
Abstract
Background
Histone methylation occurs primarily on lysine residues and requires a set of enzymes capable of reading, writing, and erasing to control its establishment and deletion, which is essential for maintaining chromatin structure and gene expression. Histone methylation and demethylation are contributed to plant growth and development, and are involved in adapting to environmental stresses. The JmjC domain-containing proteins are extensively studied for their function in histone lysine demethylation in plants, and play a critical role in sustaining histone methylation homeostasis.
Results
In this study, a total of 21 JmjC domain-containing histone demethylase proteins (JHDMs) in birch were identified and classified into five subfamilies based on structural characteristics and phylogenetic relationships among Arabidopsis, rice, maize, and birch. Although the BpJMJ genes displayed significant schematic variation, their distribution on the chromosomes is relatively uniform. Additionally, the BpJMJ genes in birch have never experienced a tandem-duplication event proved by WGD analysis and were remaining underwent purifying selection (Ka/Ks < < 1). A typical JmjC domain was found in all BpJMJ genes, some of which have other essential domains for their functions. In the promoter regions of BpJMJ genes, cis-acting elements associated with hormone and abiotic stress responses were overrepresented. Under abiotic stresses, the transcriptome profile reveals two contrasting expression patterns within 21 BpJMJ genes. Furthermore, it was established that most BpJMJ genes had higher expression in young tissues under normal conditions, with BpJMJ06/16 having the highest expression in germinating seeds and participating in the regulation of BpGA3ox1/2 gene expression. Eventually, BpJMJ genes were found to directly interact with genes involved in the “intracellular membrane” in respond to cold stress.
Conclusions
The present study will provide a foundation for future experiments on histone demethylases in birch and a theoretical basis for epigenetic research on growth and development in response to abiotic stresses.
Similar content being viewed by others

Background
In eukaryotes, the histone octamer is composed of two copies of H2A, H2B, H3, and H4 histones wrapped in 146-bp double-stranded DNA [1,2,3,4]. In Arabidopsis thaliana, numerous studies identified post-translational modifications (PTMs) of histones on peptides residue tails such as methylation, acetylation, phosphorylation, ubiquitination, and sumoylation [5, 6]. Different modification occurs at various histones of nucleosomes. For example, the lysine is methylated, acetylated, and ubiquitinated; arginine is methylated; and phosphorylation of threonine and serine. Histone methylation has powerful impacts on gene transcription and chromatin structure and could be pass on to the next generation through mitosis and meiosis [7].
Histone methylation is a reversible dynamic regulation process that involves methylation and demethylation. Histone methylation takes place primarily at the N-terminal arginine (R) and lysine (K) sites of histones H3 and H4, namely K4, K9, K27, and K36 for H3 and K20 for H4 [8]. S-adenosylmethionine was used as a methyl donor to target the N-terminal lysine (K) and arginine (R) of histone H3 and H4 through histone methyltransferases (HMTs) [9]. The protein family of SET or PRMT domains, which are primarily responsible for the methylation of histone lysine and arginine residues, has received considerable attention so far in plant histone methyltransferases studies [10]. Lysine residues can be monomethylated (Kme1), dimethylated (Kme2), or trimethylated (Kme3), while arginine can only be monomethylated (Rme1), symmetric dimethylated (Rme2s), or asymmetrically dimethylated (Rme2a) [11]. The methylation of histones has different impacts on gene expression; for example, H3K9 (H3K9me2/3) and H3K27 (H3K27me3) methylation are associated with transcriptional inhibition, while H3K4 (H3K4me2/3) and H3K36 (H3K36me3) methylation are associated with transcriptional activation [12]. These modifications serve as a kind of “histone code” for chromatin functions and transcriptional activities [13, 14]. As a result, histone alteration homeostasis is essential for plants’ normal growth and development [15].
Histone demethylation is primarily catalyzed by histone demethylase 1 (LSD1) and the protein containing the JmjC domain through amine oxidation and hydroxylation [16, 17]. Furthermore, the cofactors required by these two classes of histone demethylases show the different catalytic mechanisms: KDM1/LSD1 is a flavin adenine dinucleotide (FAD)-a dependent enzyme that can only remove single/double lysine residue methylation. However, JmjC domain-containing proteins that use Fe (II) and -ketoglutarate (KG) as cofactors will catalyze the removal of mono/di/tri-lysine residue methylation [18].
Proteins containing JmjC domain can be divided into 8 categories according to sequence similarity and catalytic specificity, namely KDM5/JARID1 group, KDM4/JHDM3 group, KDM3/JHDM2 group, JMJD6 group, JmjC domain-only group, KDM6/JMJD3 group, KDM2/JHDM1 group and PHF group [19]. However, only the first five subfamilies were found in the plant so far. Different JHDM subfamilies can catalyze the demethylation of different histones. For example, KDM5/JARID1 group proteins have been found to catalyze the demethylation of H3K4me1/2/3 [20,21,22,23,24], KDM4/JHDM3 family proteins are shown to demethylate histone H3K9me2/3 actively, and H3K36me2/3 [25,26,27], KDM3A/JHDM2 can demethylate H3K9me2 and H3K9me1 but not H3K9me3 [28], JMJD6 group can demethylate H3R2me2 and H4R3me2 [29], and JmjC domain-only subfamily can remove the methylation of H3K27me3 [30].
The JHDM gene family in different species shows various tissue-specific expression patterns. For example, in cotton, the JHDM gene family is mainly expressed in 20 and 30 days post-anthes (DPA) fibers [31]. In maize, the expression level in roots, stems, and leaves are relatively low, and also different genes exhibit various expressions in different tissues. For example, ZmJMJ7 and ZmJMJ17 are lower in silks and seedings, while ZmJMJ16 is higher in shoot tips [32]; In soybeans, the JHDM genes show a relatively low expression level in four tissues (pods, pod seeds, roots, and seeds) but a higher expression in the flower, leaf, and shoot meristem [33]. Similarly, in Rosa chinensis, it was worth noting that most of the JHDM genes were higher expressed in reproductive tissues, especially in floral meristem and closed flowers, than vegetative tissues [34].
