
Abstract
Background
Heart failure (HF) is a prevalent cause of mortality and morbidity. The molecular drivers of HF are still largely unknown.
Results
We aimed to identify circulating proteins causally associated with HF by leveraging genome-wide genetic association data for HF including 47,309 cases and 930,014 controls. We performed two-sample Mendelian randomization (MR) with multiple cis instruments as well as network and enrichment analysis using data from blood protein quantitative trait loci (pQTL) (2,965 blood proteins) measured in 3,301 individuals. Nineteen blood proteins were causally associated with HF, were not subject to reverse causality and were enriched in ligand-receptor and glycosylation molecules. Network pathway analysis of the blood proteins showed enrichment in NF-kappa B, TGF beta, lipid in atherosclerosis and fluid shear stress. Cross-phenotype analysis of HF identified genetic overlap with cardiovascular drugs, myocardial infarction, parental longevity and low-density cholesterol. Multi-trait MR identified causal associations between HF-associated blood proteins and cardiovascular outcomes. Multivariable MR showed that association of BAG3, MIF and APOA5 with HF were mediated by the blood pressure and coronary artery disease. According to the directional effect and biological action, 7 blood proteins are targets of existing drugs or are tractable for the development of novel therapeutics. Among the pathways, sialyl Lewis x and the activin type II receptor are potential druggable candidates.
Conclusions
Integrative MR analyses of the blood proteins identified causally-associated proteins with HF and revealed pleiotropy of the blood proteome with cardiovascular risk factors. Some of the proteins or pathway related mechanisms could be targeted as novel treatment approach in HF.
Similar content being viewed by others
Background
Despite significant advances in the treatment of heart failure (HF) in the last decade, the life expectancy for patients with this condition is still limited [1]. Only a small fraction of HF cases is related to a monogenic cardiomyopathy [2]. A recent genome-wide association study (GWAS) leveraging 47,309 cases and 930,014 controls has identified 11 loci associated to HF [3]. HF is characterized by an altered left ventricular (LV) function and may result from different causes. Notably, coronary artery disease (CAD), high blood pressure, atrial fibrillation and some other cardiometabolic risk factors are associated with the development of HF [4]. The molecular processes and key factors promoting the development of HF are still largely unknown. The identifications of molecules promoting the development of HF could lead to novel therapy. Epidemiological studies measuring biomarkers are subject to bias and reverse causality [5]. Hence, only a small proportion of trials are successful and lead to new licensed drugs [6, 7].
Genetic association data provide a rich resource to identify molecules and pathways involved in the development of disorders. Gene variants acting in cis and associated with intermediate phenotypes such as the expression of genes or the level of proteins in circulation can be leveraged as instrumental variables (IVs) in Mendelian randomization (MR) [8]. Since alleles are randomly allocated before the development of outcomes, MR technique is not prone to reverse causality [9]. Hence, the use of multiple IVs in MR is a robust method to evaluate causal associations. Studies have underscored that molecular targets supported by genetics have a higher chance for being licensed [10].
Disorders are complex systems characterized by the interaction of several molecules. The assessment of complex system in network is a non-biased method to probe pathways and to prioritize molecules [11, 12]. Studies have consistently underscored that molecules highly connected in network, often referred to as hubs, are enriched in signaling pathways and drug targets [13]. Herein, we implemented an integrative approach to identify causally associated blood proteins with HF and we performed network and pathway analyses to prioritize molecules and find novel druggable candidates.
Results
Mendelian randomization of blood proteins in heart failure
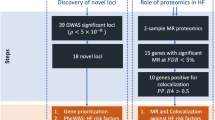
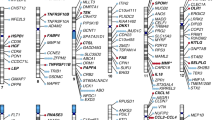
We conducted a two-sample MR analysis to identify causally associated blood proteins with HF. Summary level data from INTERVAL [14], a study including pQTLs for 2,965 different blood proteins measured in 3,301 individuals, were leveraged to identify cis-acting gene variants as instrumental variables (IVs). A minimum of 3 independent (r2 < 0.1) gene variants within a window of 500 kb were selected to identify IVs (P value < 1E-03) for the blood proteins (exposure). GWAS data from the Heart Failure Molecular Epidemiology for Therapeutic Targets Consortium (HERMES) [3], a meta-analysis including 47,309 HF cases and 930,014 controls of European ancestry, were assessed as the outcome. There were enough instruments to perform 822 cis-MR analyses. In inverse variance weighted (IVW) MR, we identified at a false discovery rate (FDR) of 5% nineteen blood proteins that were significantly associated with HF (ABO, BAG3, FLT4, TDGF1, FUT3, FSTL1, ALDH3A1, GLCE, PTHLH, CDON, FCGR2A, RGMB, AMH, MIF, IL15RA, B3GAT3, CCDC126, ST3GAL6, APOA5) (Fig. 1) (Suppl. Table 1 and Suppl. Table 2). In order to check for weak instruments, we calculated F-statistic [15] for all identified instruments for each protein significantly associated with HF. The F-statistic was > 10 for each variant confirming the validity of our selected IVs [15,16,17] (Suppl. Table 1). Among those proteins, (OR per 1 SD) ABO (OR: 1.03, 95%CI: 1.02–1.04, PIVW = 5.89E-13), BAG3 (OR: 0.79, 95%CI: 0.74–0.85, PIVW = 2.59E-09) and FLT4 (OR: 1.08, 95%CI: 1.04–1.12, PIVW = 3.34E-05) were significant after a Bonferroni correction (P < 6.08E-05, 0.05/822). We carried out the Cochran’s Q and Egger intercept tests to detect horizontal pleiotropy [18, 19]. Both the Cochran’s Q and intercept tests did not reveal heterogeneity or horizontal pleiotropy for the nineteen blood proteins (Suppl. Table 1). Thus, the nineteen blood proteins (FDR < 0.05) were considered as causal molecular candidates for downstream analyses. Among the causal candidates, 7 (ABO, FLT4, PTHLH, MIF, IL15RA, B3GAT3, CCDC126) were positively associated with HF, whereas 12 (BAG3, TDGF1, FUT3, FSTL1, ALDH3A1, GLCE, CDON, FCGR2A, RGMB, AMH, ST3GAL6, APOA5) were negatively associated with the risk of HF. The blood proteins with the largest negative and positive effect sizes on HF were BAG3 (OR: 0.79, 95%CI: 0.74–0.85, PIVW = 2.59E-09) and MIF (OR: 1.19, 95%CI: 1.08–1.32, PIVW = 5.53E-04), respectively.

