a molecular mass similar to that of an Hcp protein (≈20 kDa), indicated by the arrow, were observed at a growth temperature of 28°C. MWM: molecular weight markers are indicated. B: Quantitative co-culture assays were performed. Prey cells (MFN1032 carrying pSMC21-gfp) were or were not mixed at ratio of 1:5 with P. fluorescens MFE01, MFE01Δhcp1, MFE01Δhcp 1Δhcp2 and MFE01Δhcp2; after 4 h at 28°C, MFN1032 cfu were counted (n = 4, the error bars represent standard error of the mean). * Indicates a significant difference in MFN1032 cfu (p-value <0.05) relative to the MFN1032/MFE01 assay; ns means no significant difference. ♦ indicates a significant difference in MFN1032 cfu (p-value <0.05) relative to the MFN1032 alone control assay.
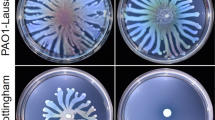
P. fluorescens MFE01 inhibits the motility of P. fluorescens strain MFN1032
The killing activity mediated by T6SS during swarming in P. mirabilis involves self-recognition (the Dienes effect), and we therefore studied the behaviour of MFE01 in conditions permitting its motility [7,8]. In swimming conditions (in 0.3% agar), P. fluorescens MFN1032 was spotted into the centre of a ring formed by MFE01 or its mutants (Figure 6A). MFE01, MFE01∆hcp1+hcp1 and MFE01∆hcp2 inhibited MFN1032 motility, but swimming MFN1032 spread through the MFE01∆hcp1 and, MFE01∆hcp1+pPSV35 mutants (Figure 6B). The hcp1 mutation thus abolished MFE01 confining MFN1032. Introduction of the native hcp1 gene in trans restored the inhibitory activity on MFN1032 swimming. Thus, in these swimming conditions, hcp1 expression seems required for P. fluorescens MFE01 to control the swimming of P. fluorescens MFN1032. In these conditions, MFE01∆hcp1 lacked motility whereas MFE01 was motile (Figure 4B). The confinement of MFN1032 could be simply due to the formation of a physical barrier by the motile MFE01 whereas non-motile MFE01∆hcp1 could not form this barrier.

Effects of P. fluorescens MFE01 and its mutants on competitive bacteria. A: Schematic drawing of the assay: P. fluorescens MFN1032 was inoculated into the centre of a ring formed by MFE01 or a mutant on LB medium in 0.3% agar. On this medium MFN1032 and ME01 can swim. B: Swimming of MFN1032 in competition with MFE01 or mutants. Results after incubation at 28°C, 24 h. The images shown are representative of three assays. C: Co-cultures were performed on swarming medium (LB in 0.6% agar) at 28°C, 24h. On this medium MFN1032 can swarm but ME01 cannot. Prey cells (MFN1032) were or were not mixed with P. fluorescens MFE01 or its derivatives at ratio 1:5. The images shown are representative of three assays. D: Quantitative black histograms represent survival of MFN1032 and white squares the MFN1032 swarm diameter (n = 4, error bars represent standard error of the mean). * Indicates a significant difference of the MFN1032 cfu (p-value <0.05) relative to the MFN1032/MFE01 assay. ns: no significant difference.
So we therefore conducted co-cultures overnight on 0.6% LB agar, conditions allowing the swarming of MFN1032. On these plates, MFE01 and mutants were unable to swarm because MFE01 lacks surfactants essential for P. fluorescens swarming [44]. MFE01 and MFE01∆hcp2 clearly inhibited MFN1032 swarming, whereas MFE01∆hcp1 and MFE01∆hcp1∆hcp2 had much less effect on MFN1032 swarming (Figure 6C,D). This indicates that the inhibition of motility observed was not due to MFE01 or its derivatives forming a physical barrier. Also, diameter of the MFN1032 swarming was reduced, albeit to a lesser extent, by the double mutant MFE01∆hcp1∆hcp2 providing further evidence for another unidentified inhibitory factor. These experiments are uninformative about whether the decrease in swarming diameter is due to a killing activity or an immobilization activity.
