
Abstract
Background
The dried stem of Cistanche, is a famous Chinese traditional medicine. The main active pharmacodynamic components are phenylethanol glycosides (PhGs). Cistanche tubulosa produces higher level of PhGs in its stems than that of Cistanche deserticola. However, the key genes in the PhGs biosynthesis pathway is not clear in C. tubulosa.
Results
In this study, we performed the full-length transcriptome sequencing and gene expression profiling of C. tubulosa using PacBio combined with BGISEQ-500 RNA-seq technology. Totally, 237,772 unique transcripts were obtained, ranging from 199 bp to 31,857 bp. Among the unique transcripts, 188,135 (79.12%) transcripts were annotated. Interestingly, 1080 transcripts were annotated as 22 enzymes related to PhGs biosynthesis. We measured the content of echinacoside, acteoside and total PhGs at two development stages, and found that the content of PhGs was 46.74% of dry matter in young fleshy stem (YS1) and then decreased to 31.22% at the harvest stage (HS2). To compare with YS1, 13,631 genes were up-regulated, and 15,521 genes were down regulated in HS2. Many differentially expressed genes (DEGs) were identified to be involved in phenylpropanoid biosynthesis pathway, phenylalanine metabolism pathway, and tyrosine metabolism pathway.
Conclusions
This is the first report of transcriptome study of C. tubulosa which provided the foundation for understanding of PhGs biosynthesis. Based on these results, we proposed a potential model for PhGs biosynthesis in C. tubulosa.
Similar content being viewed by others

Introduction
Cistanche tubulosa (Schenk) R. Wight, a valuable Chinese herb medicine, is a perennial parasitic plant specifically parasitic on the roots of Tamarix ramosissima. Besides growing in the desert area, T. ramosissima is also widely distributed in the saline-alkali area in eastern China, which provides the opportunity for planting C. tubulosa in saline-alkali land. In recent years, C. tubulosa has been successfully introduced into the saline areas of the Yellow River Delta in China [1]. Growing C. tubulosa either in the desert area or in saline-alkali land could bring great values both economically and ecologically.
Phenylethanol glycosides (PhGs), oligosaccharides, and iridoids are the main active pharmacodynamic components in C. tubulosa. PhGs, in particular, draw the most attention of the researchers because of their promising pharmacological characteristics. Studies have shown that PhGs has outstanding biological activities such as kidney tonifying, bloodand essence invigorating, intestines moistening and defecation, anti-aging, learning ability improvement, nerve and liver protection [2,3,4,5,6]. C. tubulosa accumulates more PhGs than C. deserticola, in particular echinacoside and verbascoside, which is also the more suitable species for cultivation due to its high yield [7, 8]. The biosynthesis of PhGs is highly regulated by both environment stimuli and development progress. The processing condition after harvest also significantly affect the content of PhGs. The difference in PhGs contents of different samples are significant, even for the same species. Previous studies indicated that the total contents of seven PhGs components of C. tubulosa from south of Xinjiang were approximately six times those of Kuitun and Hami in China [9]. The accumulation of PhGs varies in different part of the fresh succulent stem or different growth stages of C. tubulosa, and the young stem accumulate more PhGs than that of the old stem [10,11,12]. The content of active components in different parts of C. tubulosa was also different in different development stages. The content of PhGs in the base part of C. tubulosa fleshy stem was the highest, and in the top part was the lowest. The echinoside content of the base part was 2 ~ 8 times as higher as the top part [13, 14].
Three major components of PhGs are organic acid, saccharide, and phenylethanol aglycon [17]. Referring to the studies of Rehmannia glutinosa and C. deserticola, PhGs biosynthetic pathway is related to phenylpropanoid biosynthesis pathway (PS, Ko00940), phenylalanine metabolism pathway (PM, Ko00360), and tyrosine metabolism pathway (TM, Ko00350) [17]. However, the regulation of PhGs biosynthetic pathway and key enzymes in C. tubulosa is unclear. To uncover the biosynthesis and regulation of PhGs, to stabilize and improve the content of PhGs are all important research areas in Cistanche. However, little molecular information is available in C. deserticola except few studies on chloroplast genome sequencing and transcriptome analysis [15,16,17]. There is even less genomic and molecular information available in C. tubulosa. The limited genomic and molecular information of this species hinders the understanding of the molecular mechanisms of PhGs biosynthesis and regulation. The third-generation single-molecule real-time sequencing platform (SMRT) can capture the full-length transcripts and avoid the assembly process [18]. SMRT sequencing has been successfully used in many plant species for high quality full-length transcripts identification and transcriptome analysis [19,20,21,22]. Full-length transcriptome analysis is a powerful way for revealing the dynamics of gene expression, and understanding the molecular mechanism for complex biological processes.
In this study, the PacBio SMRT Sequel platform was employed to generate a full-length transcriptome of C. tubulosa. In addition, RNA-seq technology was used to investigate the gene expression dynamics of C. tubulosa in different developmental stages. Our study generated a high-quality transcriptome reference sequence of C. tubulosa, and identified the key metabolic enzyme genes for PhGs biosynthesis. These results lay the foundation for understanding the molecular mechanism for PhGs biosynthesis.