In Arabidopsis thaliana, the AtJMJ14, AtJMJ15, and AtJMJ18 can demethylate H3K4 and participate in the regulation of flowering time and female gametophyte development [35,36,37]. AtJMJ11/ELF6 (EARLY FLOWERING 6) and AtJMJ12/REF6 (RELATIVE OF EARLY FLOWERING 6) play an opposite role in the flowering inhibition pathway, respectively [38, 39]. Additionally, AtJMJ30 and AtJMJ32 can regulate histone demethylation at FLC sites to prevent premature flowering under high-temperature treatment [40]. AtJMJ20 and AtJMJ22 can promote the expression of GA3ox1/2 by changing the histone methylation status of H3K4 and H3R4, thereby affecting the germination of seeds [41]. In response to stress, AtJMJ17 can directly bind to OST1 (OPEN STOMATA 1) and adjust the expression level of OST1 by removing the methylation of H3K4me3 in response to drought [42]. Overexpression of AtJMJ15 in Arabidopsis directly results in a significant increase in salt tolerance [43]. It was also found in soybeans that salt treatment would increase the H3K4 trimethylation level and decrease the H3K9 methylation level of some genes [44]. In Medicago truncatula, MtJMJ05 can produce alternative splicing in response to cold stress [45]. Above all, different abiotic stresses can modulate JHDM gene expression to control the growth and development of the plant under unfavorable conditions.
The present study is focused on a perennial woody tree (birch), which is mainly distributed in high latitudes and has important ecological and economic value. Previous studies on the JHDM gene family mainly focused on annual herbaceous plants, but little is known in perennial woody plants. Here, we performed a comprehensive analysis of the JHDM family genes in birch, including classification and architecture, chromosomal location, duplication events, Ka/Ks analysis and a functional analysis of cis-acting elements, tissue-specific expression patterns, and transcriptome profiling under different abiotic stress conditions.
Results
Identification and classification of the \( JHDM \) gene family in brich
In order to identify all possible homologs of the JHDM gene family in birch, the full-length amino acid sequences of the JHDM proteins determined in Arabidopsis, rice, and maize were performed to query the protein sequence database of the Betula pendula genome with blastp program. After de-forgery and de-redundancy, 21 JHDM genes were identified and named BpJMJ01 to BpJMJ21 (Table S1).
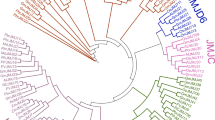
To examine the evolutionary history of these protein families in these four species and to establish the phylogenetic relationship among JHDM family genes, the phylogenetic tree was constructed with MEGA6.0 based on the neighbor-joining algorithm with full-length JHDM protein sequences of birch (21), Arabidopsis (21), rice (20), and maize (21) (Fig. 1). Based on the branching characteristics and bootstrap values in the phylogenetic tree, a total of 21 BpJMJ genes are classified into five major subfamilies: JARID1/KDM5 (4 genes), JHDM3/KDM4 (4 genes), JHDM2/KDM3 (8 genes), JHDM6 (3 genes) and JmjC-domain only subfamily (2 genes), respectively (Table S1). Interestingly, more than 70% of BpJMJ proteins share the highest protein sequence similarity with Arabidopsis homolog (Blastp P-value = 0) (Table S2), showed that the birch is a dicotyledonous tree species with 5 JHDM superfamilies’ simultaneously and has a closer genetic relationship with Arabidopsis, and away from rice and maize as monocotyledonous species.

Phylogenetic analysis of JHDM proteins in maize, rice, Arabidopsis, and birch. The rooted neighbor-joining (NJ) phylogenetic tree of JHDM family was clustered with bootstrap values shown for each clade in a different color. The BpJMJ family have been highlighted for each group with a red \( \mathrm{circle} \). Five clades are marked as JARID1, JHDM3, JHDM2, JHDM6, and JmjC-only, respectively
Furthermore, the 21 filtered JHDM amino acid sequences were analyzed in the Expasy website, including isoelectric point (PI) and molecular mass (kDa). The results show that the amino acid length of BpJMJ proteins ranges from 404 amino acids of BpJMJ19 to 2205 amino acids of BpJMJ13. In addition, the molecular mass ranges from 45,702.96 to 246,534.29 Da, indicating that the protein properties of JHDMs in birch have a wide change (Table S1).
The structures of identified JHDM genes and protein in birch
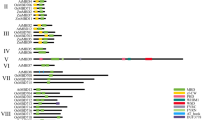
The structural heterogeneity of individual BpJMJ genes by exon-intron and corresponding domain architecture was investigated, and the variation among them is unprecedented. The average exon count for BpJMJ family is 13.52 per gene; the exon count varies considerably, ranging from 3 exon of BpJMJ06 to 36 exon of BpJMJ10 (Fig. 2A). The BpJMJ10 gene has the highest exon (36) and the most extended nucleic acid sequence in the JARID1/KDM5 subfamily (29,078 bp). The BpJMJ07, BpJMJ08, and BpJMJ17, BpJMJ18 share a similar exon-intron structure in JHDM2/KDM3, except for BpJMJ05, BpJMJ09, and BpJMJ20. Furthermore, transposon insertion is likely to cause BpJMJ genes with very long introns, such as BpJMJ08, BpJMJ09, BpJMJ10, BpJMJ14, and BpJMJ17, which may influence the expression of the corresponding gene and cause variable splicing.

Schematic structure of JHDM genes in birch. The \( \mathrm{nucleic} \) or protein sequence is represented as a black line; the N terminus is left for each. The length of each JHDM gene can be estimated using the scale at the bottom. A Exons and introns are indicated by red boxes and single lines, respectively. The boxes in the dark blue represent the untranslated regions (UTRs). B Different classes of motifs are shown with boxes in different colors. Only the top 20 most frequently occurring motifs are displayed. C Conserved domains are identified in each BpJMJ genes subfamily. The location and size of domains are shown by different color rectangles annotated in the legend
Further, the JHDM protein sequences were submitted to the MEME website with the default parameters, and up to 20 motifs (motif01-motif20) with a width between 6 and 50 were analyzed. The width, sites, and E-value of predicted conserved motifs for each BpJMJ protein are provided in Fig. S1, and Table S3. According to the sequence motif similarity, BpJMJ family could be classified into three major groups. The first group consists of the JARID1 and part JHDM3 families with 4.75 motifs on average, and the second group consists of a single family of JHDM2 proteins with 13 motifs on average. The third group, including JHDM6 and JmjC-domain family, shares an average of 1.4 motifs (Fig. 2B). The result is similar to the classification of JHDM families in the phylogenetic tree.