Identification of blood proteins potentially implicated in HF. Manhattan plot depicting blood proteins associated with heart failure (HF) in cis-MR analysis. The localization of the gene encoding the blood protein is represented on the x-axis, whereas the y-axis represents the -log10P value for the association in MR. Red and blue dashed lines are the Bonferroni and FDR 5% threshold values respectively. Red dots are genes positively associated with the development of HF, and green dots are genes negatively associated with the development of HF
As MR is subject to pleiotropy of the IVs, we performed several sensitivity analyses. In weighted median MR, which is robust to invalid instruments [20] (variants with horizontal pleiotropy), 17 blood proteins (ABO, BAG3, CDON, APOA5, CCDC126, FLT4, IL15RA, ALDH3A1, PTHLH, RGMB, AMH, GLCE, TDGF1, FSTL1, FCGR2A, B3GAT3, MIF) remained significantly associated with HF (Suppl. Table 1). The directional effects were concordant between weighted median MR and IVW MR. As an additional measure we implemented MR by using cis-IVs with a more stringent P value (P < 1E-05). By using this approach, there were enough instruments to perform multiple instruments MR for 12 causal candidate proteins. In MR, the 12 proteins (ABO, TFGF1, GLCE, CCDC126, IL15RA, FCGR2A, CDON, FUT3, ST3GAL6, B3GAT3, ALDH3A1, FLT4) remained significantly associated with HF (FDR < 0.05) with concordant directional effect (Suppl. Table 3). Among the remaining 7 proteins, 5 had one available instrument with a P value < 1E-05, for which we performed MR with the Wald ratio. MR with the Wald ratio showed that 4 proteins (BAG3, PTHLH, RGMB, APOA5) were significantly (PWald test < 0.05) associated with HF and again with concordant directional effects (Suppl. Table 4). Hence, these analyses showed that 16 blood proteins out of 19 were replicated by using a more stringent selection of IVs. We also conducted reverse MR analysis as an additional sensitivity measure. We selected 11 genome-wide significant instruments at HF risk loci. In reverse MR analysis, we found no significant association (FDR > 5% for the nineteen causal candidate proteins) (Suppl. Table 5). These data indicate that the causal candidate blood proteins were not subject to reverse causality.
Additionally, we conducted a replication two-sample MR analysis by leveraging as exposure the data from the deCODE study [21]. This study included pQTLs for 4,719 blood proteins measured in 35,559 Icelanders of European ancestry. Enough instruments were available to perform 17 cis-MR analysis (P value IVs < 1E-03) (Suppl. Table 6). Among the 17 candidate blood proteins available in the deCODE study, 14 were replicated at FDR < 5% (ABO, GLCE, IL15RA, RGMB, FSTL1, CDON, ALDH3A1, ST3GAL6, TDGF1, FUT3, CCDC126, FCGR2A, PTHLH, B3GAT3) (Suppl. Table 6). By using IVs with a P value < 1E-05, both multiple instruments MR and the Wald ratio showed that 16 proteins (ABO, FLT4, GLCE, IL15RA, TDGF1, B3GAT3, RGMB, FSTL1, ALDH3A1, CDON, ST3GAL6, FUT3, FCGR2A, CCDC126, PTHLH, MIF) were replicated in deCODE (Suppl. Table 7 and Suppl. Table 8). For the replicated blood proteins, the directional effects were concordant between deCODE and INTERVAL.
Enrichment and network pathway analysis
We aimed to identify the functional and pathway enrichments of candidate causal blood proteins. Among the causal candidates, 5 blood proteins (ABO, FUT3, GLCE, B3GAT3 and ST3GAL6) were classified as molecules involved in glycosylation in the Comprehensive GlycoEnzyme Database (GlycoEnzDB) (fold-enrichment 13.1, P = 1.40E-06, hypergeometric test). Of note, FUT3 (OR: 0.97, 95%CI: 0.96–0.98, PIVW = 8.68E-05) and ST3GAL6 (OR: 0.97, 95%CI: 0.96–0.99, PIVW = 1.06E-03) were both negatively associated with the risk of HF. FUT3 and ST3GAL6 are key enzymes leading to the generation of sialyl Lewis x, a glycan moiety decorating membrane and circulating proteins [22, 23] (Suppl. Figure 1). We next hypothesized that some of the causal candidate proteins may be involved in different ligand-receptor interactions. By using a comprehensive repository of ligand-receptor interactions reported by Shao et al. [24], we found that blood proteins associated with HF in MR were enriched in ligand and receptors (fold-enrichment 5.7, P = 4.0E-07, hypergeometric test). These molecules may contribute to 43 different ligand-receptor pairs (Suppl. Table 9). The 43 ligand-receptor pairs were enriched in Gene Ontology (GO) (molecular function) for transmembrane receptor protein serine/threonine kinase activity (P = 3.68E-12), G protein-coupled receptor activity (P = 1.08E-10), patched binding (P = 5.43E-07), activin-activated receptor activity (P = 1.30E-06) and transforming growth factor beta-activated receptor activity (P = 3.38E-06) (Suppl. Figure 2) (Suppl. Table 10).
We next performed a pathway analysis by using a network approach. Protein interaction data from InnateDB, which includes more than 19,800 curated protein interactions, was leveraged to infer a blood protein network [25]. The nineteen causal candidate proteins were used as seeds to generate a network including 155 nodes (proteins) and 160 edges (interactions) (Fig. 2A) (Suppl. Table 11). The causally associated blood proteins were overrepresented in the nodes (proteins) with the highest degree (≥ 90th percentile) (fold-enrichment 3.6, P = 7.26E-06, hypergeometric test). The top nodes (proteins) acting as hub molecules include MIF, BAG3, FSTL1, FCGR2A, TDGF1, FLT4, PTHLH, IL15RA, AMH and ALDH3A1. We interrogated the Kyoto Encyclopedia of Genes and Genomes (KEGG) [26] to perform a pathway enrichment analysis of the network. The highest enrichments were pathways in cancer (P = 1.03E-17), NF-kappa B signaling (P = 2.99E-13), TGF-beta signaling (P = 3.83E-13), lipid and atherogenesis (P = 9.53E-13) as well as fluid shear stress and atherosclerosis (P = 9.96E-12) (Fig. 2B) (Suppl. Table 12).

Network and enrichment pathway analysis of causal blood protein candidates. A) HF causal blood protein candidates were used as seeds to generate a protein interaction network inferred from InnateDB [25] (database of 19,800 curated proteins interactions). B) Pathway enrichment analysis of the network by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) [26]
Cross-phenotype analysis
Cross-phenotype association analysis were performed for the genetic association data of HF by using the interactive cross-phenotype analysis of GWAS database (iCPAG) [27] which provides enrichment and similarity metrics between traits by using an exhaustive list of ancestry LD-specific association data from the NHGRI-EBI GWAS catalog [28]. After a Bonferroni correction, this analysis showed that 75 disorders and traits were significantly associated to HF (Suppl. Table 13). Figure 3 shows the highest enrichments between HF and traits-disorders. According to iCPAG, the highest enrichments were for beta blocking agent use (P = 1.36E-24), coronary artery disease (P = 2.70E-23), low-density cholesterol (P = 9.09E-21), myocardial infarction (P = 4.87E-20), apolipoprotein B (P = 1.24E-19) and parental longevity (P = 1.28E-19).