We consequently assayed the bactericidal activity of MFE01 and its mutants by counting MFN1032 cells in each swarming condition (Figure 6D). The MFN1032 population underwent 5-logs drops after co-culture with wild-type MFE01. Co-culture with MFE01∆hcp1 and MFE01∆hcp2 did not significantly decrease the MFN1032 population. We assumed that when co-cultured with MFE01∆hcp1, MFN1032 cells could swarm and thereby escape from the killing activity mediated by Hcp2, which requires close contact. When in co-culture with MFE01∆hcp2, MFN1032 could not be able to escape but the killing activity is abolished by hcp2 mutation. In antibacterial activity assays on solid media (Figure 5B), the MFN1032 cells were immobilized on a filter, and therefore unable to escape the killing activity of the hcp1 mutant.
These results suggest that MFE01 needs Hcp1 and Hcp2 to kill MFN1032 in conditions of motility: Hcp1 could reduce the motility of prey cells and thereby facilitate killing by Hcp2.
Phenotypes assigned to Hcp1 and Hcp2 are associated to a T6SS
Analysis of MFE01 draft genome is under way. Preliminary results indicated that this genome contains only one T6SS cluster exhibiting all core component genes but hcp genes. The two hcp genes, hcp1 and hcp2, are separately located outside this cluster and are associated with vgrG genes. Into this unique T6SS we identified a gene coding for a putative protein corresponding to a protein member of EvpB/VC_A108 family (TIGR03355). This putative protein matched with the WP_016772499.1 sequence (MULTISPECIES: type VI secretion protein [Pseudomonas], with 99% identity and coverage of 100%). These proteins are described as T6SS needle sheath proteins TssC and are essential components of this system. MFE01∆tssC, a mutant disrupted in this T6SS core component gene, was non motile in swimming conditions and non-mucoid at 28°C as well as MFE01∆hcp1 (Figure 7A and B) whereas MFE01∆hcp2 conserved the wild type phenotype (mucoid and motile). During co-culture on LBG containing X-gal at 28°C with E.coli containing pUC19 (E.colipUC19), MFE01∆hcp2 and MFE01∆tssC allowed E.colipUC19 growth (blue spots) whereas MFE01∆hcp1 and MFE01 inhibited E.colipUC19 growth (white spots) (Figure 7C). ME01∆tssC phenotype seems similar to a patchwork of MFE01∆hcp1 and MFE01∆hcp2 phenotypes. These results provide a strong confirmation of Hcp1 and Hcp2 involvement in a T6SS.

Phenotypes of the MFE01∆tssC mutant. A: Swimming assay on LB in 0.3% agar at 28°C during 24 h. MFE01 and MFE01Δhcp2 were motile contrary to MFE01Δhcp1 and MFE01ΔtssC strains (n = 3). B: Representative images of mucoidy of MFE01 and MFE01ΔtssC on LB agar at 28°C, 24 h (n = 3). +: mucoid, −: non mucoid. C: Effects of P. fluorescens MFE01 and its mutants on competitive E.coli on solid medium. Co-cultures were performed on LB medium supplemented with X-Gal (40 μg/ml) at 28°C during 24 h. Prey cells (E.coli DH5αmcr containing pUC19) were or were not mixed with P. fluorescens MFE01 or its derivatives at ratio 1:1. Blue is due to X-Gal degradation by E.colipUC19. The images shown are representative of three assays.