To know more about the BpJMJ protein domain architecture diversity. All BpJMJ protein sequences were submitted in the PFAM and SMART websites with default parameters. Based on the evolutionary relationship of JHDM family in the previous phylogenetic tree, the BpJMJ family can be divided into five subfamilies, including (a) JARID1/KDM5 subfamily (4), (b) JHDM3/KDM4 subfamily (4), (c) JHDM2/KDM3 subfamily (8), (d) JHDM6 subfamily (3) and (e) JmjC-only domain subfamily (2) (Fig. 1). In the (a) JARID1/KDM5 subfamily and (b) JHDM3/KDM4 subfamily, except that BpJMJ03 lacks a JmjN domain and BpJMJ13 lack a zf-C5H2, all other protein sequences share similar domain architecture, mainly including JmjN, JmjC, and zf-C5H2. In addition, for the JARD1 subfamily, BpJMJ04 and BpJMJ14 also have additional FYRN and FYRC domains, which may have the ability to bind chromatin in the trithorax/ALL1 protein family [19]. BpJMJ10 has ARID, PHD, and PLU domain which shows high similarity with ZmJMJ10 in maize (Fig. 2C, and Table S4). And also, the BpJMJ13 has four zf-CCCH domains at the N-terminal as well, which is a short zinc-binding domain that has the pattern of three cysteines and one histidine to coordinate the zinc ion. In the (c) JHDM2/KDM3 subfamily, a total of 8 proteins mainly share only the JmjC domain and zf-4CXXC_R1 domain, but not the JmjN domain. All members, in both the (d) JHDM6 subfamily and (e) JmjC-domain only subfamily, share the same Cupin-8 domains, but there will be another JmjC domain in (d) JHDM6 subfamily and Cupin-4 in (e) JmjC-domain subfamily only (Fig. 2C). In summary, the JmjC domain is the most widespread architecture of the protein domain, which belongs to the Cupin superfamily and maybe a protein hydroxylase that catalyzes histone lysine demethylation [46].
In addition, two cofactors, Fe (II) iron and alpha-KG, mainly interacting with five key amino acid residues, are essential sites for the process of histone demethylation. The first group, including three amino acid residues (His188, Glu190, and His276), is indispensable for Fe (II) iron-binding, and the second group including two amino acid residues (Thr185 and Lys206) required for αKG binding.
To further confirm whether these conserved residues bound by cofactors had diverged among the BpJMJ proteins, all JHDM proteins from Arabidopsis, rice, and birch were performed with homologous alignment. The JHDM proteins can be divided into 2 categories depending on the retained amino acid residues of the alignment results: the first category includes the JARID1/KDM5 and the JHDM3/KDM4 subfamily, primarily His (H), Glu (E), and His (H) for Fe (II) binding, and the JHDM2/KDM3, JHDM6 and JmjC-only domain subfamily, mainly Phe (F) and Lys (K) for alpha-KG binding (Fig. S2); the second category includes JARID1/KDM5 subfamily and JHDM3/KDM4 subfamily, mainly His (H), Asp (D) and His (H) for Fe (II) binding, and in JHDM2/KDM3, JHDM6 and JmjC-only domain subfamily, mainly Thr (T) and Lys (K) for alpha-KG binding (Fig. S3).
These two categories of conserved amino acid residues for Fe (II) and α-KG binding can both exert histone demethylation function [47, 48]. The alignment results show that the most conserved amino acid residues for Fe (II) and α-KG binding sites among JHDM proteins in each category are same. However, there are still some JHDM protein whose amino acid residues are variable. For example, in the protein sequence of BpJMJ03, the first and third amino residues of the Fe (II) binding site are changed from His (H) to K (Lys) and Y (Tyr) separately, in BpJMJ01 and BpJMJ06, the first amino acid residue of the binding site of alpha-KG is changed from T (Thr) to S (Ser) and A (Ala) (Fig. S2, and Fig. S3). Taken together, the cofactors binding sites (Fe (II) and α-KG) composed of the five amino acid residues are quite important for the function of histone lysine demethylation in birch.
Chromosomal location and gene duplication of JHDM gene family in birch
The physical position of 21 BpJMJ genes, 19 of which are more evenly spread over the whole chromosome, was explored to elucidate the spatial distribution of the BpJMJ genes family in each chromosome. Unexpectedly, the other two genes (BpJMJ04 and BpJMJ09) were annotated on the contigs, which are not assembled into the genomic chromosomes (Fig. 3A).

Chromosomal localizations, gene duplication events, and Ka/Ks analysis of BpJMJ genes. A. The distribution of the 21 BpJMJ genes was mapped on the 14 chromosomes and 2 contigs. The chromosome numbers are indicated at the top of each vertical gray bar. The gene names on each chromosome’s right side correspond to the approximate locations of each BpJMJ gene. The scale on the left is in megabases. B The synteny analysis of the BpJMJ genes between birch and five representative plant species. Grey lines indicate the collinear blocks between birch and other plant genomes, while the red lines indicate the syntenic BpJMJ gene pairs. The plant named different prefixes ‘B. pendula’, ‘A. thaliana’, ‘G. max’, ‘P. trichocarpa’, ‘O. sativa’ and ‘Z. mays’ represent Betula. pendula, Arabidopsis thaliana, Glycine max, Populus trichocarpa, Oryza sativa, and Zea mays. C The ratio between Ks and Ka for paralogous BpJMJ gene pairs in birch
Moreover, gene duplication events are frequently associated with the plant evolution process and contribute to the gene family’s expansion [49, 50]. Here, the evolutionary patterns of the BpJMJ genes were surveyed in MCScanX software, and amazingly we found that there are no tandem duplications for BpJMJ genes (Table S5), speculating that the BpJMJ genes family in the long evolution of brich has never experienced a tandem-duplication event.
To explore the potential evolution relationship and further compare the JHDM gene family collinearity among different species, a comparative analysis between the BpJMJ protein and homologues from the other five representative plants, including Arabidopsis thaliana, Glycine max, Populus trichocarpa, Oryza sativa, and Zea mays, was performed. The results show that there are 6(28.5%), 15(71.4%), 16(76.2%), 4(19.0%), and 1(4.8%) BpJMJ proteins show high homology to the members from the other five species, respectively (Fig. 3B). Conclusively, the JHDM gene family in birch has more collinearity with dicotyledonous plants (Arabidopsis thaliana, Glycine max, and Populus trichocarpa), but shows a little intersection with monocotyledonous plants (Oryza sativa and Zea mays), indicating that the JHDM genes family has undergone evolutionary divergence in monocotyledonous and dicotyledonous plants.