Cross-phenotype analysis. Cross-phenotype association analysis of HF performed by using the summary statistics data of GWAS from HERMES and the interactive cross-phenotype analysis of GWAS database (iCPAG), which includes data from the NHGRI-EBI GWAS catalog. Significance of traits was determined by the Fisher exact test
Multi-trait and multivariable MR analyses
Considering the cross-phenotype analysis showing a genetic overlap between HF and several cardiovascular disorder related traits, we performed a multi-trait MR analysis for the nineteen causal blood candidate proteins. We leveraged 31 different GWAS covering 7 disease categories (atopic, autoimmune, cancer, cardiovascular, infectious, metabolic and neurologic) as outcomes (Suppl. Table 14). Figure 4 illustrates the MR analysis including the directional effect and the significance (-logP) between the nineteen blood proteins (exposure) and the different disorder-traits (outcomes). Some blood proteins such as ABO and FCGR2A show significant association with several traits often with opposite directional effects (antagonistic pleiotropy) with HF. However, some proteins such as BAG3, MIF and APOA5 show concordant associations between HF, the blood pressure (BAG3) and coronary artery disease (MIF and APOA5). BAG3 is significantly and negatively associated with systolic and diastolic blood pressure, whereas MIF and APOA5 are associated with coronary artery disease. We performed mediation analysis by using multivariable MR corrected for the exposure to cardiovascular traits. Following the correction for the diastolic blood pressure, the association between BAG3 and HF was not significant (Suppl. Table 15). Also, after correction for coronary artery disease the associations between MIF and APOA5 with HF were no longer significant (Suppl. Table 15). Taken together, these data suggest that, at least in part, the protective effect of circulating BAG3 is mediated by a reduction of the diastolic blood pressure. On the other hand, the impact of MIF and APOA5 on HF are likely mediated by the risk of coronary artery disease, a leading cause of HF [29, 30].

Multi-trait MR. Balloon plot illustrating the multi-trait MR analysis of the 19 blood proteins as exposures, and the 31 diseases and traits as outcomes. Red and green indicates positive and negative directional effects respectively. * Indicates significance at a Bonferroni threshold; † indicates significance at FDR 5% threshold
Drug target analysis
We leveraged several resources to carry out a drug target analysis. We investigated the blood proteins in order to document if they represent targets for licensed, in-development small molecules or biologics. In the Therapeutic Target Database (TTD) [31], 6 blood proteins (ABO, FLT4, FCGR2A, AMH, MIF, IL15RA) are reported as either clinical trial target or successful targets (Suppl. Table 16). In the Drug Gene Interaction Database (DGIdb) [32], a total of 111 drug-target pairs were reported for FLT4, ALDH3A1, PTHLH, FCGR2A, AMH, MIF, IL15RA and APOA5 (Suppl. Table 17). Several kinase inhibitors targeting FLT4, a blood protein positively associated with the risk of HF, are approved for the treatment of cancer. In the Open Targets database [33, 34], MIF, PTHLH, FLT4 and TDGF1 were reported as targets of approved and in-development drugs as well as antibodies (Suppl. Table 18). In the blood, MIF is positively associated with the risk of HF and is a target for Imalumab and Iguratimod, respectively an antibody and a small molecule inhibitor. Iguratimod is licensed in Japan for the treatment of rheumatoid arthritis [35], whereas Imalumab is a phase 1 monoclonal antibody destined for patients with solid tumors [36]. According to Open Targets, 15 blood proteins (MIF, CCDC126, IL15RA, FCGR2A, CDON, ALDH3A1, FSTL1, APOA5, AMH, BAG3, B3GAT3, ST3GAL6, FUT3, GLCE, RGMB) are deemed tractable for the development of antibodies (Suppl. Table 18). Taken together, these data suggest that according to the directional effect MIF, FLT4, PTHLH, ABO, CCDC126, IL15RA, and B3GAT3 (blood proteins positively associated with HF) are potential targets for HF as they are the object of approved, in-development inhibitors or deemed tractable for the development of novel inhibitors (antibodies).
Discussion
In this work, we undertook a MR analysis of the circulating proteome to identify circulating proteins causally associated with HF. A comprehensive analysis of the blood proteome identified, by using multiple cis-acting variants as IVs, nineteen proteins causally associated with HF. Causal candidate proteins were enriched in glycosylation and ligand-receptor molecules. A blood protein network showed that causal candidates were enriched as hub molecules involved in pathways related to NF-kappa B, TGF-beta and atherogenesis. Cross-phenotype and multi-trait MR showed that some blood proteins associated with HF were pleiotropic and involved in cardiovascular traits. The assessment of the druggable genome identified causal candidates for which drug repurposing or drug development could lead to novel therapies.
The present MR analysis identified several novel genes associated with HF. In this regard, among the candidate molecules only ABO and BAG3 have been previously associated with HF. Among the causal blood candidate proteins, BAG3 (OR: 0.79, 95%CI: 0.74–0.85, PIVW = 2.59E-09) and MIF (OR: 1.19, 95%CI: 1.08–1.32, PIVW = 5.53E-04) had the highest effect size in MR analysis. Mutations in BAG3 have been associated with dominant forms of myopathy affecting skeletal muscle and the heart [37]. The role of intracellular BAG3 has been intensively investigated as the protein binds to heat shock proteins and control protein folding and aggregation. Herein, we documented, to our knowledge, for the first time that secreted and circulating BAG3 may be protective on the risk of HF. A previous study showed that BAG3 administered to rodents reduced the blood pressure through a nitric oxide pathway [38]. The putative receptor whereby extracellular BAG3 reduced the blood pressure is presently unknown. These experimental data are supported by the present results, which showed in mediation analysis that the protective effect of circulating BAG3 on HF was, at least in part, mediated by a reduction of the diastolic blood pressure. Functional follow-up studies are needed to tease apart the mechanism whereby circulating BAG3 regulates the blood pressure and the risk of HF. MIF encodes the macrophage migration inhibitory factor, a pro-inflammatory molecule involved in rheumatoid arthritis [39]. Results of our MR analysis indicated that MIF was positively associated with the risk of HF. Multivariable analysis suggested that MIF promoted HF through the risk of CAD.