Conclusion
In this work, we further illustrate the importance and versatility of the T6SS in bacterial adaptation and fitness. We demonstrated cooperation between two different Hcp proteins in bacterial competition. The observations indicate that T6SS associated with Hcp1 secretion has pleiotropic effects on P. fluorescens MFE01: it affects mucoidy, motility and competitor inhibition in motility conditions. The relationships between T6SS and flagellin and between T6SS and mucoidy have not yet been fully elucidated. Nevertheless, we describe some of the mechanisms associated with MFE01 motility and inhibition of prey cell motility. This suggests that some effectors, that are used to inhibit competitor motility, could be accumulated in MFE01∆hcp1, in absence of Hcp1, and consequently might inhibit the mutant’s own motility. Analysis of MFE01 draft genome is under way. Numerous mutants should be constructed to analyse function of unknown putative genes surrounding these hcp genes to ensure an informative genomic annotation and to explain how this T6SS could impact mucoidy and motility.
Methods
Bacterial strains, plasmids and culture conditions
All strains and plasmids used are listed in Table 1. All bacterial strains were grown in LB medium with shaking (180 rpm). Pseudomonas fluorescens strains were grown at 28°C or 37°C and Escherichia coli at 37°C. Media were supplemented with antibiotics as appropriate: kanamycin (Km) 50 μg/ml (E. coli) or 200 μg/ml (P. fluorescens); tetracycline (Tc) 15 μg/ml; gentamicin (Gt) 15 μg/ml (E. coli), or 50 μg/ml or 100 μg/ml (P. fluorescens in liquid and solid media, respectively).
Hcp secretion analysis
Hcp secretion was studied by harvesting the supernatants by centrifuging the cultures at 5000 × g for 10 minutes at 20°C and passing them through a Millipore membrane with 0.22 μm pores. TCA (trichloroacetic acid, Sigma-Aldrich) was added to the supernatant to a final concentration of 10% and the mixture was incubated overnight at 4°C. The supernatant was removed by centrifugation at 13000 × g, for 30 minutes at 4°C. The protein pellet was washed twice with 5 ml of 20 mM Tris base (VWR) in cold acetone (Merck) and centrifuged at 13000 × g, for 30 minutes at 4°C. The dry pellet was then resuspended in distilled water. Proteins were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). Briefly, samples were mixed with an equal volume of 2 × Laemmli sample buffer (with β-mercaptoethanol), boiled for 5 min at 100°C and then cooled to room temperature before loading.
Mucoid phenotype and swimming assays
Strains were plated on LB agar with 0.1 mM IPTG and incubated for 24 h at 28°C; the colonies were then examined for the mucoid phenotype. Swimming assays were performed as previously described [45].
Swarming killing assay
The protocol described by Alteri and colleagues [8] was used with minor modifications. Aliquots of 5 μl of a mixture of strains at a ratio of 5:1, MFE01 or mutants: MFN1032 containing plasmid pSMC21-gfp [46] were spotted onto LB 0.6% agar with 0.1 mM IPTG. The plates were incubated overnight at 28°C, and the entire swarm was collected, and serially diluted. Aliquots of 10 μl of each dilution were plated on LB agar supplemented with 200 μg kanamycin and colony forming units counted.
Motility inhibition
Overnight cultures were centrifuged at 3000 × g, for 5 minutes at room temperature. The cell pellets were collected on toothpicks which was used to stab 0.3% agar LB plates, with 0.1 mM IPTG; the plates were then incubated at 28°C overnight.
Antibacterial competition assay
Antibacterial competition assays in solid media were performed as described by Decoin and coworkers [31]. To ensure pSMC21-gfp stability (presence or absence) in our conditions, MFN1032 containing pSMC21-gfp was cultivated in LB medium containing kanamycin until OD580 nm of 1. Serial dilutions were then plated on LB plates without Kanamycin during 24 h at 28°C. Colonies were scrapped and suspended in NaCl 9 g/L and adjusted to OD5580 nm of 1. Serial dilutions were plated on selective (with kanamycin) and non-selective (without kanamycin) plates and counting. Statistics were done by pairwise strain comparisons (non-parametric Mann–Whitney-two tailed Test): no significant difference was observed between the two conditions (p-value >0.05, n = 6).
