
Abstract
Background
Growing evidence has shown that gut microbiome composition is associated with breast cancer (BC), but the causality remains unknown. We aimed to investigate the link between BC prognosis and the gut microbiome at various oestrogen receptor (ER) statuses.
Methods
We performed a genome-wide association study (GWAS) to analyse the gut microbiome of BC patients, the dataset for which was collected by the Breast Cancer Association Consortium (BCAC). The analysis was executed mainly via inverse variance weighting (IVW); the Mendelian randomization (MR) results were verified by heterogeneity tests, sensitivity analysis, and pleiotropy analysis.
Results
Our findings identified nine causal relationships between the gut microbiome and total BC cases, with ten and nine causal relationships between the gut microbiome and ER-negative (ER-) and ER-positive (ER+) BC, respectively. The family Ruminococcaceae and genus Parabacteroides were most apparent among the three categories. Moreover, the genus Desulfovibrio was expressed in ER- BC and total BC, whereas the genera Sellimonas, Adlercreutzia and Rikenellaceae appeared in the relationship between ER + BC and total BC.
Conclusion
Our MR inquiry confirmed that the gut microbiota is causally related to BC. This further explains the link between specific bacteria for prognosis of BC at different ER statuses. Considering that potential weak instrument bias impacts the findings and that the results are limited to European females due to data constraints, further validation is crucial.
Similar content being viewed by others
Introduction
On a global scale, breast cancer (BC) is the most prevalent type of cancer affecting women [1]. Of concerning, accumulated data indicate that in 2020, there were 2.26 million new cases of BC [2]. Indeed, issues such as predisposition towards BC (family history), early start of the menstrual period, giving birth or occurrence of menopause at an advanced age, abnormal body weight, and exogenous hormone administration from oral contraceptives have a severe impact on the mortality rate of BC [3, 4]. Therefore, the high incidence and mortality rate of BC necessitates development of predictors for identifying BC patients. Moreover, it will assist doctors by improving precision medicine for BC.
It has gradually been accepted that BC is a complex disease with various aetiologies, development of which involves distinct entities with molecular and phenotypical backgrounds and different clinical results. Different types of BC have been categorized based on markers, such as oestrogen receptor (ER), basal or luminal expression profiles, and amplification of the human epidermal growth factor receptor 2 (HER2) gene. However, the prevalence of these cancers has not been elucidated profoundly [5].
In general, basal-like, HER2-positive, luminal-A-like, and luminal-B-like BC are the four intrinsic subtypes [6]. Their assessment is performed by immunohistochemical evaluation of expression of ER, progoesterone receptor (PR), HER2, and Ki-67 antigen [7]. In BC, ER plays a vital role, as 70% of detected cases show elevated ER expression [8]. Moreover, ER’s uniqueness differentiates it from other BC subtypes in terms of prognosis and biological characteristics, demonstrating sensitivity to antioestrogen therapy. The prognosis for BC patients with ER + status (defined as ER levels greater than or equal to 1%) is related to more favourable outcomes compared to those with ER- status (defined as ER levels less than 1%). ER has been regarded as an indicator with extreme importance in the prognosis of BC, as suggested by the guidelines of the two major networks, namely, the National Comprehensive Cancer Network (NCCN) and the European Society for Medical Oncology (ESMO) [9, 10]. Recently, there has been a growing interest in exploring the link between BC risk and the composition of the gut microbiota [11, 12]. However, the published information from relevant studies remains inadequate. Nevertheless, we did find evidence concerning a causal link between intestinal flora and other tumours [13, 14]. However, the same link has not been explored profoundly for BC. For example, in a study by Yang and colleagues, the authors discovered increased proliferation when colorectal cancer (CRC) cells were infected with Fusobacterium nucleatum. They observed an increase in the development of tumours in xenograft mouse models and CRC cell invasive activity [15].
Furthermore, Susan et al. found a direct interaction network between the intestinal microbiome and host immune cells that can promote cancer cell growth, also affecting cancer occurrence, development, and incidence [16]. Research has been conducted on the interaction between human microorganisms, especially the intestinal microbiome, and cancer [17]. Moreover, some results from epidemiological studies have shed light on the relationship between the human microbiome and health [18]. For example, Plaza-Diaz et al. conducted a retrospective study on the relationship between intestinal microbiome dysbacteriosis in the breast and the risk of BC [19].