Materials and methods
Plant materials and RNA sample preparation
The fleshy stems and flower organs of C. tubulosa at different developmental stages were collected from an experimental field of Shandong Academy of Agricultural Sciences in Weifang City, China. Four materials were collected for full-length sequencing with PacBio, including young succulent stem, succulent stem at harvest stage, succulent stem at flowering stage, and the inflorescence part above ground. After cleaning, the tissues were cut into small pieces and immediately frozen in liquid nitrogen, and stored at -80 °C until further processing. The total RNA samples were isolated using RNeasy Plus Mini Kit (Qiagen) according to the protocol of manufacturer (http://www.qiagen.com). The quantity and quality of total RNA was assessed using Agilent 2100 Bioanalyzer and Fragment Analyzer Automated CE System (http://www.agilent.com). For each sample, three biological replications were prepared.
Library preparation and high-throughput sequencing
The PacBio UMI Iso-Seq sequencing library was constructed using equally mixed RNAs from different tissues. First-strand cDNA was synthesized using UMI base PCR cDNA Synthesis Kit (BGI). After synthesis of first strand, PCR amplification was performed to generate double-strand cDNA. Then, multiple transcripts are connected end to end to generate multifold flux full-length transcriptome Sequel PacBio IsoSeq library. The problem of different size fragment preference was solved by Sequel platform. The library was subsequently sequenced using a PacBio Sequel system.
PacBio Iso-Seq data processing and bioinformatics analysis
After sequenced by PacBio sequel, large number of Circular Consensus Sequencing (CCS) reads were obtained. Reads of insert (ROI) was identified and classified into full-length (FL) and non-full-length (non-FL) reads. Then the high-quality full-length consistent sequence were obtained and evaluated [23]. The high-quality full-length sequences of two libraries were combined for clustering to de-redundancy and isoform expression quantification. The TransDecoder (https://transdecoder.github.io) was first used for recognizing the longest Open Reading Frames (ORFs), then the ORFs were further blasted with SwissProt (http://ftp.ebi.ac.uk/pub/databases/swissprot) and Pfam (http://pfam.xfam.org). Five other public databases were used to annotate the transcripts, including Nt and Nr of NCBI (http://www.ncbi.nlm.nih.gov/), GO (http://geneontology.org), KOG (http://www.ncbi.nlm.nih.gov/KOG) and KEGG ([24, 25]. HMMseach software (http://hmmer.org) was used for searching the plant transcription factors database (http://plntfdb.bio.uni- potsdam.de/v3.0/). CPC, txCdsPredict, and CNCI softwares and Pfam database were used to predict the coding and non-coding sequences.
Bioinformatics Analysis of RNA-Seq Data
For RNA-seq, the young fleshy stem (YS1) and the harvest stage stem (HS2) samples were used for gene expression analysis. Libraries were constructed using the method described in previous studies [26]. Briefly, the mRNA was first enriched and cleaved into short fragments, and then used to synthesize cDNAs. The cDNA fragments were purified and enriched through PCR amplification to construct cDNA library. Sequencing was conducted using BGISEQ-500 platform (BGI, China). The clean reads were obtained by removing the reads with poly N > 10%, reads containing adaptor sequences, and low-quality reads through SOAPnuke (version 1.5.2) [27]. Then, all clean reads were mapped with the full-transcripts of C. tubulosa using Bowtie2 (version 2.2.5) software [28]. The gene expression level was calculated using RSEM, and normalized using FPKM (Fragments Per Kilobase of transcript per Million fragments mapped) method [29]. The relative gene expression level between different samples was calculated using R package DEGseq [30]. Differentially expressed genes (DEGs) were identified following the criteria of fold change > = 2 and Q-value < = 0.05. To further understand the function of the DEGs, GO analysis was performed and enriched GO terms were identified using Blast2GO using hypergeometric test comparing with the whole transcript background [26, 31]. In addition, enriched KEGG pathways were identified by comparing the ratio of DEGs with the whole transcript background.
Verification of RNA-seq data by qRT-PCR
qRT-PCR was used to verify the expressionlevels of 10 selected genes. RNA samples were those used for high-throughput sequencing and the reverse transcription was performed using PrimeScript II 1st Strand cDNA Synthesis Kit (TaKaRa). The gene-specific primers were designed using PerlPrimer software and were listed in Additional Table S7. qRT-PCR reaction was performed on ABI7500 Real Time System (Applied Biosystems) using TB Green™ Premix Ex Taq™ II (TaKaRa). The parameters of thermal cycle were 94℃ for 10 min, followed by 30 cycles of 94℃ for 15 s and 60c for 1 min in a 20 µl volume. Three biological replications were performed for each reaction with actin gene as internal reference. The relative expression level of each gene between YS1 and HS2 was calculated by 2−△△Ct method.
Preparation and quantification of PhGs
The freshly harvested C. tubulosa was washed and sliced into thin sections, then steamed at 100℃ temperature for 1 min, dried in the oven, and then ground into powder. To extract PhGs, 50 mL of 50% methanol was added into 1.0 g of C. tubulosa powder, and then ultrasonic treatment was conducted for 30 min (500 W, 40 kHz). Add 50% methanol to supplement the weight loss, shake well, stand, and collect the supernatant. The supernatant was filtered through 0.22 μm microporous membrane to obtain the phenylethanol glycosides extracts.
The contents of echinacoside and acteoside were determined by HPLC using a column of Phenomenex Luna 5 μm C18(A) column (4.6 × 250 mm, 5 μm). The isocratic mobile phase consisted of mobile phase A (methanol/acetonitrile, 1:6, v/v), and mobile phase B (0.1% formic acid). The gradient program is as followed: 90% ~ 82% B at 0 ~ 8 min; 82% ~ 76% B at 8 ~ 15 min; 76% ~ 74% B at 15 ~ 20 min; 74% ~ 70% B at 20 ~ 28 min; 70% ~ 65% B at 28 ~ 35 min. The total elution time was 35 min, with the flow rate of 1 ml/min. Column temperature was kept constant at 30 °C. Ultraviolet detection wavelength was 330 nm.