For E.coli growth inhibition, Pseudomonas fluorescens MFE01 and mutants were cultivated at 28°C in LB medium with shaking at 180 rpm overnight. Escherichia coli DH5αmcr transformed with pUC19 (E.colipUC19), allowing blue detection on medium containing X-Gal, was cultivated at 37°C in LB with ampicillin (50 μg/ml) with shaking at 180 rpm overnight. The OD580 nm was adjusted to 0.5 and the strains were mixed at a 1:1 ratio. A volume of 20 μl was spotted in LB agar plates supplemented with X-Gal (40 μg/ml) and incubated overnight at 28°C.
Disruption of the hcp1 gene in P. fluorescens MFE01
MFE01∆hcp1 was generated by deletions in the 5′ (44 bp) and 3′ extremities (18 bp) of hcp1. 5′ and 3′ truncated hcp1 was generated by PCR with the Hcp3F and Hcp4R primers (see Table 2). The resulting truncated hcp1 construct was introduced between the EcoRI and HindIII sites (blunt-ended) of the transferable suicide plasmid pME3087 (6.9 Kb) [47]. The resulting plasmid, pME3087∆hcp1, was verified by sequencing and was then transferred into MFE01 by biparental mating: E. coli S17-1 [48] containing pME3087∆hcp1 and recipient MFE01 cells were mixed and spotted onto LB agar plates and incubated overnight at 37°C. The mating mixture was then suspended in 1 ml of sterile NaCl 9 g/L and 0.1 ml aliquots were spread on LB agar plates supplemented with tetracycline (12 μg/ml), to select for the presence of the integrated plasmid, and rifampicin (25 μg/ml), to kill the E. coli S17-1 donor bacteria and to ensure the selection of MFE01. The resulting hcp1 mutant (containing two truncated hcp1 genes) was named MFE01∆hcp1.
Disruption of the hcp2 gene in P. fluorescens MFE01∆hcp1 and disruption of the tssC gene in P. fluorescens MFE01
A markerless hcp2 mutation was introduced into MFE01∆hcp1 strain by the protocol described by Decoin and co-workers [31]. The resulting strain was named MFE01∆hcp1∆hcp2. The same protocol with PCR modifications was used to introduce a markerless tssC mutation into MFE01 strain. This tssC deletion was achieved by PCR with the muta1tssC and muta2tssC primers (<650 bp product) or the muta3tssC and muta4tssC primers (<650 bp product) (Table 2). The PCR products obtained corresponded to the upstream and downstream parts, respectively, of the tssC gene of MFE01, each carrying an overlapping sequence at the end. PCR parameters were as follows: annealing temperature, 62°C, extension time, 45 s; 35 cycles. A third PCR was then carried out in which the overlapping sequences of the two first products were hybridized together allowing the tssC deletion after PCR with the muta1tssC and muta4tssC primers. The PCR parameters were as follows: annealing temperature, 62°C; extension time, 1 m 45 s and 35 cycles. The mutant containing the tssC deletion was verified by DNA sequencing and named MFE01∆tssC.
Heterologous expression of mucA in MFE01
The mucA (PFL_1449) sequence was amplified with the PFL_1449mucAF and PFL_1449mucAR primers (Table 2) by standard PCR from genomic DNA from P. protegens Pf-5. The PCR conditions were as follows: an annealing temperature of 64°C, an extension time of 30 s and 25 cycles. The polymerase used was Phusion® High-Fidelity DNA polymerase (NEB). The PCR product was digested with SacI (NEB) and XbaI (NEB) and ligated into pPSV35 [49] digested with the same enzymes. E. coli was transformed with the ligation product and the resulting plasmid was checked by PCR. Fresh MFE01 colonies were washed twice in cold sterile distilled water and transformed with 5 μl of plasmid DNA by electroporation in 1 mm cuvette at 1.8 kV (Savant electroporater). LB was then added and the mix incubated for 1 h at 28°C with shaking (180 rpm). The samples were then plated on LB agar supplemented with gentamicin and with 0.1 mM IPTG.