The circulating proteins related to the risk of HF were highly enriched in molecules involved in glycosylation (fold-enrichment 13.1, P = 1.40E-06). Notably, FUT3 (OR: 0.97, 95%CI: 0.96–0.98, PIVW = 8.68E-05) and ST3GAL6 (OR: 0.97, 95%CI: 0.96–0.99, PIVW = 1.06E-03) were negatively associated with the risk of HF. FUT3 and ST3GAL6 encode for fucosyl transferase 3 and ST3 beta-galactoside alpha-2,3-sialyltransferase 6, respectively. They are involved in the synthesis of sialyl Lewis X (Siaα2,3Galβ1,4[Fucα1,3] GlcNAc), a glycan moeity involved in cell adhesion and the recruitment of inflammatory cells (Suppl. Figure 1). Experimental evidence suggests that circulating glycosyltransferase promotes extracellular glycosylation including the synthesis of sialyl Lewis x [40]. These data suggest that the synthesis of sialyl Lewis X in the blood could be protective in HF possibly by acting as a decoy factor that limit leukocyte adhesion. Consistent with this hypothesis, systemic administration of a sialyl Lewis x analogue in a large animal model of cardiac arrest decreased the recruitment of inflammatory cells to the myocardium and preserved the myocardial function [41]. Further work is needed to address the potential role of glycosylation in HF.
HF-associated causal proteome was enriched in ligands and receptors. Ligand-receptor pairs were overrepresented by serine/threonine kinase, G protein-coupled receptor, patched binding, activin-activated receptor binding and transforming growth factor beta-activated receptor activity. Blood IL15RA (OR: 1.02, 95%CI: 1.01–1.04, PIVW = 8.60E-04) concentrations were positively associated with the risk of HF. IL15RA encodes for the IL15R alpha-chain, which is secreted through a protease-dependent process and binds to IL15 [42, 43]. Secreted receptors may function as natural antagonist or agonist for their ligands and thus exert an important control over many biological processes. Contradicting reports for secreted IL15RA suggest that it may either antagonize or promote IL15 signaling [44, 45]. In a mouse model of myocardial infarction, the administration of IL15 improved cell death and LV function [46]. Inquiry into the function of IL15RA-IL15 on the myocardial function could help design new therapies. TDGF1 encodes for teratocarcinoma-derived growth factor 1 (also known as CRIPTO), which is acting as natural antagonist for activin receptor ActRII [47, 48]. Blood TDGF1 (OR: 0.98, 95%CI: 0.97–0.99, PIVW = 8.20E-5) concentrations were negatively associated with the risk of HF suggesting that inhibition of the activin pathway might reduce the risk of HF. Consistent with the present data, activin-mediated signaling (ActRII) in cardiomyocyte promoted the degradation of sarcoplasmic reticulum Ca2 + ATPase (ATP2A2 also known as SERCA2), an important regulator of cardiac contractility, and the development of aging-related HF in a rodent model [49]. Among the other blood proteins acting as receptors, CDON (OR: 0.95, 95%CI: 0.93–0.98, P = 2.59E-04) is involved in hedgehog (HH) signaling [50], a pathway playing a role in cardiac regeneration [51, 52], whereas AMH (OR: 0.82, 95%CI: 0.74–0.91, P = 4.71E-04) binds to AMHR2 and is related to the TGF-beta pathway [53]. AMH encodes for the anti-Müllerian hormone. Lower level of this hormone have been associated with increased cardiovascular risk in women [54].
Some of the causal blood protein candidates may represent suitable drug targets for HF. For instance, repurposing the MIF antagonist Iguratimod may lower the risk of CAD and HF. Among the other approved drugs, FLT4 inhibitors may lower the risk of HF, but long-term therapy needed to treat HF is a limiting factor as this class of drugs is associated with a high rate of side effects [55,56,57,58]. Though further follow-up studies are needed, some of the blood candidate molecules were involved in pathways, which could be targeted in HF. For instance, glycosylation and the activin pathways may offer novel opportunity for the treatment of HF. For instance, bimagrumab, a monoclonal antibody, is an antagonist of the activin type II receptors (ActRII) [59]. In patients with type 2 diabetes and obesity, a phase 2 study has shown that bimagrumab increased the lean mass and enhanced insulin sensitivity [60]. In addition, the development of new fusion proteins between a target and a peptide, such as the Fc domain, increase the biological activity of the target-protein [61]. As a case in point, considering the protective effect of secreted BAG3 on the risk of HF, the functionalization of this protein could represent a therapeutic opportunity.
The present study has some limitations. MR is subject to horizontal pleiotropy [9]. As such, some IVs may be associated with the outcome through unknown pathways, which are not related to the exposition. However, we have implemented several measures to decrease the risk of horizontal pleiotropy. The selection of cis-instruments instead of trans-instruments is known to lower the risk of pleiotropy [9, 62]. In addition, both the Cochran’s Q test for heterogeneity and the Egger-intercept tests provide further assessment of pleiotropy [18, 19]. Finally, the implementation of the weighted median MR, which is resistant to invalid instruments (i.e. IVs related to the outcome through another process not related to the exposition) also provided robustness to the results [20].
Conclusion
By using MR, we identified circulating proteins causally associated with HF. These proteins highlighted molecular pathways playing a key role in the development of this disorder. Integrative analysis showed that proteins were involved in several interactions where they were acting as hub molecules. Some of the molecules or pathways could be targeted for drug repurposing or the development of new small molecules or biologics in order to treat HF.
Methods
Genetic associations for heart failure
Summary statistics from GWAS data of a meta-analysis including 47,309 HF cases and 930,014 controls were downloaded for analyses (HERMES) [3]. The GWAS meta-analysis included participants of European ancestry from 26 cohorts. Cases included clinical cases of HF from any aetiology. GWAS meta-analysis was adjusted for age, sex and principal components and included 8,281,262 genetic variants.
Mendelian randomization
To derive the exposures, protein expression quantitative trait loci (pQTL) were leveraged from the INTERVAL study, which included data for 2,965 different blood proteins measured in 3,301 individuals [14]. Blood proteins were measured using the SOMAscan assay. The study reported 10,572,788 genetic variants. Two-sample MR using the INTERVAL and (exposure) and HERMES (outcome) studies was carried out by using at least 3 cis-instrumental variables (at P < 1E-03 and P < 1E-05) selected within a window of ± 500 kb around the transcription start site of the candidate gene. Independent (r2 < 0.1) instrumental variables (SNPs) were identified with PLINK1.9 based on genotypes from European populations from the 1000 Genome project. Horizontal pleiotropy was evaluated by using the Cochran’s Q test and the Egger-intercept test. We performed inverse variance weighted MR and as sensitivity analyses we used the weighted median MR, which allows the use of up to 50% of invalid instruments. F-statistic [15,16,17] was calculated for each IV using the formula β2/SE2. Multivariate MR using HF as the outcome was performed by correcting the exposition for selected cardiovascular traits (blood pressure and CAD). MR analyses were performed by using the Mendelian Randomization package. Multiple test correction was performed, and significance was established at FDR < 0.05. FDR was calculated by using the R package multtest with the Benjamini and Hochberg test. The Wald ratio was calculated when only one IV with a P value < 1E-05 was available. The Wald ratio was calculated with the TwoSampleMR library in R.