The contents of total PhGs were quantitated by ultraviolet spectrophotometry. Weigh 10 mg of echinosiden and put it in a 50 ml volumetric flask and dissolved in 50% methanol to obtain the standard solution. 0.2 mL, 0.4 ml, 0.6 ml, 0.8 ml, and 1.0 ml of standard solution were accurately aliquot to a 10 ml volumetric flask, diluted with 50% methanol to a final volume of 10 ml, and shaken well. The absorbance was measured at 330 nm to make the standard curve. The absorbance of the sample was measured at 330 nm, and the content of total PhGs was calculated according to the standard curve.
Results
Full-length transcriptome sequencing and de novo assembly
Two PacBio IsoSeq libraries (F01, H01) were constructed by combining equal amount of total RNAs from four types of tissues including young succulent stem, succulent stem in harvest stage, succulent stem at flowering stage and the inflorescence above ground. PacBio sequencing generated a total of 21.87 Gb dataset including 4,321,117 sub-reads. A total of 254,813 Circular Consensus Sequencing (CCSs) reads were generated with the mean read length of 5,348 bp and 6,485 bp in the two libraries. SMRT analysis identified a total of 871,064 reads of full length non-chimeric transcripts (FLs) from CCSs. After removing the redundant sequences, a total of 237,772 unique isoforms (transcripts) were obtained. The transcript lengths were ranged from 199 bp to 31,857 bp, with a N50 of 1,797 bp. The GC content is 41.90% (Fig. 1a; Supplementary Table S1). BUSCO assessment showed that the transcripts are with high quality (Supplementary Figure S1).

Characteristics of unique transcript isoforms of C. tubulosa. a Length distribution of the transcripts, b GO annotation of the transcripts, c Species mapped with the transcripts
To further test the integrity of the protein coding transcripts, the TransDecoder software was used for identifying the candidate coding regions in isoforms, through further blasted with the SwissProt and Pfam Database. In total, 127,241 transcripts (53.51%) showed significant homology to known proteins and contained the complete coding sequence (CDS). The PacBio sequencing provided large number of full-length transcripts of C. tubulosa, an important genomic resource for herb Cistanche research community.
Functional annotation of the transcripts
To acquire the comprehensive annotation for these transcripts, the sequences were used for aligning with seven functional databases. The results showed that 174,391 (73.34%), 142,528 (59.94%), 122,202 (51.39%), 132,164 (55.58%), 130,770 (55.00%), 105,386 (44.32%), and 125,067 (52.60%) transcripts could match the Nr, Nt, SwissProt, KEGG, KOG, Pfam and GO databases, respectively (Supplementary Table S1). In total, there were 188,135 (79.12%) transcripts were annotated by more than one public databases. There were 29,622 transcripts were identified actively expressed with FPKM more than 0.5.
GO analysis grouped these genes into three categories: biological process, cell composition and molecular function. In terms of molecular function, catalytic activity and binding were the most abundant classes; in terms of cell, cell part, membrane and membrane part and organelle were the main classes; in terms of biological process, cellular process, and metabolic process were the most abundant classes (Fig. 1b). Nr annotation showed that 64,315 (36.88%), 35,698 (20.47%), and 17,491 (10.03%) transcripts matched the species of Sesamum indicum, Handroanthus impetiginosus, and Erythranthe guttata, respectively. The other 5,947 (3.41%) transcripts were matched with Chenopodium quinoa and the rest of 50,940 (29.21%) transcripts had similarity with other plant species (Fig. 1c).
We found that the coding mRNA and the non-coding lncRNA accounted for 7,587(32.63%), and 128,218 (53.92%), respectively.(Fig. 2; Supplementary Table S2). In this study, a total of 3,862 (1.62%) transcripts encoding 57 types of TFs were identified through blasting with PlnTFDB database (Supplementary Table S3). Among these TFs, MYB, C3H, bHLH, AP2-EREBP, WRKY, NAC, and mTERF accounted for 50% of the total TFs, representing the most abundant TF families. The annotation of these transcripts provides a framework for future gene identification and gene transcriptionregulation in C. tubulosa.

Venn diagram showed the prediction results of coding ability. Different colors represent different prediction methods
Determination of PhGs content in succulent stem
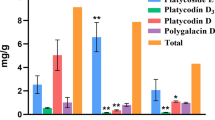
PhGs, for example, acteoside, echinoside, salidroside, and isoacteoside are known to be the primary active molecules in Cistanche. We measured the content of echinacoside, acteoside, and total PhGs in YS1 and HS2 (Fig. 3). The results showed that PhGs content was 46.74% of dry matter in YS1, while it was 31.22% in HS2. The content of echinoside and acteoside was 28.82% and 9.17% in YS1, while 17.26% and 7.33% in HS2, respectively.

C. tubulosa samples and phenylethanol glycosides content of stem at different developmental stages. a YS1 and HS2 stem of C. tubulosa, b The contents of echinacoside, acteoside and total PhGs
Identification of enzyme genes in PhGs biosynthesis
PhGs biosynthetic pathway is related to PS, PM, and TM pathways, We analyzed the genes involved in these three pathways. Using the annotated full-length transcriptome data, we identified 1080 transcripts which encod 22 enzymes involved in PhGs biosynthesis (Table 1; Supplementary Table S4). In most cases, more than one unigenes were annotated as the same enzyme, and the transcripts number encoding UDP-glycosyltransferase were the most (185 transcripts) and followed by peroxidase (138 transcripts). Among these genes, the average length of copper amine oxidase, phenylalanine ammonia-lyase, and cinnamyl alcohol dehydrogenase transcripts were 2212.89 bp, 1977.84 bp, and 955.84 bp, respectively.