In this context, Mendelian randomization (MR) is a popular technique for determining whether exposure and complicated results are causally related without any potentially detrimental intervention. By using genome-wide association study (GWAS) summary statistics, the MR technique was applied for exploration of a causal relationship between ER + and ER-BC risk and intestinal microbiota composition in this study.
Materials and methods
Research design
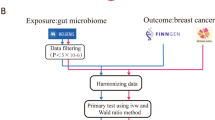
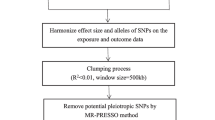
The causal relationship between gut microbial genera and the different statuses of ER BC was explored using a two-sample MR method. MR incorporates summary data from GWASs, minimizing the influence of confounding variables. Figure 1 illustrates the outline of the research design. Three critical assumptions were made to contend with the decrease in the impact of bias on the obtained results when using the MR technique. Initially, the intestinal microbiome should correlate strongly with instrumental variables (IVs), and the same IVs cannot be related to any potential confounders in later analyses. Last, the IVs should influence the outcome independently of exposure factors and other pathways [20].

Schematic description of MR analysis in the discovery phase. Assumption 1: genetic variants are robustly associated with exposure. Assumption 2: genetic variants are not associated with confounders. Assumption 3: genetic variants affect outcomes only through the exposure of interest. LD, linkage disequilibrium
Database sources and tools
Human gut microbiota
Single-nucleotide polymorphisms (SNPs) related to the human gut microbiome composition were selected as IVs from the MiBioGen consortium (https://mibiogen.gcc.rug.nl/). The MiBioGen consortium contains 16 S RNA gene sequencing profiles and genotyping data from 18,340 samples, among which the correlation between a patient’s gut microbiota and genetic variation was studied [21]. The MiBioGen group collected data from 25 cohorts in 11 nations of European ancestry, including the exposure dataset, which had 122,110 SNPs of 211 taxa (from genus to phylum level). The selection criteria of IVs included the following: (1) association threshold setting of P < 5 × 10− 5 [22]; (2) performing a window of linkage disequilibrium (LD) for all IVs (r2 < 0.001, distance = 10 MB); (3) exclusion of SNPs that were less than three; and (4) an instrumental variable with mean F-statistics higher than 10 (F = R2 × (N − 2)/(1 − R2), where R2 represents the proportion of the variability of the exposure explained by each IV, and N means the sample size [23]).
Breast cancer
The outcome sources were the different ER statuses of BC, including total BC, ER + BC, and ER- BC. The Breast Cancer Association Consortium (BCAC) provided the BC data used [24], which comprised 10,680,257 SNPs and 228,951 samples (N = 122,977 cases and 105,974 controls). Of these, 127,442 samples (N = 21,468 cases; 105,974 controls) and 10,680,257 SNPs were related to ER- BC survival. On the other hand, 175,475 samples (N = 69,501 cases; 105,974 controls) and 10,680,257 SNPs were linked to ER + BC survival. The remaining three MR assumptions were satisfied by eligible IVs with a significance level of P < 5 × 10− 5, the specifics of which are detailed elsewhere. All information was retrieved from consortia providing publicly available summary statistics of European ancestry. Each employed GWAS received ethical approval from the relevant institutions.
Statistical analysis
Inverse-variance weighted (IVW) is a method that combines multiple site effects of all SNPs when using MR analysis in a meta-analysis model. The premise of applying IVW is that all SNPs are valid IVs and completely independent of each other [25]. The IVW approach was primarily employed for varied ER statuses to calculate the causal link between gut microbiota and BC. We also performed the IVW method to examine the heterogeneity of each SNP (P<0.05) separately. The OR and 95% confidence interval (CI) represented the effect size. The leave-one-out strategy was applied in sensitivity analysis to validate the reliability of the data. Utilization of the MR‒Egger, MR pleiotropy residual sum and outlier (MR-PRESSO) tests (R package “MRPRESSO”) and intercept analysis in pleiotropy analysis confirmed the accuracy of the results obtained for the causal link of the gut microbiota to BC. At the same time, the Bonferroni approach was used to corrected P-values, in addition we provide the corrected p-values in Tables S1, S2 and S3 using the Benjamini-Hochberg (FDR) method. All statistical analyses were implemented using the “TwoSampleMR” package in R Version 4.2.2.