Insertion of the hcp1 gene into pPSV35 and construction of strain MFE01∆hcp1+hcp1
The hcp1 gene was amplified from the P. fluorescens MFE01 genome with the Hcp1F and Hcp2R primers (Table 2). The PCR conditions were as follows: an annealing temperature of 59°C, an extension time of 30 s and 25 cycles. The polymerase used was the Phusion® High-Fidelity DNA polymerase (NEB). The amplified fragments were inserted into the pPSV35 shuttle vector at SmaI site by blunt-end ligation, and the resulting pPSV35-hcp1 was used to transform E. coli DH5αmcr cells by electroporation. Plasmid DNA was isolated using the QIAprep Spin Miniprep Kit (Qiagen) and checked by PCR and asymmetric digestion with BamHI (NEB) to verify the orientation of the insert. MFE01∆hcp1 was transformed with pPSV35-hcp1 by electroporation as described in the section “Heterologous expression of mucA in MFE01”. The resulting strain was called MFE01∆hcp1+hcp1.
Preparation and purification of extracellular matrix (ECM) containing exopolysaccharides (EPS)
Strains were tested for EPS production on LB agar at 28°C. ECM was extracted as described by Dignac and colleagues with modifications [50]. Biomass was harvested and suspended in NaCl 9 g/L then sonicated at 37 Watts for 30 sec on ice and centrifuged (20,000 × g for 30 min). Clear supernatants were collected and three volumes of ice-cold ethanol were added; the samples were incubated for 24 h at 4°C, and EPS were collected by centrifugation (7000 × g, 30 min). The EPS-containing pellets were resuspended in water and lyophilised.
Analysis of the monosaccharide composition of EPS by gas chromatography – flame ionization
Lyophilised EPS were hydrolysed by treatment with 2M trifluoroacetic acid for 2 hours at 110°C. Monosaccharides were then derivatised: methanol-1M HCl (Supelco) was added and the samples incubated at 80°C overnight; then a mix of hexamethyldisiloxan:trimethyldisiloxan:pyridine (3:1:9, Supelco) was added and the samples incubated at 110°C for 20 minutes. The resulting derivatives were dried then dissolved in 1 ml of cyclohexane and injected into the 3800 GC system equipped with a CP-Sil5-CB column (Agilent Technologies). Elution was performed with the following gradient of temperature: 120°C to 160°C at a rate of 10°C per minute, 160°C to 220°C at a rate of 1.5°C per minute, 220°C to 280°C at a rate of 20°C per minute. Quantification was based on the internal standard and response factor determined previously for each monosaccharide.
Biofilm formation
Pure culture biofilms were grown in continuous-culture three-channel flow cells (channel dimensions, 1 by 4 by 40 mm). The system was assembled and prepared as follows: system sterilization during 4 h with bleach at 1.2% and rinsing with sterile NaCl 9 g/L. Before flow chamber inoculation, LB medium was injected into the system at 28°C. Overnight cultures were centrifuged at 8 000 × g during 5 min at room temperature. Pellets were recovered and washed twice with 2 mL of sterilized physiological water. Channels were inoculated with 1 mL of pure bacterial suspension at DO580nm = 0.1. Bacteria were allowed to attach to the glass surface (microscope coverslip, VWR International, Fontenay sous Bois, France) during 2 h at 28°C under static conditions. Biofilm growth was then performed under a constant flow of LB medium with antibiotics at appropriate concentration if necessary (Wakson Marlow 205S, 2.5 mL/h) for 48 h at 28°C.