Reverse Mendelian randomization
Two sample reverse MR was carried out by using the HERMES study as the exposure and the blood proteins from the INTERVAL study as the outcome. IVs were selected by using a window of 500 kb at risk loci and by using the lead GWAS significant variant (P < 5E-08). In total, we leveraged 11 independent cis-instrumental variables in the HERMES study. We performed inverse variance weighted MR and the weighted median MR. Horizontal pleiotropy was evaluated using the Egger-intercept test and the Cochran’s Q test.
Replication in deCODE
For replication we used the deCODE study [21], which included data for 4,719 different blood proteins measured in 35,559 Icelandic individuals. Blood proteins were measured by using the SOMAscan version 4 assay. The study reported 27,2 million genetic variants. Analysis was conducted for the nineteen causal candidate proteins and MR was performed as described above for the INTERVAL study.
Enrichment analyses
To perform enrichment analyses, we downloaded data from the Comprehensive GlycoEnzyme Database (GlycoEnzDB) [63] and the ligand-receptor repository from Shao et al. [24]. Hypergeometric test was performed by using R. Pathway and gene ontology enrichments were performed by using enrichR [64, 65]. EnrichR Pathways and Gene Ontology were generated on 2021/10/20. The Comprehensive GlycoEnzyme Database was downloaded on 2021/10/22.
Network analyses
Candidate causal blood proteins were used as seeds to generate a network based on data from InnateDB, which includes more than 19,800 curated protein interactions [25]. Edge list of association pairs was analyzed by using NetworkAnalyst [66]. We identified hub nodes (genes) as those with a degree ≥ 90th percentile. Network of protein / protein interactions was generated on 2021/10/20 using Generic PPI / IMEx parameters.
Cross-trait analysis
GWAS for HF was evaluated with the Cross-Phenotype Analysis of GWAS database (iCPAGdb) [27]. iCPAGdb uses ancestry LD-specific association data across 3,793 traits-disorders, which were selected from the NHGRI-EBI GWAS catalog to compute cross-phenotype enrichment analyses. iCPAGdb reports pairwise traits and shared signal. Output data are reported as Fisher exact test with adjustment (Benjamini–Hochberg and Bonferroni) and the Chao-Sorenson similarity index. Results of cross-trait analysis were generated on 2021/10/21.
Multi-trait Mendelian randomization analysis
Multi-trait Inverse Variance Weighted MR were performed by using data of pQTL from the INTERVAL study (exposure) and 31 traits-disorders (outcomes). Traits were selected from 7 disease categories (atopic, autoimmune, cancer, cardiovascular, infectious, metabolic and neurologic). Data were downloaded from the NHGRI-EBI GWAS catalog and UK Biobank data previously processed by the Neale lab (see Data availability). Association from the MR analyses were deemed significant after applying the Bonferroni correction (P < 1.61E-03, 0.05/31). Results were illustrated as a balloon plot. Graphs were generated with ggplot2 in R.
Drug target analysis
We assessed the druggability of each protein candidates by using the following resources: Therapeutic Target Database (TTD) [31], Drug Gene Interaction Database (DGIdb) [32] and Open Targets [33, 34]. For each repository, we reported the drug-gene pairs by using approved and non-approved drugs. In the Open Targets database, we also reported the tractability index for the development of antibodies. Drug target analysis were generated on the 29 to 30/11/2021 using DGIdb v 4.2.0; Open targets v 21.11 and TTD (update of 2021/09/29).
Availability of data and materials
All data are available in the manuscript including in supplementary tables and figures. Summary statistics for GWAS HF and GWAS pQTL INTERVAL are publicly available. URLs are provided in the manuscript Data availability section.
Abbreviations
- CAD:
-
Coronary Artery Disease
- DBP:
-
Diastolic Blood Pressure
- FDR:
-
False Discovery Rate
- GO:
-
Gene Ontology
- GWAS:
-
Genome Wide Associated Study
- HF:
-
Heart Failure
- IVs:
-
Instrumental variables
- IVW:
-
Inverse Variance Weighted
- LD:
-
Linkage Disequilibrium
- LVF:
-
Left Ventricular function
- MR:
-
Mendelian Randomization
- Multi-trait MR:
-
Multi trait Mendelian Randomization
- Multivariable MR:
-
Multivariable Mendelian Randomization
- pQTL:
-
Protein Quantitative Trait Loci
- SBP:
-
Systolic Blood Pressure
- SNP:
-
Single Nucleotide Polymorphism
References
Glynn PA, Ning H, Bavishi A, Freaney PM, Shah S, Yancy CW, et al. Heart failure risk distribution and trends in the United States population, NHANES 1999–2016. Am J Med. 2021;134:e153–64.
Czepluch FS, Wollnik B, Hasenfuß G. Genetic determinants of heart failure: facts and numbers. ESC Heart Fail. 2018;5:211–7.
Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11:163.
Meijers WC, de Boer RA. Common risk factors for heart failure and cancer. Cardiovasc Res. 2019;115:844–53.
Sattar N, Preiss D. Reverse causality in cardiovascular epidemiological research: more common than imagined? Circulation. 2017;135:2369–72.
Drucker E, Krapfenbauer K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J. 2013;4:7.
Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19–34.
Lee K, Lim C-Y. Mendelian randomization analysis in observational epidemiology. J Lipid Atheroscler. 2019;8:67–77.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89-98.
King EA, Davis JW, Degner JF. Are drug targets with genetic support twice as likely to be approved? revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019;15:e1008489.
Choobdar S, Ahsen ME, Crawford J, Tomasoni M, Fang T, Lamparter D, et al. Assessment of network module identification across complex diseases. Nat Methods. 2019;16:843–52.
Mitra K, Carvunis A-R, Ramesh SK, Ideker T. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet. 2013;14:719–32.
Farkas IJ, Korcsmáros T, Kovács IA, Mihalik Á, Palotai R, Simkó GI, et al. Network-based tools for the identification of novel drug targets. Sci Signal. 2011;4:pt3.
Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–9.
Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey SG. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–63.
Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. 2012;21:223–42.
Georgakis MK, Gill D, Rannikmäe K, Traylor M, Anderson CD, Lee J-M, et al. Genetically determined levels of circulating cytokines and risk of stroke. Circulation. 2019;139:256–68.