Results
All raw results were listed in Table S1, S2 and S3 the manuscript, the overview of the relationship between gut microbiome and BC, ER- BC, and ER + BC were shown in Tables S1, S2 and S3, respectively.
Causal effects of the gut microbiome on BC
The results provided in Table 1 from IVW analysis showed that genus Parabacteroides ID 954 (odds ratio (OR) = 0.87, 95% CI, 0.79–0.96, P = 0.007), genus Adlercreutzia ID 812 (OR = 0.92, 95% CI, 0.87–0.98, P = 0.014), genus Ruminococcaceae UCG-013 ID 11,370 (OR = 0.92, 95% CI, 0.86–0.99, P = 0.027), genus Lactobacillales ID 1800 (OR = 0.94, 95% CI, 0.88-1.00, P = 0.041), genus Desulfovibrio ID 3173 (OR = 0.94, 95% CI, 0.88-1.00, P = 0.043), genus Ruminiclostridium 6 ID 11,356 (OR = 0.94, 95% CI, 0.89-1.00, P = 0.046) and family Rikenellaceae ID 967 (OR = 0.92, 95% CI, 0.85-1.00, P = 0.047) acted as preventative measures for total BC. Alternatively, the genera Ruminococcaceae ID 2050 (OR = 1.11, 95% CI, 1.00-1.22, P = 0.035) and Sellimonas ID 14,369 (OR = 1.05, 95% CI, 1.01–1.09, P = 0.013) appeared to be linked to a greater risk of total BC (Table 1).
Gut microbiome causal impacts on ER- BC
Table 1 provides evidence suggesting that the genus Desulfovibrio ID 3173 (OR = 0.84, 95% CI, 0.75–0.93, P = 0.001, IVW method), genus Eubacterium Xylanophilum Group ID 14,375 (OR = 0.82, 95% CI, 0.71–0.95, P = 0.007), genus Lachnospiraceae NK4A136 Group ID 11,319 (OR = 0.88, 95% CI, 0.79–0.97, P = 0.010), genus Dorea ID 1997 (OR = 0.84, 95% CI, 0.73–0.97, P = 0.014), genus Clostridium sensustricto1 ID 1873 (OR = 0.86, 95% CI, 0.75–0.99, P = 0.033), genus Parabacteroides ID 954 (OR = 0.82, 95% CI, 0.68–0.98, P = 0.034), genus Olsenella ID 822 (OR = 0.93, 95% CI, 0.87–0.99, P = 0.034) and genus Candidatus Soleaferrea ID 11,350 (OR = 0.91, 95% CI, 0.83-1.00, P = 0.040) reduced the risk of ER- BC. On the other hand, the family Ruminococcaceae ID 2050 (OR = 1.15, 95% CI, 1.00-1.33, P = 0.049) and unknown genus ID 826 (OR = 1.13, 95% CI, 1.01–1.26, P = 0.027) presented a risk for the development of ER- BC (Table 2).
Causal effects of the gut microbiome on ER + BC
Using the IVW method, we discovered that the genus Adlercreutzia ID 812 (OR = 0.88, 95% CI, 0.81-0.95, P = 0.001), genus Parabacteroides ID 954 (OR = 0.87, 95% CI, 0.77–0.98, P = 0.024), Marvinbryantia ID 2005 (OR = 0.92, 95% CI, 0.85–0.99, P = 0.039) and family Streptococcaceae ID 1850 (OR = 0.92, 95% CI, 0.84-1.00, P = 0.042), as well as family Rikenellaceae ID 967 (OR = 0.91, 95% CI, 0.83–0.99, P = 0.029), were linked to a decreased incidence of ER + BC (Table 3). Conversely, the genus Sellimonas ID 14,369 (OR = 1.08, 95% CI, 1.03–1.13, P = 0.001), genus Ruminococcus Gauvreauii Group ID 11,342 (OR = 1.11, 95% CI, 1.01–1.20, P = 0.022), family Ruminococcaceae ID 2050 (OR = 1.15, 95% CI, 1.04–1.26, P = 0.005), and order Bacillales ID 1674 (OR = 1.05, 95% CI, 1.00-1.10, P = 0.042) were linked to a high risk of ER + BC (Table 3). We did not find significant correlations between the other taxa, 211 in number, and BC. Of note, when applying the Bonferroni method to adjust for multiple comparisons across various classification levels, none of the results in this study achieved a significance level that survived the Bonferroni-corrected threshold. Moreover, considering that Bonferroni correction may lead to false-negatives, the results of this study were not subjected to correction.