Differentially expressed genes (DEGs) between different developmental stages
RNA-seq was performed to quantify the transcript abundance in YS1 and HS2. Six samples were sequenced using DNBSEQ-500 platform, and on average 21.59 Mb clean reads were generated from each sample. About 87.87% of the reads were mapped to the full-length transcriptome. A total of 175,162 unigenes were identified expressing in these two stages. We used a stringent cutoff of FPKM ≥ 1 to define transcripts that were robustly expressed in specific tissue. Among these transcripts, 75,719 (43.23%) in YS1 and 75,214 (42.94%) in HS2 were detected.
Between these two stages, 29,152 DEGs were identified (Supplementary Table S5). To compare with YS1, there were 13,631 and 15,521 up- and down-regulated genes in HS2. The DEGs were classified into three main GO categories: biological process, cellular component, and molecular function (Fig. 4). Cellular process and metabolic process were the top two terms in the biological process. In cellular component category, DEGs were mainly distributed in terms of cellular anatomical entity and intracellular. The most abundant terms in the molecular function were catalytic activity and binding. KEGG analysis classified the DEGs into 136 metabolic pathways (Supplementary Table S6). ABC transporters, DNA replication, phenylpropanoid biosynthesis, plant-pathogen interaction, plant hormone signal transduction were observed in the top 20 enriched pathways (Fig. 4). In addition, phenylalanine, tyrosine, and tryptophan biosynthesis pathway appeared in the top 30 enriched pathways, which were responsible for generating phenylethanol glycosides.

GO classification and KEGG pathway enrichment analysis
To further validate the RNA-seq results, 10 genes (four involved in PS pathway, three in TM pathway, two UGT genes and one GuAO gene) with different expression levels and functions were selected for qRT-PCR analysis. The qRT-PCR expressions of these genes were in accordance with the RNA-Seq results, with the correlation coefficient of 0.8665 (Fig. 5 and Supplementary Table S7).

Verification of DEGs by qRT-PCR. a Transcript levels of 10 genes related to PhGs synthesis. Data are means of three replicates, and error bars represent ± SE (n = 3). b Pearson’s correlation of gene expression ratios between RNA-seq and qRT-PCR. The correlation of the fold change was analyzed by RNA-seq (x-axis) with qRT-PCR (y-axis) data
DEGs encoding transcription factors
Previous studies have shown that WARK, MYB, and bHLH transcription factors play key roles in regulation of phenylpropane synthesis pathways [32,33,34]. In our results, 27 Gene encoding WARK transcription factor were downregulated while only 6 genes were upregulated at HS2 stage. Most of genes that encode MYB and bHLH transcription factors were down-regulated at HS2 stage. AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals. The expression level of 34 AP2-EREBP transcription factor genes decreased and 7 genes upregulated at HS2 stage (Supplementary Table S8).
DEGs involved in phenylethanol glycosides biosynthesis
In C. tubulosa, many DEGs involved in PS, PM, and TM pathways were identified between YS1 and HS2 stages (Table 2; Supplementary Table S4). In some gene families, different genes exhibited varied expression levels, and many genes expressed in a very low level. We only selected the genes with relative high expression level (FPKM level over 10) for further analysis.
Phenylpropanoid biosynthesis is an important pathway of secondary metabolism in plants. Cinnamic acid, coumaric acid, caffeic acid, and ferulic acid are intermediates of this pathway. These acids can be further converted into coumarin, chlorogenic acid, flavonoids, and lignin. In phenylpropanoid metabolic pathway, most genes families encoding the catalytic enzymes were down-regulated in HS2 stages, including phenylalanine ammonia-lyase, cinnamic acid 4-hydroxylase, and shikimate O-hydroxycinnamoyl-transferase. All four genes encoding Phenylalanine ammonia-lyase (PAL), the first key enzyme of PS pathway, were down-regulated in HS2. In some gene families, the expression level of part of the genes were elevated, while other members were decreased, for example, coumarate 3-hydroxylase, 4-coumarate-CoA ligase, caffeoylshikimate esterase, and caffeic acid 3-O-methyltransferase. However, it is worth noting that in these gene families, only one of them has increased expression, while others have decreased expression. For example, there were six transcripts with relative high expression level of C3H gene family, among which 4 genes decreased by more than two fold in HS2 to compare with that in YS1, but only one transcript was up-regulated. In CSE gene family, there were nine transcripts, among which the expression of four genes were greatly reduced while one transcript was increased. Three genes showed elevated expression in HS2, including 5-O-(4-coumaroyl)-D-quinate 3’-monooxygenase, caffeoyl-CoA O-methyltransferase, and Cinnamoyl-CoA reductase genes. CYP98A3 functions to catalyze p-Coumaroyl quinic acid to caffeoyl quinic acid, or p-Coumaroyl shikimic acid to caffeoyl shikimic acid, respectively. Four CYP98A3 genes were found in C. tubulosa, two of them had elevated expression in HS2 stage. CCR and CCoAOMT are key enzymes of PS pathway and related to lignin synthesis. Two CCR genes were expressed highly at HS2, while almost undetectable at YS1. The expression changes of these genes suggested that the synthesis of phenylethanoside precursors, such as caffeic acid and ferulic acid, were channeled to lignin synthesis in HS2.