Kulinskaya E, Dollinger MB. An accurate test for homogeneity of odds ratios based on Cochran’s Q-statistic. BMC Med Res Methodol. 2015;15:49.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45:1961–74.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14.
Ferkingstad E, Sulem P, Atlason BA, Sveinbjornsson G, Magnusson MI, Styrmisdottir EL, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53:1712–21.
Okajima T, Fukumoto S, Miyazaki H, Ishida H, Kiso M, Furukawa K, et al. Molecular cloning of a novel α2,3-sialyltransferase (ST3Gal VI) that sialylates Type II lactosamine structures on glycoproteins and glycolipids*. J Biol Chem. 1999;274:11479–86.
Mondal N, Dykstra B, Lee J, Ashline DJ, Reinhold VN, Rossi DJ, et al. Distinct human α(1,3)-fucosyltransferases drive Lewis-X/sialyl Lewis-X assembly in human cells. J Biol Chem. 2018;293:7300–14.
Shao X, Liao J, Li C, Lu X, Cheng J, Fan X. Cell TalkDB: a manually curated database of ligand-receptor interactions in humans and mice. Brief Bioinform. 2021;22:bbaa269.
Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, et al. InnateDB: systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–33.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–61.
Wang L, Balmat TJ, Antonia AL, Constantine FJ, Henao R, Burke TW, et al. An atlas connecting shared genetic architecture of human diseases and molecular phenotypes provides insight into COVID-19 susceptibility. Genome Med. 2021;13:83.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–12.
Lala A, Desai AS. The role of coronary artery disease in heart failure. Heart Fail Clin. 2014;10:353–65.
Rush CJ, Berry C, Oldroyd KG, Rocchiccioli JP, Lindsay MM, Touyz RM, et al. Prevalence of coronary artery disease and coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. JAMA Cardiol. 2021;6:1130–43.
Zhou Y, Zhang Y, Lian X, Li F, Wang C, Zhu F, et al. Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022;50:D1398–407.
Freshour SL, Kiwala S, Cotto KC, Coffman AC, McMichael JF, Song JJ, et al. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021;49:D1144-51.
Koscielny G, An P, Carvalho-Silva D, Cham JA, Fumis L, Gasparyan R, et al. Open targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 2017;45:D985–94.
Ochoa D, Hercules A, Carmona M, Suveges D, Gonzalez-Uriarte A, Malangone C, et al. Open targets platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res. 2021;49:D1302–10.
Xie S, Li S, Tian J, Li F. Iguratimod as a new drug for rheumatoid arthritis: current landscape. Front Pharmacol. 2020;11:73.
Mahalingam D, Patel MR, Sachdev JC, Hart LL, Halama N, Ramanathan RK, et al. Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours. Br J Clin Pharmacol. 2020;86:1836–48.
Meister-Broekema M, Freilich R, Jagadeesan C, Rauch JN, Bengoechea R, Motley WW, et al. Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat Commun. 2018;9:5342.
Carrizzo A, Damato A, Ambrosio M, Falco A, Rosati A, Capunzo M, et al. The prosurvival protein BAG3: a new participant in vascular homeostasis. Cell Death Dis. 2016;7:e2431–e2431.
Bilsborrow JB, Doherty E, Tilstam PV, Bucala R. Macrophage migration inhibitory factor (MIF) as a therapeutic target for rheumatoid arthritis and systemic lupus erythematosus. Expert Opin Ther Targets. 2019;23:733–44.
Lee-Sundlov MM, Ashline DJ, Hanneman AJ, Grozovsky R, Reinhold VN, Hoffmeister KM, et al. Circulating blood and platelets supply glycosyltransferases that enable extrinsic extracellular glycosylation. Glycobiology. 2017;27:188–98.
Schermerhorn ML, Tofukuji M, Khoury PR, Phillips L, Hickey PR, Sellke FW, et al. Sialyl LewisX oligosaccharide preserves cardiopulmonary and endothelial function after hypothermic circulatory arrest in lambs. J Thorac Cardiovasc Surg. 2000;120:230–7.
Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Rα recycles and presents IL-15 In trans to neighboring cells. Immunity. 2002;17:537–47.
Rubinstein MP, Kovar M, Purton JF, Cho J-H, Boyman O, Surh CD, et al. Converting IL-15 to a superagonist by binding to soluble IL-15R. Proc Natl Acad Sci. 2006;103:9166–71.
Mortier E, Bernard J, Plet A, Jacques Y. Natural, proteolytic release of a soluble form of human IL-15 receptor alpha-chain that behaves as a specific, high affinity IL-15 antagonist. J Immunol. 2004;173:1681–8.
Rubinstein MP, Kovar M, Purton JF, Cho J-H, Boyman O, Surh CD, et al. Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha}. Proc Natl Acad Sci U S A. 2006;103:9166–71.
Ameri K, Bayardorj D, Samurkashian R, Fredkin M, Fuh E, Nguyen V, et al. Administration of interleukin-15 peptide improves cardiac function in a mouse model of myocardial infarction. J Cardiovasc Pharmacol. 2020;75:98–102.
Kelber JA, Shani G, Booker EC, Vale WW, Gray PC. Cripto is a noncompetitive activin antagonist that forms analogous signaling complexes with activin and nodal. J Biol Chem. 2008;283:4490–500.
Gray PC, Harrison CA, Vale W. Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc Natl Acad Sci. 2003;100:5193–8.
Roh JD, Hobson R, Chaudhari V, Quintero P, Yeri A, Benson M, et al. Activin type II receptor signaling in cardiac aging and heart failure. Sci Transl Med. 2019;11:eaau8680.
Song JY, Holtz AM, Pinskey JM, Allen BL. Distinct structural requirements for CDON and BOC in the promotion of Hedgehog signaling. Dev Biol. 2015;402:239–52.
Dunaeva M, Waltenberger J. Hh signaling in regeneration of the ischemic heart. Cell Mol Life Sci. 2017;74:3481–90.
Kawagishi H, Xiong J, Rovira II, Pan H, Yan Y, Fleischmann BK, et al. Sonic hedgehog signaling regulates the mammalian cardiac regenerative response. J Mol Cell Cardiol. 2018;123:180–4.
Hart KN, Stocker WA, Nagykery NG, Walton KL, Harrison CA, Donahoe PK, et al. Structure of AMH bound to AMHR2 provides insight into a unique signaling pair in the TGF-β family. Proc Natl Acad Sci USA. 2021;118:e2104809118.
de Kat AC, Verschuren WM, Eijkemans MJC, Broekmans FJM, van der Schouw YT. Anti-müllerian hormone trajectories are associated with cardiovascular disease in women: results from the doetinchem cohort study. Circulation. 2017;135:556–65.