Sensitivity analysis
The accuracy of the results between the gut microbiome and different statuses of ER of BC were evaluated by sensitivity analysis. No significant heterogeneity was identified (Tables 1, 2 and 3). Among the bacteria, the MR‒Egger and MR-PRESSO intercept tests showed no evidence of pleiotropy (P > 0.05) (Tables 1, 2 and 3). The F value was greater than 20, indicating no weak IV bias (Table 1, 2 and 3). Moreover, the MR estimation results predicted by leave-one-out analysis were not driven by specific SNPs.
Discussion
In this two-sample MR study, we identified nine causal relationships between the gut microbiome and total BC, nine between the gut microbiome and ER + BC, and ten between the gut microbiome and ER- BC. We also discovered that the family Ruminococcaceae and genus Parabacteroides appeared in three categories, while the genus Desulfovibrio was proven to participate in the link between total BC and ER- BC. Concerning the association between ER + BC and total BC, the genera Sellimonas and Adlercreutzia and family Rikenellaceae exhibited significant implications. These findings provide insight into the clinical and experimental investigation of bacterial targets.
BC
The analysis showcased causal relationships between the genus Parabacteroides, genus Sellimonas, genus Adlercreutzia, genus Ruminococcaceae UCG-013 within this family, genus Desulfovibrio, genus Ruminiclostridium 6, order Lactobacillales, family Rikenellaceae and total BC. Of note, when we set the threshold for statistical significance at a p value of 5 × 10− 8, no causal relationship between gut microbiota and the three types of BC was identified. Subsequently, we applied Bonferroni correction to account for multiple comparisons at different taxonomic ranks, with varying adjusted p values (adjusted p value < 3.1 × 10–3 for phylum, adjusted p value < 5.6 × 10–3 for class, adjusted p value < 2.5 × 10–3 for order, adjusted p value < 1.4 × 10–3 for family, and adjusted p value < 3.8 × 10–4 for genus), considering the number of bacterial traits within each specific gut microbiota rank. However, after applying this correction, none of the results in our study reached the adjusted p value threshold (Table S4). As a result, we did not perform correction on our findings. It is worth noting that application of Bonferroni correction may lead to false-negatives. This can be attributed to the complex interplay typically observed in the gut-cancer axis, which is often influenced by multiple factors. Furthermore, the individual contribution of a single microbiota genus in causing disease may not hold as significant a role as previously hypothesized.
There is limited research on the relationship between Adlercreutzia and BC. A previous study showed that stromal tissue of the breast had a high percentage of fat and a low percentage of fibrosis in malignant versus benign breast disease. At the same time, Adlercreutzia (Actinobacteria) exhibited a positive association with fibrosis percentage at the order level [26]. Adlercreutzia was also found to have an increased abundance after all dietary treatment groups in which those nutritional compounds showed an inhibiting effect on tumour growth [27]. Both studies agree with our findings.
Surprisingly, Lactobacillales was mainly studied in animal experiments in which the research focused on BC. A laboratory trial showed that giving milk fermented by Lactobacillus casei CRL431 (belonging to order Lactobacillales) maintained improved anticancer response, decreased tumour development, and reduced lung metastases and tumour vascularity in mice [28]. In another study, the authors gave the same application to mice and had similar results [29], thus supporting our results.
Using 16 S rRNA sequencing to compare the gut microbiome between individuals with nonpuerperal mastitis and healthy individuals, the outcomes demonstrated a positive correlation between the family Ruminococcaceae in breast tissue and differential expression of immune-related genes [30]. However, Flores et al. examined the level of nonovarian systemic oestrogens, which might contribute to higher BC risk in postmenopausal women. They found a strong association with the faecal Ruminococcaceae family [31]. Another finding suggested that Desulfovibrio might be used as a diagnostic marker because premenopausal BC patients have a decreased abundance of short-chain fatty acid-producing bacteria compared to healthy premenopausal women [32]. The research suggests that the family Ruminococcaceae and genus Desulfovibrio separately play a protective role in nonpuerperal mastitis patients and postmenopausal BC patients, which was consistent with our overall study findings. Contrary to our results, the family Ruminococcaceae presented a risk for postmenopausal women. Its function might be related to women with BC at different times.