Confocal laser scanning microscopy (CLSM) and image analyses
Microscopic observations were performed with a LSM 710 system, Zeiss, Germany by using a 63x oil immersion objective. Biofilms were observed after a 15 min incubation with 5 μM Syto 9® green fluorescent nucleic acid stain (life technologies) (excitation and emission λ, 488 nm and 510 nm, respectively). Biofilm stacks were analysed with COMSTAT software. The calculated parameter was the biovolume, which is the volume of bacteria (in μm3) per μm2 of glass surface. The results were the mean of at least five independent experiments.
Mass spectrometry analysis
Mass spectroscopy (MS) analyses were performed with a MALDI-TOF AutoflexIII (Brucker) in positive ion mode as described by Barbey et al. [51]. Statistical analyses of the sequences involved determining the probability based on Mowse score with MASCOT software (peptide tolerance = 100 ppm and mass values = MH+). A p-value of less than 0.05 was considered significant. The criteria used to accept a protein identification based on peptide mass fingerprinting (PMF) data included a score probability greater than a score threshold defined by MASCOT software.
Statistical analysis
Non-parametric Mann–Whitney Tests (two tailed) with GraphPad Prism version 6.0 (La Jolla, CA) were used for statistical analyses. A p-value <0.05 was considered to be statistically significant.
References
Jani AJ, Cotter PA. Type VI secretion: not just for pathogenesis anymore. Cell Host Microbe. 2010;8(1):2–6.
Records AR. The type VI secretion system: a multipurpose delivery system with a phage-like machinery. Mol Plant Microbe Interact. 2011;24(7):751–7.
Basler M, Mekalanos JJ. Type 6 secretion dynamics within and between bacterial cells. Science. 2012;337(6096):815.
Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe. 2010;7(1):25–37.
Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J Bacteriol. 2011;193(21):6057–69.
Brunet YR, Espinosa L, Harchouni S, Mignot T, Cascales E. Imaging type VI secretion-mediated bacterial killing. Cell Rep. 2013;3(1):36–41.
Gibbs KA, Urbanowski ML, Greenberg EP. Genetic determinants of self identity and social recognition in bacteria. Science. 2008;321(5886):256–9.
Alteri CJ, Himpsl SD, Pickens SR, Lindner JR, Zora JS, Miller JE, et al. Multicellular bacteria deploy the type VI secretion system to preemptively strike neighboring cells. PLoS Pathog. 2013;9(9):e1003608.
Aschtgen MS, Gavioli M, Dessen A, Lloubes R, Cascales E. The SciZ protein anchors the enteroaggregative Escherichia coli Type VI secretion system to the cell wall. Mol Microbiol. 2010;75(4):886–99.
Aschtgen MS, Thomas MS, Cascales E. Anchoring the type VI secretion system to the peptidoglycan: TssL, TagL, TagP… what else? Virulence. 2010;1(6):535–40.
Pukatzki S, McAuley SB, Miyata ST. The type VI secretion system: translocation of effectors and effector-domains. Curr Opin Microbiol. 2009;12(1):11–7.
Silverman JM, Agnello DM, Zheng H, Andrews BT, Li M, Catalano CE, et al. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol Cell. 2013;51(5):584–93.
Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7(5):745–54.
Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312(5779):1526–30.
Ventre I, Goodman AL, Vallet-Gely I, Vasseur P, Soscia C, Molin S, et al. Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc Natl Acad Sci U S A. 2006;103(1):171–6.
Moscoso JA, Mikkelsen H, Heeb S, Williams P, Filloux A. The Pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ Microbiol. 2011;13(12):3128–38.
Hassan KA, Johnson A, Shaffer BT, Ren Q, Kidarsa TA, Elbourne LD, et al. Inactivation of the GacA response regulator in Pseudomonas fluorescens Pf-5 has far-reaching transcriptomic consequences. Environ Microbiol. 2010;12(4):899–915.
Records AR, Gross DC. Sensor kinases RetS and LadS regulate Pseudomonas syringae type VI secretion and virulence factors. J Bacteriol. 2010;192(14):3584–96.