TM pathway was the main pathway for phenylethanol aglycon synthesis in Rehmannia glutinosa [35]. And PM pathway was speculated the pathway for synthesizing phenylethanol aglycon in C. deserticola [17]. Aromatic-L-amino-acid decarboxylase (AADC), copper amine oxidase (CuAO), and aryl-alcohol dehydrogenase are present in both TM and PM pathways. We found two AADC genes and seven CuAO genes were all down-regulated in HS2. In Rehmannia glutinosa and Petroselinum crispum, tyrosine/DOPA decarboxylase catalyzes (TyDC) decarboxylation of tyrosine to Tyramine [35, 36]. In C. tubulosa, the expression levels of five members of tyrosine/DOPA family were all very low, and there was no significant difference between two stages. Therefore, we suggested that AADC plays an important role in this decarboxylation process in C. tubulosa, rather than TyDC. The similar situation may happen with the deamination of tyrosine, because the tyrosine aminotransferase and histidinol-phosphate aminotransferase expressed lowly and unchanged between two stages.
Glycosylation is one of the final steps involved in the biosynthesis of many plant secondary metabolites. UDP-glycosyltransferase (UGT) can transfer sugar moieties from active sugar molecules (e.g. UDP-glucose) to various acceptor molecules. There were 28 UGT genes identified highly expressed in this study, and 13 of which were down-regulated and three were up-regulated in HS2. The shikimate O-hydroxycinnamoyltransferase was inferred involved in catalyzing the acyl-transfer from coenzyme A-activated acids to phenylethanol aglycon [36]. We identified 13 unigenes encoding shikimate O-hydroxycinnamoyltransferase, and two of which were down-regulated in HS2.
Discussion
The dried succulent stems of Cistanche were widely used in traditional Chinese medicines. C. deserticola and C. tubulosa are the two main medicinal plants [37]. The genomic and transcriptomic resources of Cistanche are mainly from C. deserticola [15,16,17]. In the current study, we analyzing the stem full-length transcriptome of C. tubulosa using PacBio SMRT Sequel platform, and obtained 237,772 unique transcripts. The proportion of the annotated transcripts using the seven software and several public databases is only 79.12%, while in the case of general plants, the proportion is about 90% [23].
Due to the biological activity of PhGs, the biosynthetic pathway has been extensively studied in order to obtain PhGs rich medicinal materials. The putative PhGs biosynthesis pathway was established based on precursor feeding experiments. Feeding tyrosine and phenylalanine to cell suspension culture of Cistanche can increase the accumulation of acteoside, echinoside or 2’-acetyl acteoside [38, 39]. Isotope labeled feeding studies in Syringa vulgaris and Olea europaea showed that tyrosine to 3,4-dihydroxytyrosol (DHPA) conversion was through dopamine or tyramine pathway, and the conversion ofphenylalanine to coffeyl moiety was through phenylpropane pathway [40, 41]. It recognized that the organic acid acyl moiety (coffeyl, feruloyl or coumaroyo) of PhGs were generated from phenylpropanoid synthesis pathway, while the synthesis of phenylethanol moiety has several possible choices. In Rehmannia glutinosa, phenylethanol part was considered to be started from the tyrosine precursors by the tyrosine-derived pathway [35, 42, 43]. In C. deserticola, the synthesis of phenylethanol part is reported presumably from two pathways. One is the caffeic acid or ferulic acid pathway, which is part of the PS pathway. The other is the PM pathway, in which the phenylethanol was converted to phenylethanol aglycon [17].
Studies indicated that tyrosine could be converted to 4-hydroxyphenyl- acetaldehyde by aminotransferase and 4-hydroxyphenylacetaldehyde synthase though 4-hydroxyphenylpyruvate [35, 44]. In this pathway, tyrosine aminotransferase (TAT), histidinol-phosphate aminotransferase (hisC), and aspartate aminotransferase (GOT) are the main active enzymes. We found that both TAT and hisC genes expressed lowly and unchanged in YS1 and HS2. In GOT gene family, one gene was up-regulated and one gene was down-regulated in HS2, the changing trend of expression was not obvious. Meanwhile, we didn’t found 4-hydroxyphenylpyruvate decarboxylase (4HDPC) coding gene in the full-length transcripts of C. tubulosa. Therefore, we suggested that the 4-hydroxyphenylpyruvate pathway maybe not the primary one in C. tubulosa. Based on these results, we proposed a potential model of PhGs biosynthesis regulation in C. tubulosa (Fig. 6). In summary, the phenylethanol part is derived from two parallel pathways, the PM pathway and TM pathway in C. tubulosa. In the PM pathway, phenylalanine is decarboxylated under the action of aromatic-L-amino-acid decarboxylase to produce phenylethylamine, and then generate phenylethylalcohol under copper amine oxidase and aryl-alcohol dehydrogenase. In the TM pathway, tyrosine or its oxidation product L-DOPA were first decarboxylated to produce tyramine or dopamine, then to tyrosol or hydroxytyrosol under the action of CuAO and AADH, respectively. However, the acyltransferase corresponding gene is still uncertain, which transfers caffeoyl- group to phenylethanol aglycon in the downstream of PhGs synthesis. Inferred from transcriptome data, shikimate O-hydroxycinnamoyltransferase may play the role, but the specific catalytic function requires further study.

The proposed pathways and genes involved in the biosynthesis of PhGs and lignins in C. tubulosa.