However, no relevant study has investigated the relationship between Parabacteroides, Sellimonas, Ruminiclostridium 6, Rikenellaceae, and total BC.
ER- BC
We further found evidence of causal relationships between the genus Desulfovibrio, genus Eubacterium Xylanophilum Group, genus Lachnospiraceae NK4A136 Group, genus Dorea, unknown genus, genus Clostridium sensustricto1, genus Parabacteroides, genus Olsenella, genus Candidatus Soleaferrea, and family Ruminococcaceae with ER-BC.
Recently, authors have reported the categorization of patients with HER2 + BC who received trastuzumab into nonresponsive (NR) and complete response (R) groups. Compared to R patients, NR patients had lower α-diversity and lower abundance of Lachnospiraceae. Additionally, transfer of faecal microbiota into HER2 + BC mice from NR and R recapitulated the response to trastuzumab observed in patients [33]. An interesting study also showed that BC survivors had a higher risk of cancer recurrence with a lower relative abundance of Lachnospiraceae [34]. The corresponding research showed that Lachnospiraceae can serve as a protective factor against BC.
A clinical trial on BC survivors sequencing faecal microbes demonstrated an abundance of Dorea, which was negatively associated with physical functioning, vitality, and mental health subscales. Alternatively, BC survivors without obesity had a significantly higher relative abundance of the genus Ruminococcus (belonging to the family Ruminococcacea) [35]. The study indicated that Dorea and Ruminococcus were linked to BC risk, though our results did not indicate this for the former. However, Dorea and uncultured Ruminococcus, which were considered to be signature bacteria to distinguish neoadjuvant chemotherapy (NAC) effectual group patients from the NAC noneffectual group, were increased in the NAC effectual group, according to analysis of the relationship between the gut microbiome and BC patients’ responses to NAC efficacy [36]. This diversity suggests that more in-depth investigation is warranted. It also suggests that the intestinal flora may be a target for therapy in different BC subtypes. Interestingly, an unknown genus, 826, acted as a precursor to the development of ER-BC, though more research on its biology is needed. We did not find evidence for a genetic correlation between other members of the gut microbiome and ER- BC.
ER + BC
Our findings suggested that the genera Adlercreutzia, Sellimona, Ruminococcus gauvreauii group, Parabacteroides, and Marvinbryantia; the families Ruminococcacea, Rikenellaceae, and Streptococcaceae; and the order Bacillales had a causal relationship with ER + BC.
Notably, in a clinical study on postmenopausal women with ER + BC and 153 faecal samples, a significantly lower richness of Marvinbryantia, Ruminococcaceae NK4A214, and Ruminococcaceae UCG-005 was found after treatment [37]. The results for Ruminococcaceae agree with ours, yet those for Marvinbryantia do not.
Unfortunately, there is a great lack of data exploring the relationship between other intestinal bacterial genera and ER + BC. The order Bacillales exhibited a positive association with fibrosis percentage in para-carcinoma tissue [26]. Analysis of the microbial composition of the breast revealed that Streptococcaceae and Bacillaceae (belonging to Bacillales) were stepwise enhanced in healthy to prediagnostic and postdiagnostic (including adjacent normal and tumour) tissues [38]. The findings suggest a potential signal in early BC diagnosis.
Most potential targets in BC
As appeared in the three classifications, the related intestinal flora, Ruminococcaceae and Parabacteroides, might have a vital role in clinical therapy. Many findings verify that the gut microbiome greatly impacts patients who receive immunotherapy [39]. Additionally, several published papers indicate a differential abundance of Ruminococcaceae in patients who respond to anti-PD-1 immunotherapy [40, 41], as well as of Parabacteroides distasonis [42]. Desulfovibrio, which showed a causal relationship with total BC and ER-BC, was investigated experimentally in colon cancer [43] and acute gastrointestinal injury [44]. Sellimonas, Adlercreutzia, and Rikenellaceae were causally related to total BC and ER + BC. However, due to inadequate evidence, including clinical trials and mechanical experiments on BC, further investigation is essential.