The Ka/Ks ratios of the paralogous genes measured in the JHDM gene subfamily are all less than 1 (Fig. 3C), indicating that the JHDM gene family undergoes purifying selection during evolution, which helps to understand the evolution of the birch JHDM gene family in dicotyledonous plants. Furthermore, the divergent period of the JHDM gene family in birch (woody plant) has been traced back to 52.19 MYA (Table S6), even earlier than in Arabidopsis (herb) (9.6–16.1 MYA) [51].
The cis-acting regulatory elements in the promoter of JHDM genes in birch
To elucidate the JHDM gene family regulatory mechanism about abiotic or biotic stress in birch, the genomic sequence of 2 kb upstream promoters of birch JHDM family genes was used to query the Plant Care database to search for cis-regulatory elements (CREs). The 13 classes of CREs related to hormone (ABRE, CGTCA-motif, TGACG-motif, ERE, and TCA-element), light reaction (G-box, GT1-motif, TCT-motif, and Box-4), and stress (ARE, W-box, and LTR) were detected (Fig. 4, and Table S7). Each predicted CRE exists in at least 9 JHDM genes, and each gene has at least 6 CREs distribution (Fig. S4). In detail, hormone-related CREs, including ABRE (abscisic acid-responsive), CGTCA-motif (MeJA-responsive), TGACG-motif (MeJA-responsive), TGA (salicylic acid-responsive), ERE (ethylene-responsive) and TCA-motif (salicylic acid-responsive), has largest proportion and distributed in 20, 17, 14, 12 and 9 BpJMJ genes, respectively. Further, followed by CREs related to light response, including G-box, GT1-motif, TCT-motif, and Box-4, are distributed in 18, 17, 16, and 14 JHDM genes, respectively. The last is stress-related CREs, including ARE (anaerobic induction), W_box (wounding and pathogen responsive), and LTR (low-temperature responsive) are distributed among 19, 13, and 10 JHDM genes, respectively (Fig. S5). Various environmental variables, such as hormones, light, and low-temperature stress, disrupt the transcriptional control of BpJMJ genes.

Cis-elements in the promoter sequences of the 21 BpJMJ genes. Distribution of cis-acting elements related to hormone, light, and stress in the promoter sequences of the 21 BpJMJ genes. Putative ABRE, W-box, TGA-element, ERE, TCA-element, CGTCA-motif, G-box, GT1-motif, ARE, TGACG-motif, TCT-motif, Box 4, and LTR core sequences are represented by different symbols as indicated in legend at the right. The 2 kb sequences upstream of the initiation codon (ATG) of the JHDM genes can be estimated using the scale per 300 bp above
Transcriptome analysis and role of JHDM genes in seed germination of birch
To better understand the potential roles of the BpJMJ genes family in plant growth and development, a comprehensive expression analysis was accomplished based on tissue-specific transcriptome data of two-year-old birch released by Chen et al. 2019 [52]. It can be seen from the heatmap that, except for BpJMJ11, the expression of the other 20 BpJMJ genes in various tissues is quite different. The majority of BpJMJ genes are heavily expressed in young tissues, such as birch roots and flowers (Fig. 5A, and Table S8). All members of the JHDM2 subfamily, as well as the vast majority of the JHDM3 subfamily (BpJMJ02, BpJMJ13, and BpJMJ15), the JARID1 subfamily (BpJMJ03 and BpJMJ14), and the JHDM6 subfamily (BpJMJ06 and BpJMJ16) are highly expressed in floral tissues and partially in leaf tissues. Interestingly, the JmjC-only subfamily has a high level of expression in mature tissues. For instance, BpJMJ17 is highly expressed in the xylem, while BpJMJ19 is highly expressed in the leaves.

Tissue-specific expression profiles of BpJMJ genes in birch. A. The hierarchical clustering of expression profiles of 21 BpJMJ genes in four tissues. The color scale representing gene TPM (Transcripts Per Million) is shown on the right. The cell in yellow represents low gene expression, and the blue one indicates high gene expression. The different tissues are noted on the bottom of each line. Cluster dendrograms are shown on the left and above, respectively. B. The tissue-specific expression pattern of BpJMJ genes and their positive correlation with intron number. The significance of the difference was determined using the t.test. C. Tissue-specific relative expression of BpJMJ06/16 and GA3ox1/2 examined by qRT-PCR. The qRT-PCR data was normalized using the birch actin gene and are shown relative to the stem. The X-axes show different tissues (stem, root, leaf, ungerminated seeds (Seed#1), and germinating seeds (Seed#2)), and y-axes are scales of relative expression level. Bars represent standard deviations (SD) of three technical replicates
Furthermore, the JHDM genes have a huge diversity in their gene structures within the same subfamily, which is often displayed in the number of introns (Fig. 2A). Previous research has shown that increasing the number of introns will improve mRNA stability and promote gene expression [53]. As a result of evaluating the relationship between the number of introns in each JHDM family gene and their expression levels in various tissues, the number of introns in the JHDM gene was strongly positively correlated with gene expression in the roots and flowers (Table S9). The importance of variations in the expression of JHDM genes with different numbers of introns was investigated, and it was revealed that the JHDM gene with more introns expressed higher in the young tissues (Fig. 5B). For example, the BpJMJ07, BpJMJ09, BpJMJ10, and BpJMJ18, with 14, 13, 15, and 15 introns respectively, are relatively highly expressed in roots and flowers.
On the other hand, previous studies have shown that histone modifications can regulate plant growth and development in response to changes in the external environment. Cho et al. (2007) [41] have reported that JMJ20/22 of Arabidopsis could regulate the expression of GA3ox1/2 in response to light, thereby affecting seed germination. The BpJMJ06/16 in birch are the exact homologous genes of AtJMJ20/22. The expression of BpJMJ06/16 was quantified using qRT-PCR in various tissues, including stem, root, leaves, ungerminated seeds, and germinating seeds. The results show that the BpJMJ06/16 were highly expressed in germinating seeds and maintained a relatively low expression in other mature tissues (Fig. 5C). Additionally, the BpGA3ox1/2 shows a similar tissue-specific expression pattern with BpJMJ06/16, suggesting that BpJMJ06/16 could promote the germination of birch seeds which may affect the expression of BpGA3ox1/2 by regulating histone methylation modification.