 The grey box represents the organic acid acyl moiety of PhGs derived from PS pathway;
The grey box represents the organic acid acyl moiety of PhGs derived from PS pathway;
 The orange box represents the phenylethanol moiety or phenylethanol glycosides that derived from TM/PM pathways;
The orange box represents the phenylethanol moiety or phenylethanol glycosides that derived from TM/PM pathways;
 The pink box represents metabolites in tyrosine/phenylalanine metabolism pathways;
The pink box represents metabolites in tyrosine/phenylalanine metabolism pathways;
 The green box represents lignins derived from the PS pathway
The green box represents lignins derived from the PS pathway
There were significant differences in gene expression between YS1 and HS2 stages in C. tubulosa, and the expression level of many catalyzing enzyme genes related to PhGs synthesis were decreased. The expression level of catalyzing enzyme genes that control the flow to lignin were increased in HS2 (CCR and CCoAOMT) (Fig. 6). The result indicated that the synthesis of PhGs was weakened while the synthesis of lignin was activated at HS2 stage, which supported the rationality of harvest the succulent stem of C. tubulosa at this stage in practice. The regulators of PhGs biosynthetic pathway are still uncertain, however, some transcription factors have been found to regulate the expression of key enzyme genes in phenylpropanoid pathway [30,31,32, 45, 46]. In Arabidopsis, overexpression of the MYB transcription factor resulted in activation of genes across the entire phenylpropanoid pathway, including genes such as AtPAL1 and At4CL [30,31,32]. In Plagiochasma appendiculatum, the overexpression of bHLH transcription factor also upregulated PaPAL and Pa4CL1 [45]. In WRKY1 transgenic tomato, researchers found that transcript of key genes from phenylpropanoid pathway accumulated [46]. Our results showed that the expression of WARK, MYB, and bHLH transcription factors changed significantly, the dramatic gene expression changes in these transcription factors suggested their potential regulatory functions in PhGs synthesis pathway.
Conclusions
In the present study, the full-length transcriptome and gene expression profiling of C. tubulosa stem in different developmental stages were achieved. Key genes of catalyzing enzymes for PhGs biosynthesis were identified, and a model for PhGs biosynthesis of C. tubulosa was proposed. The expression patterns of key genes were consistent with the accumulation of PhGs. These results laid the foundation for further studies on molecular mechanism of PhGs biosynthesis and regulation in C. tubulosa.
Availability of data and materials
Raw sequence reads of the RNA-seq data were available in NCBI Short Read Archive (SRA) Database (Bioproject: PRJNA786765). The assembled full length transcriptome data have been deposited in NCBI Transcriptome Shotgun Assembly (TSA) Database (TSA submission: SUB10777875).
Ethical approval and consent to participate.
The Cistanche tubulosa used in this study was cultivated. The materials were collected from plants grown in the experimental station of Shandong Academy of Agricultural Sciences.
Abbreviations
- PhGs:
-
Phenylethanol glycosides
- PS:
-
Phenylpropanoid synthesis pathway
- TM:
-
Tyrosine metabolism pathway
- PM:
-
Phenylalanine metabolism pathway
- PhGs:
-
Phenylethanol glycosides
- PAL:
-
Phenylalanine ammonia-lyase
- C4H:
-
Cinnamic acid 4-hydroxylase
- C3H:
-
Coumarate 3-hydroxylase
- CSE:
-
Caffeoylshikimate esterase
- CYP98A3:
-
5-O-(4-coumaroyl)-D-quinate 3’-monooxygenase
- HCT:
-
Shikimate O-hydroxycinnamoyltransferase
- 4CL:
-
4-Coumarate-CoA ligase
- COMT:
-
Caffeic acid 3-O-methyltransferase
- CCoAOMT:
-
Caffeoyl-CoA O-methyltransferase
- F5H:
-
Ferulate-5-hydroxylase
- PER:
-
Peroxidase
- CCR:
-
Cinnamoyl-CoA reductase
- CAD:
-
Cinnamyl alcohol dehydrogenase
- AADC:
-
Aromatic-L-amino-acid decarboxylase
- TyDC:
-
Tyrosine/DOPA decarboxylase
- GOT:
-
Aspartate aminotransferase
- hisC:
-
Histidinol-phosphate aminotransferase
- TAT:
-
Tyrosine aminotransferase
- PPO:
-
Copper amine oxidase (CuAO) Polyphenol oxidase
- AADH:
-
Aryl-alcohol dehydrogenase
- UGT:
-
UDP-glycosyltransferase
- 4HDPC:
-
4-Hydroxyphenylpyruvate decarboxylase
- DEGs:
-
Differentially expressed genes
- YS1:
-
Young fleshy stem
- HS2:
-
Harvest stage
- FPKM:
-
Fragments Per Kilobase of transcript per Million fragments
References
Hou L, Wang YT, Fan ZX, Zhao SZ, Li CS, Wang XJ. Study on Feasibility Inoculating Cistanche tubulosa to Wild Tamarix chinensis in Yellow River Delta. Mod Chin Med Apr. 2018;20(4):437–40.
Lin WY, Yao C, Cheng J, Kao ST, Tsai FJ, Liu HP. Molecular pathways related to the longevity promotion and cognitive improvement of Cistanche tubulosa in Drosophila. Phytomedicine. 2017;26:37–44.
Zhang K, Ma X, He WJ, Li HX, Han SY, Jiang Y, Wu H, Han L, Ohno T, Uotsu N, Yamaguchi K, Ma Z, Tu P. Extracts of Cistanche deserticola can antagonize immunosenescence and extend life span in senescence-accelerated mouse prone 8(SAM-P8) mice. Evid Based Complement Alternat Med. 2014;2: 601383.
Peng XM, Gao L, Huo SX, Liu XM, Yan M. The mechanism of memory enhancement of acteoside (verbascoside) in the senescent mouse model induced by a combination of D-gal and AlCl3. Phytother Res. 2015;29(8):1137–44.