Miyamoto S, Kakutani S, Sato Y, Hanashi A, Kinoshita Y, Ishikawa A. Drug review: pazopanib. Jpn J Clin Oncol. 2018;48:503–13.
Jones RL, Ratain MJ, O’Dwyer PJ, Siu LL, Jassem J, Medioni J, et al. Phase II randomised discontinuation trial of brivanib in patients with advanced solid tumours. Eur J Cancer. 2019;120:132–9.
Imbulgoda A, Heng DYC, Kollmannsberger C. Sunitinib in the treatment of advanced solid tumors. Recent Results Cancer Res. 2014;201:165–84.
Wells SA, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–41.
Spitz RW, Dankel SJ, Bell ZW, Wong V, Abe T, Kang M, et al. Blocking the activin IIB receptor with bimagrumab (BYM338) increases walking performance: a meta-analysis. Geriatr Gerontol Int. 2021;21:939–43.
Heymsfield SB, Coleman LA, Miller R, Rooks DS, Laurent D, Petricoul O, et al. Effect of bimagrumab vs placebo on body fat mass among adults with type 2 diabetes and obesity: a phase 2 randomized clinical trial. JAMA Netw Open. 2021;4:e2033457.
Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med. 2012;4:1015–28.
Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40:740–52.
Liu G, Puri A, Neelamegham S. Glycosylation network analysis toolbox: a MATLAB-based environment for systems glycobiology. Bioinformatics. 2013;29:404–6.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90-97.
Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:W234-41.
Acknowledgements
Not applicable.
Data availability—URLs
Summary statistics GWAS HF: http://www.broadcvdi.org/
INTERVAL: https://app.box.com/s/u3flbp13zjydegrxjb2uepagp1vb6bj2
deCODE: https://www.decode.com/summarydata/
iCPAG: http://cpag.oit.duke.edu/explore/app/
PLINK: http://zzz.bwh.harvard.edu/plink/
MR: https://rdrr.io/cran/MendelianRandomization/
NetworkAnalyst: https://www.networkanalyst.ca/
Enrichr: https://amp.pharm.mssm.edu/Enrichr/
DGIdb: https://www.dgidb.org/
Therapeutic Target Database: http://db.idrblab.net/ttd/
Open Targets: https://www.opentargets.org/
Data availability for GWAS in multi-trait Mendelian randomization
Asthma_Valette_K: https://www.ebi.ac.uk/gwas/publications/34103634
Dermatitis_Paternoster_L: https://www.ebi.ac.uk/gwas/publications/22197932
Rheumatoid_arthritis_Okada_Y: https://www.ebi.ac.uk/gwas/publications/24390342
SLE_Bentham_J: https://www.ebi.ac.uk/gwas/publications/26502338
IBD_de_Lange_KM: https://www.ebi.ac.uk/gwas/publications/28067908
T1D_Forgetta_V: https://www.ebi.ac.uk/gwas/publications/32005708
Colorectal_CA_Tanikawa_C: https://www.ebi.ac.uk/gwas/publications/29471430
Breast_CA: http://bcac.ccge.medschl.cam.ac.uk/bcacdata/icogs-complete-summary-results/
Prostate_CA_Schumacher_FR: https://www.ebi.ac.uk/gwas/publications/29892016
CAD: https://data.mendeley.com/datasets/gbbsrpx6bs/1
AF: http://csg.sph.umich.edu/willer/public/afib2018/
Stroke_Traylor_M: https://www.ebi.ac.uk/gwas/publications/29531354
Heart_failure_Shah_S: https://cvd.hugeamp.org/dinspector.html?dataset=GWAS_HERMES_eu
SBP_Evangelou_E: https://www.ebi.ac.uk/gwas/publications/30224653
DBP_Evangelou_E: https://www.ebi.ac.uk/gwas/publications/30224653
Candidiasis: https://docs.google.com/spreadsheets/d/1kvPoupSzsSFBNSztMzl04xMoSC3Kcx3CrjVf4yBmESU/edit#gid=227859291
Chronic_viral_hepatitis: https://docs.google.com/spreadsheets/d/1kvPoupSzsSFBNSztMzl04xMoSC3Kcx3CrjVf4yBmESU/edit#gid=227859291
Herpes_simplex: https://docs.google.com/spreadsheets/d/1kvPoupSzsSFBNSztMzl04xMoSC3Kcx3CrjVf4yBmESU/edit#gid=227859291
COVID19_severe_Pairo-Castineira_E: https://www.ebi.ac.uk/gwas/publications/33307546
Tuberculosis_progression_Luo_Y: https://www.ebi.ac.uk/gwas/publications/31434886
Gout_Tin_A: https://www.ebi.ac.uk/gwas/publications/21768215
T2D_Mahajan_A: https://www.ebi.ac.uk/gwas/publications/29632382
Alzheimer_Jansen_IE: https://www.ebi.ac.uk/gwas/publications/30617256
Parkinson_Nalls_MA:https://drive.google.com/drive/folders/10bGj6HfAXgl-JslpI9ZJIL_JIgZyktxn
Depression_Major_Cai_N: https://www.ebi.ac.uk/gwas/publications/32231276
ALS_van_Rheenen_W: https://www.ebi.ac.uk/gwas/publications/27455348
Funding
Work of the authors is supported by the Canadian Institutes of Health Research grants to P.M. (FRN148778, FRN159697) and the Quebec Heart and Lung Institute Fund. Y.B. holds a Canada Research Chair in Genomics of Heart and Lung Diseases. S.T. and B.J.A. hold a scholarship from Fonds de Recherche du Québec-Santé (FRQS). P.M. is the recipient of the Joseph C. Edwards Foundation granted to Université Laval.
Author information
Authors and Affiliations
Contributions
L.H.M.M and P.M. conceived and designed experiments. S.M, L.H.M.M, M.S.S and P.M. performed MR analyses. L.H.M.M and P.M. performed network, enrichment and cross-trait analyses. L.H.M.M prepared the figures and tables. L.H.M.M and P.M. drafted the manuscript. All the authors critically reviewed the manuscript and provided intellectual inputs. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All datasets, including GWAS summary statistics were publicly available and did not require ethical approval.
Consent for publication
Not applicable.
Competing interests
No competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Suppl. Figure 1.
Glycosylation pathway for Sialyl-lewis x. FUT3 and ST3GAL6 encode for fucosyl transferase 3 and ST3 beta-galactoside alpha-2,3-sialyltransferase 6, respectively. They are involved in the synthesis of sialyl Lewis x. Suppl. Figure 2. Gene Ontology of ligand-receptor pairs derived from the blood candidate proteins. Ligand-receptor pairs were identified through publicly available database[24]. Gene Ontology enrichment analysis performed using the 43 identified ligands/receptors pairs and the GO molecular function database.