Transcriptome analysis of birch JHDM gene family in response to abiotic stress
To investigate the JHDM gene family’s expression in response to abiotic stress. The Spearman-correlation analysis between the number of 13 cis-acting elements in the BpJMJ gene promoter regions and their expression in different tissues revealed that LTR exhibits a significant correlation among the flowers, leaves, and roots (Table S10). Furthermore, BpJMJ genes with LTR elements have significantly lower expression in birch leaves than non-LTR BpJMJ genes (Fig. 6A), implying that the LTR factor will react to the external low temperature by down-regulating gene expression.

The expression profiles of BpJMJ genes response to abiotic stress. A. The tissue-specific expression pattern of BpJMJ genes with/without LTR elements. The t. test was used to determine the significance of the difference, and the dashed box indicates the two groups of genes with larger differences for subsequent verification. B. The BpJMJ gene expression with cold stress. The color scale representing gene TPM (Transcripts Per Million) is shown on the right. The cell in yellow represents low gene expression, and the blue one indicates high gene expression. The horizontal axis represents the cold treatment time of birch, including 0.5 h, 1.0 h, 1.5 h, 2.0 h, 2.5 h, and 3.0 h, and the vertical axis represents 21 BpJMJ genes clustered by their TPM. C. The BpJMJ gene expression with salt stress. The JHDM genes with significant differences are shown above. The red bar represents control, the green bar represents 200 mM NaCl salt treatment, and the ordinate represents TPM. “*” means that the P-value calculated by the t-test is less than 0.05
To further validate this hypothesis, the cold-treated (6 °C) transcriptome of the two-month-old birch from the State Key Laboratory of Northeast Forestry University was used for analyzing the gene expression pattern. The findings revealed that low-temperature stress-induced all JHDM genes in birch with no quantitative preference, but there were two opposing expression patterns: (I) JHDM gene up-regulation (11 genes) and (II) JHDM gene down-regulation (10 genes) in 1 h (Fig. 6B, and Table S11). Surprisingly, genes containing LTR elements, such as BpJMJ12, BpJMJ16, BpJMJ21, and BpJMJ06, were down-regulated, while genes without LTR elements, BpJMJ07, BpJMJ19, and BpJMJ20, were high expression under cold stress conditions (Fig. 6B). Taken together, the BpJMJ gene containing LTR elements could be down-regulated in response to low temperature.
On the other hand, in the JARID1/KDM5 subfamily and JmjC-domain only subfamily, the genes included BpJMJ03, BpJMJ04, BpJMJ10, BpJMJ14, BpJMJ17, and BpJMJ19 are belong to the up-regulated genes, the JARID1/KDM5 subfamily genes are involved in the H3K4me1/2/3 demethylase. In the JHDM3/KDM4 subfamily, all genes (BpJMJ02, BpJMJ11, BpJMJ12, and BpJMJ13) are inhibited by cold stress. The above-mentioned two JHDM gene expression patterns may eventually result in the down-regulation of the whole genome H3K4me1/2/3 level and the up-regulation of the H3K9me2/3 level. There are differences in expression patterns with low-temperature stress in the JHDM2/KDM3 subfamily and the JHDM6 subfamily. Four genes (BpJMJ05, BpJMJ07, BpJMJ09, BpJMJ20) and one gene (BpJMJ01) are up-regulated, and the other four genes (BpJMJ08, BpJMJ15, BpJMJ18, BpJMJ21) and 2 genes (BpJMJ16, BpJMJ06) expressions are inhibited by cold stress, which may lead to down-regulation and up-regulation of H3K9me1/2 and H3K27me2/3 in the whole genome of birch. Above all, the gene expression of the JHDM family is severely affected by low-temperature stress.
Moreover, the salt treatment can also affect the expression of the JHDM subfamily. By sequencing the transcriptome of the leaves after salt treatment, it was found that a total of 7 BpJMJ genes expression was inhibited by 200 mM salt treatment, which was distributed among JARID1 (BpJMJ04, BpJMJ10), JHDM3 (BpJMJ02, BpJMJ12), and JHDM2 (BpJMJ07, BpJMJ08) subfamily (Fig. 6C, and Table S12). There were no significantly up-regulating for BpJMJ genes during salt treatment (Fig. S6). In summary, salt treatment could have inhibited the expression of some BpJMJ genes in leaves of birch.
Co-expression of low-temperature stress genes in the \( BpJMJ \) gene family
To further study the mechanism of the birch JHDM gene family in response to low-temperature stress, the 16 transcriptome profiles under cold stress were used to construct a gene co-expression network by WGCNA. There were a total of 28 expression pattern modules identified (Fig. S7). A total of 6 BpJMJ genes equally participate in three co-expression network modules with a weight threshold of Pearson correlation coefficient (greater than 0.1). The largest co-expression network (network1) contains 2020 genes and 851,957 interactions, with 877 (43.42%) genes having close links to BpJMJ04 and BpJMJ20 (Fig. S8). The second interaction network (network2) comprises 377 genes and 7663 interactions, with 81 genes (21.49%) related to BpJMJ01 and BpJMJ07. Lastly, the smallest co-expression network (network3) consists of 255 genes and 9663 interactions, with 112 genes (43.92%) related to BpJMJ10 and BpJMJ13 (Fig. 7A).

The co-expression network for the low-temperature response BpJMJ genes and GO enrichment analysis. A. Co-expression network. On the left is the complete gene expression network, and on the right is the network of genes directly related to BpJMJ genes. The green dots indicate genes that are not directly related to BpJMJ genes, and the red dots indicate genes directly related to BpJMJ genes. B. GO enrichment analysis. The ordinate represents the abundance of enrichment, the color of the histogram represents the -log10(P-value), the terms in blue, red and green color represent the GO enrichment result of BpJMJ-directly genes from network1, network2 and network3, respectively
Furthermore, gene ontology (GO) analysis was performed on the co-expression genes directly related to the BpJMJ genes in each of the three networks. By dissecting the co-expressed genes, 600 co-expression genes were found to show a significant relationship with GO term “intracellular part” (Fig. 7B), of which 156 genes were in the cellular components of network2 and network3, and 444 genes are involved in intracellular transport in network1. The trait of resistance to cold has been identified in particular concerning intracellular signal transduction, which can effectively improve the cold resistance of plants by promoting the stability of intracellular membranes [54]. In addition, the term “calcium ion channels” was found in go enrichment result of the network1, which has been extended to have a response to cold stress (Fig. S9). Similar to network3, some terms related to the proteasome and ubiquitin degradation were also detected (Fig. 7B). Lastly, some co-expression genes were shown to be related to seed dormancy, lipid storage, chromosomal reorganization, response to freezing, and inhibition of gene expression in network2.