Sex hormone dysregulation is acknowledged as one of the main risk factors for development of BC. Female individuals with ER + tumours are considered to benefit from endocrine therapy [45], and female sex hormone levels influence microbiota composition, though the suggestion is bidirectional [46, 47]. A collection of bacteria within the gastrointestinal tract, referred to as the oestrobolome [31], was proven to be involved in production of beta-glucuronidase enzymes and to affect modulation of oestrogen metabolism, circulation, and excretion [48]. Genes encoding ß-glucuronidase are present in several species and bacterial genera in the human gut, including Edwardsiella, Bacteroides, Bifidobacterium, Collinsella, Alistipes, Clostridium, Citrobacter, and Marvinbryantia [49]. The last genus acts as a potential target for ER + BC. Further exploration is needed.
Comparison with other studies
Wei et al. published an MR analysis based on five common cancers, in which they highlight that an increased abundance of Sellimonas predict a higher risk of ER + BC. This finding is consistent with ours. Another MR study conducted by Long et al. investigated the causal relationship between eight cancers and indicated that Actinobacteria and Bifidobacterium are risk factors for BC. Both bacterial taxa show significant importance, though they were not included in our findings. Overall, the underlying mechanism of the intestinal microbiome warrants future investigation. In this study, taxa of the gut microbiome were analysed ranging from the genus to phylum level, establishing a conceptual basis for experimentally exploring specific bacterial strains. Furthermore, the results of our study validated the cause-and-effect connection between the gut microbiome and BC and innovatively put forth a viable and achievable therapeutic approach.
Novel applications
In general, the prognosis of ER-BC is poorer than that of ER + BC. Females diagnosed with ER- are more likely to undergo chemotherapy [50]. Chemotherapies, which mainly involve paclitaxel (PTX), continue to be the most popular and economical treatment, despite the two main drawbacks: toxicity and no specificity in target. Interestingly, the Chinese medicine Ganoderma lucidum, when combined with PTX, enhances tumour suppression by restoring the gut microbiota [51]. As some traditional Chinese medicines have been found to regulate the gut microbiota in treating CRC [52], they might provide a novel therapeutic approach for BC.
Strengths and limitations
This study has some strengths. First, a two-sample MR study was carried out to test the potential causal relationship between the gut microbiota and BC. The genetic variants obtained from a large-scale GWAS led to eliminating the confounding bias caused by the inverse causal problem and supported creditable evidence. Furthermore, gut microbiome taxa were examined from the genus to phylum level, providing a theoretical foundation for experimental investigation of particular bacterial strains. Additionally, the study confirmed the causal relationship of the gut microbiome with BC and further creatively proposes a viable and feasible therapeutic method.
However, there are some significant limitations. First, similar to other MR studies, the effect of weak instrument bias on our findings could not be excluded. Second, the results apply solely to European females because the summary data from the GWAS were subjected to geographical constraints, and more validation experiments to confirm the results presented in this paper are needed. Third, stratified analysis on PR and HER2 was not conducted due to the lack of secondary data. Moreover, p < 5 × 10-8 is an accepted cut-off in GWASs; when the GWAS p value was set at 5 × 10-8 during the screening process, three types of gut microbiota were identified for each BC subtype. However, the p values were all greater than 0.05, indicating no significant causal relationship. Therefore, we set the GWAS p value threshold as 5 × 10-5 in the present study. None of the results in this study achieved a significance level that survived the Bonferroni-corrected threshold, and considering that Bonferroni correction may lead to false-negatives, the results of this study were not subjected to correction. Finally, the gut microbiota might be impacted by environmental or genetic factors that lead to the lower variance explained by genetic instruments.
Conclusion
In summary, our MR study confirmed that gut microbiota has a causal relationship with BC. It also allowed for explanation of the link between specific bacteria and prognosis for BC with different ER statuses. In addition, some bacteria are regarded as potential targets for clinical treatment. However, further investigations are required to elucidate the underlying mechanism by which the gut microbiota profoundly impacts BC.
Data Availability
Statement.
The data used in this study are publicly available.
GWAS data.
https://www.nature.com/articles/s41588-020-00763-1.
MiBioGen consortium.
BC.
https://gwas.mrcieu.ac.uk/datasets/ieu-a-1126/.
ER- BC.
https://gwas.mrcieu.ac.uk/datasets/ieu-a-1128/.
ER + BC.
References
Katsura C, et al. Breast cancer: presentation, investigation and management. Br J Hosp Med (Lond). 2022;83(2):1–7.
Wilkinson L, Gathani T. Understanding breast cancer as a global health concern. Br J Radiol. 2022;95(1130):20211033.