Additional file 2: Suppl. Table 1.
Mendelian randomization INTERVAL (P < 1E-03). Results of MR analysis for the nineteen causal candidate proteins in INTERVAL. Table includes: the aptamer and corresponding id code along with the protein name; the number of SNPs used as IVs in MR for each protein; the median, minimum and maximum F-statistics value for IVs; the beta, standard error and p-values for Inverse Variance Weighted and Weighted Median methods; heterogeneity tests (Cochrane’s Q test and intercept Egger test). Suppl. Table 2. Mendelian randomization INTERVAL (P < 1E-03) (all results). Results of MR analysis for all the proteins with enough IVs in INTERVAL. Table includes: the aptamer and corresponding id code along with the protein name; the number of SNPs used as IVs in MR for each protein; the median, minimum and maximum F-statistics value for IVs; the beta, standard error and p-values for Inverse Variance Weighted and Weighted Median methods; heterogeneity tests (Cochrane’s Q test and intercept Egger test). Suppl. Table 3. Mendelian randomization INTERVAL (P < 1E-05). Results of MR analysis for the nineteen causal candidate proteins in INTERVAL. Table includes: the aptamer and corresponding id code along with the protein name; the number of SNPs used as IVs in MR for each protein; the median, minimum and maximum F-statistics value for IVs; the beta, standard error and p-values for Inverse Variance Weighted and Weighted Median methods; heterogeneity tests (Cochrane’s Q test and intercept Egger test). Suppl. Table 4. Mendelian randomization INTERVAL (Wald ratio; P < 1E-05). Results of mendelian randomization with Wald ratio, using INTERVAL as the exposure. Wald ratio was calculated using the most significant SNP for proteins not presenting enough instruments for previously described two-sample mendelian randomization. Table includes: the aptamer and corresponding id code along with the protein name; the corresponding lead SNP identification; the beta, standard error and p-values for the GWAS outcome, the GWAS pQTL and for the Wald ratio. Suppl. Table 5. Reverse Mendelian Randomization for the nineteen causal candidates. Results of reverse MR for the nineteen causal candidate proteins identified in the study. Table includes: the aptamer and corresponding id code along with the protein name; the number of SNPs used as IVs in MR for each protein; the beta, standard error and p-values for Inverse Variance Weighted and Weighted Median methods; heterogeneity tests (Cochrane’s Q test and intercept Egger test). Suppl. Table 6. Mendelian randomization deCODE (P < 1E-03). Results of mendelian randomization for all nineteen causal candidate proteins, using deCODE [21] as the exposure. Table includes: the aptamer and corresponding id code along with the protein name; the number of SNPs used as IVs in MR for each protein; the median, minimum and maximum F-statistics value for IVs; the beta, standard error and p-values for Inverse Variance Weighted and Weighted Median methods; heterogeneity tests (Cochrane’s Q test and intercept Egger test). Suppl. Table 7. Mendelian randomization deCODE (P < 1E-05). Results of mendelian randomization for all nineteen causal candidate proteins, using deCODE [21] as the exposure. Table includes: the aptamer and corresponding id code along with the protein name; the number of SNPs used as IVs in MR for each protein; the median, minimum and maximum F-statistics value for IVs; the beta, standard error and p-values for Inverse Variance Weighted and Weighted Median methods; heterogeneity tests (Cochrane’s Q test and intercept Egger test). Suppl. Table 8. Mendelian randomization deCODE (Wald ratio; P < 1E-05). Results of mendelian randomization with Wald ratio, using deCODE [21] as the exposure. Wald ratio was calculated using the most significant SNP for protein not presenting enough instruments for previously described two-sample mendelian randomization. Table includes: the aptamer and corresponding id code along with the protein name; the corresponding lead SNP identification; the beta, standard error and p-values for the GWAS outcome, the GWAS pQTL and for the Wald ratio. Suppl. Table 9. Ligand-receptor pairs generated from causal blood candidate proteins. Ligand-receptor interactions for causal candidate blood proteins by using the comprehensive repository reported by Shao et al. [24]. Bold are causal candidate blood proteins. Suppl. Table 10. Gene Ontology (molecular function) for ligand-receptor pairs generated from the causal candidate proteins. Enrichment of all ligand-receptor pairs in Gene Ontology (GO Molecular Function). Suppl. Table 11. Network nodes (proteins) and degree. Degree for each protein in the network. Suppl. Table 12. Pathway enrichment (KEGG) for the network. Summary results of enrichment for all proteins in the network by using the Kyoto Encyclopedia of Gene and Genomes (KEGG) database. Suppl. Table 13. Cross-phenotype association analysis of HF by using iCPAG. Summary of iCPAG results. Trait 1 is the GWAS data from the HERMES study; Trait2 is the trait-disorder, which is compared to trait1 for sharing a similar genetic architecture. Reported in the table: p-value, FDR and Bonferroni adjusted p-value for the Fisher’s exact test; the Chao-Sorensen similarity index between trait 1 and trait 2; the list of SNPs in common between trait 1 and 2; links to corresponding experimental factor ontology (EFO) in EMBL-EBI database. Suppl. Table 14. Multi-trait MR. Table summarizing the 31 traits and diseases used in the multi-trait MR. Suppl. Table 15. Results of multivariable MR corrected for cardiovascular traits. Summary of multivariable MR and univariate MR for BAG3, MIF and APOA5. Univariate exposure is HF, whereas multivariate exposure is HF corrected for the selected trait in parenthesis. Reported are: estimate (beta), se (standard error) and p-value. DBP: diastolic blood pressure; SBP: systolic blood pressure; CAD: coronary artery disease. Suppl. Table 16. Therapeutic Target Database (TTD) for the causal candidate proteins. Summary results of druggable genome in the Therapeutic Target Database (TTD) for the causal candidate proteins. Reported are: the gene symbol and name; target type; disease for which there is an indication; drugs associated to the target. NA: not available. Suppl. Table 17. Drug Gene interaction Database (DGIdb) for the causal candidate proteins. Summary of druggable genome in the Drug Interaction database (DGIdb) for the causal candidate proteins. Reported are: the gene symbol; drug associated with the target; sources and pmids from the National Library of Medicine. Suppl. Table 18. Open Targets for the causal candidate proteins. Summary of druggable genome in the Open Targets for the causal candidate proteins. Reported are: gene symbol; drugs associated with the target; drug type (sm: small molecule, ab: antibody); category ab is the prediction confidence that the target is tractable for the development of an antibody.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Moncla, LH.M., Mathieu, S., Sylla, M.S. et al. Mendelian randomization of circulating proteome identifies actionable targets in heart failure. BMC Genomics 23, 588 (2022). https://doi.org/10.1186/s12864-022-08811-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08811-2