Discussion
Histone methylation is a post-translational modification of specific amino acids that regulates gene expression by modulating transcription factor accessibility to target genes. Histone methylation is linked to a variety of complex biological functions, including heterochromatin formation and transcriptional regulation. Histone methylation is implicated in the repression or activation of gene expression. The methylation and demethylation of histone modifications have been linked to chromatin state in plants, and this has been shown to influence plant growth and development. Histone demethylases with the JmjC domain, which is strongly conserved in plants, play an important role in preserving histone methylation homeostasis in vivo. In animals and plants, proteins with the JmjC domain belong to a large family of histone lysine demethylases that play an important role in histone modification, which is an important aspect of epigenetics. In this study, a comprehensive group of 21 JHDM genes was identified and characterized from Betula pendula genome, including phylogenetic tree, gene structure, domain and motif architecture, chromosome location, duplication events, Ka/Ks analysis, expression profiles and WGCNA analysis.
The phylogenetic study revealed that the JHDM gene family is highly conserved in both perennial woody plants and annual herbs, with 21 BpJMJ genes divided into five subfamilies: JARID1(4), JHDM3(4), JHDM2(8), JHDM6(3), and JmiC-only (2) in birch. Furthermore, since the JHDM gene family in birch has not undergone large-scale gene duplication events, its number of genes is quite similar to those of Arabidopsis (21) [55], rice (20) [56], and maize (19) [32], demonstrating the JHDM gene family’s significance in plant evolution. However, gene collinearity analysis proved that, compared with monocots, there are more homologous JHDMs among birch and dicots. This may cause by the loss of gene family, such as the absence of JHDM6 and JmiC-only subfamily in maize [32], which gives rise to the evolutionary divergence of the JHDM gene family in monocots and dicots.
We also investigated the composition of birch JHDM proteins and discovered some preliminary structural characteristics. Except that BpJMJ03 lack of a JmjN domain and BpJMJ13 lack of a zf-C5H2, the JARID1/KDM5 and JHDM3/KDM4 subfamilies share identical domain architecture, with JmjN, JmjC, and ZF-C5H2 being the most common. BpJMJ04 and BpJMJ14 both have additional FYRN and FYRC domains for the JARD1 subfamily, which may have the capacity to bind chromatin in the trithorax/ALL1 protein family [19, 57]. Through the interaction of FYRC domains with transcription factors, AtJMJ14 has been shown to play a role in Arabidopsis flowering time regulation [35, 58]. A total of 8 proteins in the JHDM2/KDM3 subfamily share only the JmjC domain but not the JmjN domain. All genes share Cupin-8 domain in the JHDM6 subfamily and JmjC-domain only subfamily, but there will be another JmjC domain in the JHDM6 subfamily and Cupin-4 in the JmjC-domain only subfamily. The JmjC domain, which belongs to the Cupin superfamily and maybe a protein hydroxylase that catalyzes histone lysine demethylation, is the most common protein domain architecture.
The divergence time for paralogous BpJMJ genes was furtherly calculated. The divergence time for paralogous gene pairs indicates that their divergence occurred much earlier than that of A. thaliana (9.6–16.1 MYA). Generally, if the value of Ka/Ks is < 1, it indicates gene pairs may have evolved from purifying selection (also called negative selection); Ka/Ks = 1 suggests neutral selection, while Ka/Ks > 1 means positive selection [51].
The JHDM gene family has been shown to have a variety of functions, including involvement in growth and development processes such as cell division and differentiation, such as flowering [59, 60], photoperiod [59], and hormone stress [61], as well as the ability to reverse methylation at multiple sites, including H3K4, H3K9, and H3K27. Recent studies have reported that several histone modifications, including H3K4me3, H3K9ac, H3K9me2, H3K23ac, H3K27ac, H3K27me3, and H4ac, along with DNA methylation response to abiotic stresses, such as drought stress, salt stress, and temperature fluctuations [62, 63]. Furthermore, similar research in maize has confirmed that most ZmJMJ gene expressions were promoted by heat stress [32]. To further investigate the response of the JHDM gene family to abiotic stress in birch, the transcriptome of low-temperature treatment was used to reveal the BpJMJ gene expression pattern. After being exposed to low-temperature stress for 1 h, the expression of 11 BpJMJ genes was significantly up-regulated, whereas the expression of the other ten genes was inhibited. These two expression categories have some links with the class of the JHDM gene subfamily. For example, the expression of all genes in the JARID1/KDM5 subfamily is up-regulated by cold stress, while the expression of all genes in the JHDM3/KDM4 subfamily is down-regulated under cold stress [64]. Similarly, the expression of the JARID1 family can be up-regulated with high-temperature stress in maize, so it is speculated that temperature-induced stress can affect the expression of JARID1 subfamily genes among different species.
The Low temperature-responsive elements (LTRE), the basic sequence is ‘CCGAAA’, act as cis-element involved in low-temperature responsive. LTREs were also observed with very relative low frequencies in gene promoter sequences that are functionally involved in condition-specific heat stress and with hexamer (CCGAAA) signature [65]. However, so far, the research on the response mechanism of LTRE has mainly focused on heat shock proteins (HSPs). Previous studies have shown that the two elements, heat shock element (HSE) and LTR, have a synergistic effect in the response of HSPs in green macroalga to cold stress [66]. However, there are relatively few studies on LTRE in other genes. Previous studies on JmjC genes found that at least in maize, cotton and cabbage, its gene expression will be affected severely by low-temperature stress. Therefore, the correlation between the LTR element in the promoter region of the BpJMJ gene and its expression under low-temperature stress was performed to analysis, and found that the existence of LTREs may inhibit the expression of BpJMJ genes under low-temperature conditions, with a similar phenomenon in cotton [32].