Luo HY, Cao RR, Wang LJ, Zhu LJ. Protective effect of Cistanchis A on ethanol-induced damage in primary cultured mouse Hepatocytes. Biomed Pharmacother. 2016;83:1071–9.
Wang T, Chen C, Yang M, Deng BW, Kirby GM, Zhang XY. Cistanche tubulosa ethanol extract mediates rat sex hormone levels by induction of testicular steroidgenic enzymes. Pharm Biol. 2016;54(3):481–7.
Song Y, Zeng K, Jiang Y, Tu P. Cistanches Herba, from an endangered species to a big brand of Chinese medicine. Med Res Rev. 2021;41(3):1539–77.
Zhu NL, Xu R, Wu HF, Ma GX, Zhu YD, Peng F, Wang X, Ren XM, Chen J, Xu XD. Fingerprint comparative analysis of Cistanche deserticola Y.C. Ma and Cistanche tubulosa (Schrenk) Wight. Chin Pharm J. 2016;51(13):1116–9.
Zhou Y, Li W, Han LF, Song XB, Li PF, Wang R, Zhang BL. Identifcation of Chinese traditional medicine Cistanches herba from diferent places by HPLC-ESI-MS and FTIR methods. Spectrosc Spectral Anal. 2015;35(4):1056–61.
Wang X, Guo Y. Study on the chemcial composition variations of Cistanche tubulosa during the whole growth period. J China Agric Univ. 2017;22(11):28–35.
Yang TX, Lu YX, Guo YH, Zhai ZX, Yu GJ. Studied of dry matter accumulation and echinacoside content of Cistanche tubulosa in Huabei plain. China J Chin Materia Med. 2006;31(16):1317–20.
Yang TX, Du YH, Liu JN, He M, Gao Q. Determination on active ingredient content of Cistanche tubulosa in different growth period and different parts. Lishizhen Med Mat Med Res. 2014;25:1191–3.
Guo XF, Wu YD, Ni H, Jia XG. Determination on active ingredient content for different parts of Cistanche tubulosa. J Xinjiang Med Univ. 2012;35(1):3.
Yang TX, Du YH, Liu JN, Ming H, Gao Q. Determination on active ingredient content of Cistanche tubulosa in different growth period and different parts. Lishizhen Med Mad Res. 2014;25(5):3.
Sun X, Li L, Pei J, Liu C, Huang LF. Metabolome and transcriptome profiling reveals quality variation and underlying regulation of three ecotypes for Cistanche deserticola. Plant Mol Biol. 2020;102(3):253–69.
Li X, Zhang TC, Qiao Q, Ren ZM, Zhao JY, Yonezawa T, Hasegawa M, Crabbe MJ, Li J, Zhong Y. Complete Chloroplast Genome Sequence of Holoparasite Cistanche deserticola (Orobanchaceae) Reveals Gene Loss and Horizontal Gene Transfer from Its Host Haloxylon ammodendron (Chenopodiaceae). PLoS ONE. 2013;8:e58747.
Li Y, Wang X, Chen T, Yao F, Li C, Tang Q, Sun M, Sun G, Hu S, Yu J, Song S. RNA-Seq Based De Novo Transcriptome Assembly and Gene Discovery of Cistanche deserticola Fleshy Stem. PLoS ONE. 2015;10(5):e0125722.
Rhoads A, Au KF. PacBio Sequencing and Its Applications. Genomics Proteomics Bioinformatics. 2015;13:278–89.
Schaarschmidt S, Fischer A, Lawas LMF, Alam R, Septiningsih EM, Bailey-Serres J, Jagadish SVK, Huettel B, Hincha DK, Zuther E. Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L. Int J Mol Sci. 2020;31(21):8148.
Li L, Liu H, Wen W, Huang C, Li X, Xiao S, Xu D. Full Transcriptome Analysis of Callus Suspension Culture System of Bletilla striata. Front Genet. 2020;11:995.
Xu Z, Peters RJ, Weirather J, Luo H, Liao B, Zhang X, Zhu Y, Ji A, Zhang B, Hu S, Au KF, Song J, Chen S. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015;82:951–61.
Hoang NV, Furtado A, Mason PJ, Marquardt A, Kasirajan L, Thirugnanasambandam PP, Botha FC, Henry RJ. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genomics. 2017;18:395.
Zhao C, He L, Xia H, Zhou X, Geng Y, Hou L, Li P, Li G, Zhao S, Ma C, Tang R, Pandey MK, Varshney RK, Wang X. De novo full length transcriptome analysis of Arachis glabrata provides insights into gene expression dynamics in response to biotic and abiotic stresses. Genomics. 2021;113(3):1579–88.
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–51.
Wang J, Zhang Q, Cui F, Hou L, Zhao S, Xia H, Qiu J, Li T, Zhang Y, Wang X, Zhao C. Genome-wide analysis of gene expression provides new insights into cold responses in Thellungiella salsuginea. Front Plant Sci. 2017;8:713.
Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–71.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–8.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Gonzalez A, Zhao M, Leavitt JM, Lloyd AM. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008;53(5):814–27.
Borevitz JO, Xia Y, Blount J, Dixon RA, Lamb C. Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell. 2000;12:2383–94.
Tohge T, Nishiyama Y, Hirai MY, Yano M, Nakajima J, Awazuhara M, Inoue E, Takahashi H, Goodenowe DB, Kitayama M, Noji M, Yamazaki M, Saito K. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005;42(2):218–35.