Mollica V, Rizzo A, Massari F. Re: Toni K. Choueiri, Daniel Y.C. Heng, Jae Lyun Lee, Efficacy of Savolitinib vs Sunitinib in Patients With MET-Driven Papillary Renal Cell Carcinoma: The SAVOIR Phase 3 Randomized Clinical Trial. JAMA Oncol. In presshttps://doi.org/10.1001/jamaoncol.2020.2218 SAVOIR: From Own Goal to Winning Goal? Eur Urol Oncol, 2020. 3(4): p. 561-562
Tamimi RM, et al. Population attributable risk of modifiable and nonmodifiable breast Cancer risk factors in postmenopausal breast Cancer. Am J Epidemiol. 2016;184(12):884–93.
Barcellos-Hoff MH. Does microenvironment contribute to the etiology of estrogen receptor-negative breast cancer? Clin Cancer Res. 2013;19(3):541–8.
Kunc M, Biernat W, Senkus-Konefka E. Estrogen receptor-negative progesterone receptor-positive breast cancer - nobody’s land or just an artifact? Cancer Treat Rev. 2018;67:78–87.
Barzaman K, et al. Breast cancer: Biology, biomarkers, and treatments. Int Immunopharmacol. 2020;84:106535.
Howlader N et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst, 2014. 106(5).
Cardoso F, et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol. 2020;31(12):1623–49.
Gradishar WJ, et al. Breast Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(4):452–78.
Wu AH, et al. Gut microbiome associations with breast cancer risk factors and tumor characteristics: a pilot study. Breast Cancer Res Treat. 2020;182(2):451–63.
Tzeng A, et al. Human breast microbiome correlates with prognostic features and immunological signatures in breast cancer. Genome Med. 2021;13(1):60.
Chambers LM, et al. Disruption of the gut microbiota confers Cisplatin Resistance in Epithelial Ovarian Cancer. Cancer Res. 2022;82(24):4654–69.
Li Q, et al. Gut microbiota: its potential roles in pancreatic Cancer. Front Cell Infect Microbiol. 2020;10:572492.
Yang Y, et al. Fusobacterium nucleatum increases proliferation of Colorectal Cancer cells and Tumor Development in mice by activating toll-like receptor 4 signaling to Nuclear Factor-kappaB, and Up-regulating expression of MicroRNA-21. Gastroenterology. 2017;152(4):851–866e24.
Erdman SE, Poutahidis T. Gut microbiota modulate host immune cells in cancer development and growth. Free Radic Biol Med. 2017;105:28–34.
Elinav E, et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13(11):759–71.
Valdes AM, et al. Role of the gut microbiota in nutrition and health. BMJ. 2018;361:k2179.
Plaza-Diaz J, et al. Association of breast and gut microbiota dysbiosis and the risk of breast cancer: a case-control clinical study. BMC Cancer. 2019;19(1):495.
Davies NM, Holmes MV, Davey Smith G. Reading mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601.
Kurilshikov A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65.
Luo Q, et al. Effects of Gut Microbiota and Metabolites on Heart failure and its risk factors: a two-sample mendelian randomization study. Front Nutr. 2022;9:899746.
Zhang Y, et al. Causal associations between gut microbiome and cardiovascular disease: a mendelian randomization study. Front Cardiovasc Med. 2022;9:971376.
Guo Q et al. Identification of novel genetic markers of breast cancer survival. J Natl Cancer Inst, 2015. 107(5).
Yuan S, Larsson SC. Coffee and caffeine consumption and risk of kidney Stones: a mendelian randomization study. Am J Kidney Dis. 2022;79(1):9–14e1.
Hieken TJ, et al. The breast tissue microbiome, stroma, immune cells and breast cancer. Neoplasia. 2022;27:100786.
Gagnon E, et al. Impact of the gut microbiota and associated metabolites on cardiometabolic traits, chronic diseases and human longevity: a mendelian randomization study. J Transl Med. 2023;21(1):60.
Aragon F, et al. Inhibition of growth and metastasis of breast Cancer in mice by milk fermented with Lactobacillus casei CRL 431. J Immunother. 2015;38(5):185–96.
Mendez Utz VE, et al. Milk fermented by Lactobacillus casei CRL431 administered as an immune adjuvant in models of breast cancer and metastasis under chemotherapy. Appl Microbiol Biotechnol. 2021;105(1):327–40.
Zhu J, et al. Interactions between the breast tissue microbiota and host gene regulation in nonpuerperal mastitis. Microbes Infect. 2022;24(3):104904.