Jacques M, Marrie TJ, Costerton JW. Review: Microbial colonization of prosthetic devices. Microb Ecol. 1987;13(3):173–91.
Terry JM, Pina SE, Mattingly SJ. Role of energy metabolism in conversion of nonmucoid Pseudomonas aeruginosa to the mucoid phenotype. Infect Immun. 1992;60(4):1329–35.
Wood LF, Leech AJ, Ohman DE. Cell wall-inhibitory antibiotics activate the alginate biosynthesis operon in Pseudomonas aeruginosa: Roles of sigma (AlgT) and the AlgW and Prc proteases. Mol Microbiol. 2006;62(2):412–26.
Ramsey DM, Baynham PJ, Wozniak DJ. Binding of Pseudomonas aeruginosa AlgZ to sites upstream of the algZ promoter leads to repression of transcription. J Bacteriol. 2005;187(13):4430–43.
Hoiby N, Ciofu O, Bjarnsholt T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010;5(11):1663–74.
Pugashetti BK, Metzger Jr HM, Vadas L, Feingold DS. Phenotypic differences among clinically isolated mucoid Pseudomonas aeruginosa strains. J Clin Microbiol. 1982;16(4):686–91.
Wiens JR, Vasil AI, Schurr MJ, Vasil ML. Iron-regulated expression of alginate production, mucoid phenotype, and biofilm formation by Pseudomonas aeruginosa. MBio. 2014;5(1):e01010–3.
Rao J, Damron FH, Basler M, Digiandomenico A, Sherman NE, Fox JW, et al. Comparisons of two proteomic analyses of non-mucoid and mucoid Pseudomonas aeruginosa clinical isolates from a cystic fibrosis patient. Front Microbiol. 2011;2:162.
Unterweger D, Kitaoka M, Miyata ST, Bachmann V, Brooks TM, Moloney J, et al. Constitutive type VI secretion system expression gives Vibrio cholerae intra- and interspecific competitive advantages. PLoS One. 2012;7(10):e48320.
Fett WF, Wells JM, Cescutti P, Wijey C. Identification of exopolysaccharides produced by fluorescent pseudomonads associated with commercial mushroom (Agaricus bisporus) production. Appl Environ Microbiol. 1995;61(2):513–7.
Scanlan PD, Buckling A. Co-evolution with lytic phage selects for the mucoid phenotype of Pseudomonas fluorescens SBW25. Isme J. 2012;6(6):1148–58.
Muhammadi AN. Genetics of bacterial alginate: alginate genes distribution, organization and biosynthesis in bacteria. Curr Genomics. 2007;8(3):191–202.
Decoin V, Barbey C, Bergeau D, Latour X, Feuilloley MG, Orange N, et al. A type VI secretion system is involved in Pseudomonas fluorescens bacterial competition. PLoS One. 2014;9(2):e89411.
Dagorn A, Chapalain A, Mijouin L, Hillion M, Duclairoir-Poc C, Chevalier S, et al. Effect of GABA, a bacterial metabolite, on Pseudomonas fluorescens surface properties and cytotoxicity. Int J Mol Sci. 2013;14(6):12186–204.
Chapalain A, Rossignol G, Lesouhaitier O, Merieau A, Gruffaz C, Guerillon J, et al. Comparative study of 7 fluorescent pseudomonad clinical isolates. Can J Microbiol. 2008;54(1):19–27.
Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, Mavrodi DV, et al. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol. 2005;23(7):873–8.
Kitaoka M, Miyata ST, Brooks TM, Unterweger D, Pukatzki S. VasH is a transcriptional regulator of the type VI secretion system functional in endemic and pandemic Vibrio cholerae. J Bacteriol. 2011;193(23):6471–82.
Miyata ST, Kitaoka M, Brooks TM, McAuley SB, Pukatzki S. Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect Immun. 2011;79(7):2941–9.