Wang F, Zhi J, Zhang Z, Wang L, Suo Y, Xie C, Li M, Zhang B, Du J, Gu L, Sun H. Transcriptome Analysis of Salicylic Acid Treatment in Rehmannia glutinosa Hairy Roots Using RNA-seq Technique for Identification of Genes Involved in Acteoside Biosynthesis. Front Plant Sci. 2017;8:787.
Torrens-Spence MP, Gillaspy G, Zhao BY, Harich K, White RH, Li JY. Biochemical evaluation of a parsley tyrosine decarboxylase results in a novel 4-hydroxyphenylacetaldehyde synthase enzyme. Biochem Bioph Res Co. 2012;418(2):211–6.
Han JP, Song JY, Liu C, Chen J, Qian J, Zhu YJ, Shi LC, Yao H, Chen SL. Identification of Cistanche species (Orobanchaceae) based on sequences of the plastid psbA-trnH intergenic region. Yao Xue Xue Bao. 2010;45(1):126–30.
Liu JY, Guo ZG, Zeng ZL. Improved accumulation of phenylethanoid glycosides by precursor feeding to suspension culture of Cistanche salsa. Biochem Eng J. 2007;33(1):88–93.
Hu GS, Jia JM, Kim DH. Effects of feeding tyrosine and phenylalanine on the accumulation of phenylethanoid glycosides to Cistanche deserticola cell suspension culture. Chin J Nat Med. 2014;12(5):367–72.
Ellis BE. Production of hydroxyphenylethanol glycosides in suspension cultures of Syringa vulgaris. Phytochemistry. 1983;22(9):1941–3.
Saimaru H, Orihara Y. Biosynthesis of acteoside in cultured cells of Olea europaea. J Nat Med-Tokyo. 2010;64(2):139–45.
Wang F, Yang X, Zuo X, Miao C, Zhang Z. Full-length transcriptome sequence and identification of genes involved in phenylethanol glycoside biosynthesis in Rehmannia glutinosa. Acta Pharmaceutica Sinica. ISSN 0513–4870, CN 11–2163/R. https://kns.cnki.net/kcms/detail/11.2163.R.20211209.0131.007.html
Alipieva K, Korkina L, Orhan IE, Georgiev MI. Verbascoside-a review of its occurrence (bio)synthesis and pharmacological significance. Biotechnol Adv. 2014;32:1065–76.
Zhou Y, Zhu J, Shao L, Guo M. Current advances in acteoside biosynthesis pathway elucidation and biosynthesis. Fitoterapia. 2020;142: 104495.
Wu YF, Zhao Y, Liu XY, Gao S, Cheng AX, Lou HX. A bHLH Transcription Factor Regulates Bisbibenzyl Biosynthesis in the Liverwort Plagiochasma appendiculatum. Plant Cell Physiol. 2018;59(6):1187–99.
Shinde BA, Dholakia BB, Hussain K, Panda S, Meir S, Rogachev I, Aharoni A, Giri AP, Kamble AC. Dynamic metabolic reprogramming of steroidal glycol-alkaloid and phenylpropanoid biosynthesis may impart early blight resistance in wild tomato. Plant Mol Biol. 2017;95(4–5):411–23.
Acknowledgements
Thanks to professor Yuhai Guo of China Agricultural University for the guidance of C. tubulosa cultivation.
Funding
This work was financially supported by grants from the Shandong provincial crop elite variety development project (2017LZN032); Shandong province agricultural technology innovation project; Innovative Public Service Platform of Shandong Province: Public Service Platform for Molecular Identification of Traditional Chinese Medicine (2018JGX111); Agricultural scientific and technological innovation project of Shandong Academy of Agricultural Sciences (CXGC2018E13); Taishan Scholar Foundation of Shandong Province (ts20190964).
Author information
Authors and Affiliations
Contributions
XJW, LH, and PFW conceived and designed the study, GHL, JJZ, JWP, RXL, and XJZ performed the experiments. QLC analyzed the data and drafted the manuscript with LH. XJW and PFW revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Consent for publication
All the authors have signed the consent form.
Competing interests
All authors read the final version of this manuscript and agreed with the journal policy. The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Figure S1. BUSCO assembly evaluation results. C (complete): matches the BUSCO database sequence; F (fragmented): only part of the sequence can be compared with the BUSCO database; D (duplicate): multiple genes are compared with the same BUSCO; M (missing): the filtered sequence.
Additional file 2:
Supplementary Figure S2. Transcription factor family classification. The X-axis represents the corresponding number of isoforms, the Y-axis represents the transcription factor family classification.
Additional file 3:
Supplementary Table S1. Seven databases showed the annotation results of the full-length transcripts.
Additional file 4:
Supplementary Table S2. Coding ability prediction results of the full-length transcripts.
Additional file 5:
Supplementary Table S3. Transcription factor families and transcription factor genes.
Additional file 6:
Supplementary Table S4. Differential expression of genes involved in PS, PM and TM pathways.
Additional file 7:
Supplementary Table S5. Differentially expressed genes between HS2 and YS1.
Additional file 8:
Supplementary Table S6. KEGG pathways of DEGs between HS2 and YS1.
Additional file 9:
Supplementary Table S7. Primers for qRT-PCR analysis.
Additional file 10:
Supplementary Table S8. Differential expression of transcription factors genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Hou, L., Li, G., Chen, Q. et al. De novo full length transcriptome analysis and gene expression profiling to identify genes involved in phenylethanol glycosides biosynthesis in Cistanche tubulosa. BMC Genomics 23, 698 (2022). https://doi.org/10.1186/s12864-022-08921-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08921-x