Flores R, et al. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med. 2012;10:253.
He C, et al. Changes of intestinal microflora of breast cancer in premenopausal women. Eur J Clin Microbiol Infect Dis. 2021;40(3):503–13.
Di Modica M, et al. Gut Microbiota Condition the therapeutic efficacy of Trastuzumab in HER2-Positive breast Cancer. Cancer Res. 2021;81(8):2195–206.
Okubo R, et al. Impact of chemotherapy on the association between fear of cancer recurrence and the gut microbiota in breast cancer survivors. Brain Behav Immun. 2020;85:186–91.
Smith KS, et al. Health-related quality of life is associated with fecal microbial composition in breast cancer survivors. Support Care Cancer. 2022;31(1):10.
Li Y, et al. Metagenomic analyses reveal distinct gut microbiota signature for Predicting the Neoadjuvant Chemotherapy responsiveness in breast Cancer patients. Front Oncol. 2022;12:865121.
Aarnoutse R, et al. Changes in intestinal microbiota in postmenopausal oestrogen receptor-positive breast cancer patients treated with (neo)adjuvant chemotherapy. NPJ Breast Cancer. 2022;8(1):89.
Hoskinson C, et al. Composition and functional potential of the human mammary Microbiota Prior to and following breast tumor diagnosis. mSystems. 2022;7(3):e0148921.
Li W, et al. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019;447:41–7.
Hakozaki T, et al. The gut Microbiome Associates with Immune Checkpoint Inhibition Outcomes in patients with Advanced Non-Small Cell Lung Cancer. Cancer Immunol Res. 2020;8(10):1243–50.
Zheng Y, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):193.
Huang J, et al. Ginseng polysaccharides alter the gut microbiota and kynurenine/tryptophan ratio, potentiating the antitumour effect of antiprogrammed cell death 1/programmed cell S4death ligand 1 (anti-PD-1/PD-L1) immunotherapy. Gut. 2022;71(4):734–45.
Kapral M, et al. Quantitative evaluation of transcriptional activation of NF-kappaB p65 and p50 subunits and IkappaBalpha encoding genes in colon cancer cells by Desulfovibrio desulfuricans endotoxin. Folia Microbiol (Praha). 2010;55(6):657–61.
Han C, et al. Intestinal microbiota and antibiotic-associated acute gastrointestinal injury in sepsis mice. Aging. 2021;13(7):10099–111.
Lippman ME, et al. The relation between estrogen receptors and response rate to cytotoxic chemotherapy in metastatic breast cancer. N Engl J Med. 1978;298(22):1223–8.
Vieira AT, et al. Influence of oral and gut microbiota in the Health of Menopausal Women. Front Microbiol. 2017;8:1884.
Parida S, Sharma D. The Microbiome-Estrogen Connection and Breast Cancer Risk Cells, 2019. 8(12).
Fuhrman BJ, et al. Associations of the fecal microbiome with urinary estrogens and estrogen metabolites in postmenopausal women. J Clin Endocrinol Metab. 2014;99(12):4632–40.
Kwa M et al. The intestinal microbiome and estrogen receptor-positive female breast Cancer. J Natl Cancer Inst, 2016. 108(8).
Tan AR, Swain SM. Adjuvant chemotherapy for breast cancer: an update. Semin Oncol. 2001;28(4):359–76.
Su J, et al. Anti-breast Cancer enhancement of a Polysaccharide from Spore of Ganoderma lucidum with Paclitaxel: suppression on Tumor Metabolism with Gut Microbiota Reshaping. Front Microbiol. 2018;9:3099.
Zhao H, et al. Colorectal Cancer, gut microbiota and traditional chinese medicine: a systematic review. Am J Chin Med. 2021;49(4):805–28.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
WH, DQ and BP participated in study design. WH, GH, and DW had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. WH and GH prepared the tables and Fig. 1. YX, JQ and XM performed the literature search. WH, GH and DW analyzed the data and wrote the paper. DQ, BP and XM jointly supervised the study. All authors read and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors of this study affirm that there were no financial or commercial ties that may be viewed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hong, W., Huang, G., Wang, D. et al. Gut microbiome causal impacts on the prognosis of breast cancer: a Mendelian randomization study. BMC Genomics 24, 497 (2023). https://doi.org/10.1186/s12864-023-09608-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09608-7