Eventually, through sequence alignment and phylogenetic relationship analysis, BpJMJ06/16 were found to be homologous genes to Arabidopsis JMJ20/22. It was documented that it could affect seed germination by modulating the dynamic balance of H3K9me3 and H3K4me3 in response to the external environment. Complete loss of PRC2 activity leads to derepression of seed maturation and dormancy genes at the seedling stage and subsequently compromises germination [67]. Here, in order to analyze whether BpJMJ06/16 plays a role in the germination of birch seeds, the gene expression in different tissues was verified by qRT-PCR. It was found that the expression level in the mature tissues was relatively low, but it was highly expressed in germinating seeds, suggesting that in young tissues, it may be necessary to enhance the expression of genes that activate seed germination by increasing the modification of H3K4me3. The genes, GA3ox1 and GA3ox2, have been expressed mainly in the embryonic axis and directly regulated by JMJ20 and JMJ22 in Arabidopsis [41]. Here, the expression of GA3ox1/2 in birch was detected by qRT-PCR, which shows a similar tissue-specific expression pattern with BpJMJ06/16, so it is speculated that it may have the same regulatory mechanism as in Arabidopsis.
Conclusion
In this study, we identified a total of 21 JHDM genes in birch, which were evenly distributed between 12 chromosomes and two contigs. Based on amino acid similarities, these BpJMJ genes were divided into five subfamilies. Despite the wide structural variation in exon/intron, functional domains and amino acid residues were more conserved among genes in the same subfamily. Phylogenetic clustering and collinearity analysis revealed that the BpJMJ genes have a closer relationship with dicots, and its divergence occurs even earlier than Arabidopsis and other cruciferous plants. The transcriptome profiles showed that the BpJMJ gene is mostly expressed in young plant tissues, including flowers and roots, and that BpJMJ20/22 is mostly expressed in germinating seeds, as confirmed by qRT-PCR. Moreover, a detailed expression profile revealed that cold stress affects most BpJMJ genes, of which those with LTR elements will be down-regulated in response to the cold stress. Finally, the gene co-expression network (WGCNA) and GO enrichment analysis reveal the main way in response to the low-temperature condition. Both of these findings, taken together, will serve as a critical foundation for future studies on the role of particular BpJMJ genes and research on histone methylation and demethylation in birch.
Methods
Plant materials and seed germination
The birch seeds were acquired from the State Key Laboratory of Tree Genetics and Breeding (TGB) at Northeast Forestry University, China, in 2020. The seeds were sown into plastic pots of 25 cm diameter and 35 cm height in June and grown in the greenhouse for 3 months. Subsequently, the roots, stems, and leaves were collected, frozen, and stored in liquid nitrogen for subsequent experimental analysis.
In the seed germination experiment, the newly collected seeds were wholly first immersed in a 10% phosphate buffer saline (PBS) buffer, shaken for 2 days, and then allowed to stand for a week in the dark 4 °C. After that, seeds were washed with distilled water three times, 1 min each, and evenly spread on the sterilized soaked filter paper, store in a constant temperature 22 °C, 16 h light/8 h dark incubator. After ten days, the seeds that had just grown out of bacon and ungerminated seeds were both collected and stored in liquid nitrogen for subsequent experiments.
Identification of JmjC domain-containing proteins in birch
To identify all potential JHDM homologous gene family in the Betula pendula genome (https://genomevolution.org/ CoGe/GenomeInfo.pl?gid=35079 and https://genomevolution.org/CoGe/ GenomeInfo.pl?gid=35080), the amino acid sequences of the JHDM genes determined in Arabidopsis (21), rice (20) and maize (19) (Table S13), were used to query the protein sequence database of Betula pendula genome with blastp program (P-value <1e-10). The JmjC domains (Pfam: PF02373, SMART: SM00558) [68, 69] were used to filter the candidates by HMM search engine (http://www.hmmer.org) [70]. Subsequently, the presence of JmjC domains in the candidate’s protein was verified by CDD https://www.ncbi.nlm.nih.gov/ (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) [71, 72] and SMART (http://smart.embl.de/) [73]. Consequently, 21 homologous BpJMJ genes were confirmed in birch genome after removing redundant transcripts, which are uniformly named as BpJMJ1-BpJMJ21 based on their genomic localization.
The basic information of the JHDM genes in birch, including the gene ID, CDS, and amino acid number, exon and intron location, and the physical location, are all extracted and calculated from the genome annotation. The isoelectric point (PI) and molecular weight (kDa) of the genes are obtained through ExPASy (http://www.expasy.org/tools/) website using default parameters [74]. The motif and functional domain of the BpJMJ genes are predicted from the MEME (https://meme-suite.org/meme/) and PFAM websites, respectively [75, 76]. In addition, the homology alignment of the BpJMJ gene with other species is performed on the Jalview (http://www.jalview.org/getdown/release/) with muscle algorithm.
Phylogenetic analysis of JHDM proteins in birch
The amino acid sequences of the JHDMs in Arabidopsis, rice, and maize are obtained from Qian et al. 2018 and Cao et al. 2008. The newly predicted JHDM proteins in birch were aligned with JHDM members in Arabidopsis, rice, and maize-based on Muscle algorithm in MEGA6.0 (https://www.megasoftware.net). Then, the phylogenetic relationship among these four species was calculated with the neighbor-joining method and 1000 bootstrap corrections and visualized in the iTOL (https://itol.embl.de) [77]. The protein sequences involved in the phylogenetic tree and the Newick file obtained after MEAG software analysis are provided in the Table S14, and Table S15.
Chromosomal localization, synteny analysis, gene duplication events, and Ka/Ks analysis of JHDM genes in birch
The physical location of 21 JHDM genes in birch was obtained from the genome annotation, and TBtools mapped their distribution in each chromosome. The gene duplication of JHDM genes was characterized using BLASTp and Multiple Collinearity Scan toolkit (MCScanX) (http://chibba.pgml.uga.edu/mcscan2/#tm) [78], and the synteny analysis of JHDM genes among the Arabidopsis thaliana, Glycine max, Populus, Oryza sativa, and Zea mays was performed in TBtools with default parameters. The protein data for Arabidopsis is downloaded from TAIR website (https://www.arabidopsis.org), and the protein data for the other species is extracted from Esemble Plants (http://plants.ensembl.org/index.html).