Pulcrano G, Iula DV, Raia V, Rossano F, Catania MR. Different mutations in mucA gene of Pseudomonas aeruginosa mucoid strains in cystic fibrosis patients and their effect on algU gene expression. New Microbiol. 2012;35(3):295–305.
Tart AH, Wolfgang MC, Wozniak DJ. The alternative sigma factor AlgT represses Pseudomonas aeruginosa flagellum biosynthesis by inhibiting expression of fleQ. J Bacteriol. 2005;187(23):7955–62.
Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4(11):811–25.
Das S, Chakrabortty A, Banerjee R, Chaudhuri K. Involvement of in vivo induced icmF gene of Vibrio cholerae in motility, adherence to epithelial cells, and conjugation frequency. Biochem Biophys Res Commun. 2002;295(4):922–8.
Hilbi H, Segal G, Shuman HA. Icm/Dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol Microbiol. 2001;42(3):603–17.
Watarai M, Derre I, Kirby J, Growney JD, Dietrich WF, Isberg RR. Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse Lgn1 locus. J Exp Med. 2001;194(8):1081–96.
de Pace F, Boldrin De Paiva J, Nakazato G, Lancellotti M, Sircili MP, Guedes Stehling E, et al. Characterization of IcmF of the type VI secretion system in an avian pathogenic Escherichia coli (APEC) strain. Microbiology. 2011;157(10):2954–62.
Rossignol G, Sperandio D, Guerillon J, Duclairoir Poc C, Soum-Soutera E, Orange N, et al. Phenotypic variation in the Pseudomonas fluorescens clinical strain MFN1032. Res Microbiol. 2009;160(5):337–44.
Rossignol G, Merieau A, Guerillon J, Veron W, Lesouhaitier O, Feuilloley MG, et al. Involvement of a phospholipase C in the hemolytic activity of a clinical strain of Pseudomonas fluorescens. BMC Microbiol. 2008;8:189.
Davey ME, Caiazza NC, O’Toole GA. Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J Bacteriol. 2003;185(3):1027–36.
Schnider U, Keel C, Voisard C, Defago G, Haas D. Tn5-directed cloning of pqq genes from Pseudomonas fluorescens CHA0: mutational inactivation of the genes results in overproduction of the antibiotic pyoluteorin. Appl Environ Microbiol. 1995;61(11):3856–64.
Simon R, Pehle A. A broad host range mobilization system for in vitro genetic engineering: transposon mutagenesis in Gram-negative bacteria. Biotechology. 1983;1:784–90.
Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2005;102(22):8006–11.
Dignac MF, Urbain V, Rybacki D, Bruchet A, Snidaro D, Scribe P. Chemical description of extracellular polymers: implication on activates sludge floc structure. Wat sci tech. 1998;8–9:45–53.
Barbey C, Crepin A, Cirou A, Budin-Verneuil A, Orange N, Feuilloley M, et al. Catabolic pathway of gamma-caprolactone in the biocontrol agent Rhodococcus erythropolis. J Proteome Res. 2012;11(1):206–16.
Acknowledgements
This study was supported by a GRR CBS Sesa grant from the Région Haute-Normandie and FEDER funds. We are grateful to Josette Guérillon and Gaëlle Rossignol, who initiated T6SS research in the LMSM lab. We also thank Alex Edelman and Awa N’Diaye Renoult for linguistic support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors declare that they have no competing interest.
Authors’ contributions
VD carried out the assays with the help of MG and CB and participated in the design of the experiments and writing of the manuscript. AM designed the study, wrote the manuscript and analysed most of the data. CDP and FL were involved in ECM analysis. NO and MF were involved in scientific discussions during the design of the study. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Decoin, V., Gallique, M., Barbey, C. et al. A Pseudomonas fluorescens type 6 secretion system is related to mucoidy, motility and bacterial competition. BMC Microbiol 15, 72 (2015). https://doi.org/10.1186/s12866-015-0405-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-015-0405-9