The Ka value, Ks value, and Ka/Ks ratios for the paralogs JmjC gene-pairs were calculated by TBtools. The paralogous genes were identified by searching the term “syntenic region” in the birch genome. The rate of divergence was calculated by using the following formula: T = Ks/2r, where Ks represents the synonymous substitutions per site and r is the rate of divergence. For dicotyledonous plants, the hypothesis is 1.5 synonymous substitutions per site of 108 years [79].
Identification of cis-acting elements of \( JHDM \) genes promoter
The 2 kb upstream genomic DNA sequence (promoter region) of the 21 JHDM family gene was retrieved from the birch genome by TBtools and submitted to the PlantCare website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) for prediction of the cis-acting elements. The 13 most frequent cis-acting elements were visualized in TBtools [80].
The tissue-specific transcriptome profile of the JHDM gene family
The RNA-seq sample datasets for each tissue, including leaf, root, flower, and xylem, were obtained from two-year-old birch at the Experimental Station of Northeast Forestry University, Harbin, P.R. China. Sequencing data were provided in Gene Expression Omnibus under accession number PRJNA535361. The bioinformatics analysis process is as follows: a) the original transcriptome data was aligned to the birch genome with hisat2 (https://daehwankimlab.github.io/hisat2/). b) the aligned results were performed to calculate TPM (Transcripts Per Million) by stringtie (http://ccb.jhu.edu/software/stringtie/). c) Finally, the BpJMJ gene expression in different tissues was scaled by setting the scale = “row” parameter in pheatmap function of R (https://cran.r-project.org), and then the hierarchical clustering of expression profiles of 21 BpJMJ genes was visualized by R-heatmap package.
The expression analysis of the JHDM gene family under abiotic stress
The high-throughput transcriptome sequencing of birch treated with low temperature (6 degrees Celsius) during five periods (0.5 h, 1 h, 1.5 h, 2 h, 2.5 h, and 3 h) was downloaded from Yan et al. (2020), [81], in which the control was planted in an environment of 25 °C without low-temperature treatment. Sequencing data were provided in Gene Expression Omnibus under accession number PRJNA532995, and the bio-informatics procedure was followed above.
With the aim of identifying the JHDM genes in response to salt in birch, transcriptome libraries from the leaves of control and plants with 200 mM NaCl treatment were provided by Shao et al. (2018) [82]. Sequencing data were provided in Gene Expression Omnibus under accession number PRJNA471213, and the bio-informatics procedure was followed above.
Genomic RNA extraction and quantitative RT-PCR analysis
The roots, stems, leaves collected from the three-month-old birch seedlings cultivated in the soil, ungerminated and germinating seeds, were ground as a fine powder by liquid nitrogen. Total RNA was isolated with Eastep® Super (Promega Beijing) according to the manufacturer’s instructions. The specific primers of genes for qRT-PCR were designed according to the birch genome and Geneious (http://www.geneious.com), which are list in Table S16. About 1 μg RNA was mixed with the 10 ul reverse-transcribed buffer in 2 ml PCR-tube, which comprising 1 ul gDNA Eraser Buffer, 2 ul 5×g DNA Eraser Buffer and the rest is RNase free ddH2O. The standard protocol was set as 2 min at 42 °C, followed by 10 min at 0 °C. The reaction product was added to a 10 ul reaction containing 1 ul Primer Script RT Enzyme, 1 ul RT Primer Mix, 4 ul 5 × Primer Script Buffer, and RNase Free ddH2O. The reverse transcribes parameters were as follows: 15 min at 37 °C followed by 5 s at 85 °C and 10 min at 0 °C. The ten-fold diluted digestion DNA products were used as a template for quantitative RT-PCR of betulin synthetic genes. The tubulin gene was identified from the birch reference genome as the internal control. PCR analysis was conducted using the Applied ABI7500 Real-Time PCR System with SYBR Premix Ex Taq™ II. Each PCR was conducted in a 15 ul reaction mixture containing 6.3 ul of 20 X diluted cDNA, 7.5 ul of SYBR Green Supermix, 0.6 ul of forward primers, and 0.6 ul of reward primers. Each gene of transcript level was calculated by the 2-(−ΔΔCt) CT method. All the qRT-PCR experiments were performed in three independent replicates.
Co-expression network construction and GO enrichment analysis of \( BpJMJ \) gene family
The WGCNA (Weighted Correlation Network Analysis) https://horvath.genetics.ucla.edu/html/ (https://horvath.genetics.ucla.edu/coexpressionnetwork/) [83] were obtained to identify co-expressed genes with the low-temperature responsive BpJMJ genes and was visualized in Cytoscape (https://cytoscape.org/) [84]. The data used as input for the co-expression network construction are the TPM derived from the time series experiment of low-temperature stress. Only genes with a TPM greater than 10 in all samples are retained. The optimalβ (soft thresholding power) value was determined to be 9 after 20 iterations. The Pearson algorithm is then used to calculate the correlation coefficient, and the result is stored as a signed co-expression matrix. The parameter “minModuleSize” value was set to 30, and the parameter “mergeCutHeight” value was set to 0.15. To ensure the reliability and readability of the results, the edges were filtered with weights below 0.1 and used Cytoscape to display genes directly/indirectly related to BpJMJ genes. The GO annotation was performed by goatools (https://github.com/tanghaibao/goatools) [85] with the fisher-exact test algorithm and visualized using R.
Availability of data and materials
All transcriptome data, including PRJNA535361 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA535361), PRJNA532995 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA532995) and PRJNA471213 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA471213) used and/or analyzed during the present study are available in NCBI.
Abbreviations
- ABRE:
-
Cis-acting element involved in the abscisic acid responsiveness
- W box:
-
Elicitation
- TGA-element:
-
Auxin-responsive element
- ERE:
-
Ethylene-responsive element
- TCA-element:
-
Cis-acting element involved in salicylic acid responsiveness
- CGTCA-motif:
-
Cis-acting regulatory element involved in the MeJA-responsiveness
- G-box:
-
Light responsiveness
- GT1-motif:
-
Light responsiveness
- ARE:
-
Anaerobic induction
- TGACG-Motif:
-
cis-acting regulatory element involved in the MeJA-responsiveness
- TCT-motif:
-
Light responsiveness
- Box 4:
-
Light responsiveness
- LTR:
-
Low-temperature responsiveness;
References
Chrun ES, Modolo F, Daniel FI. Histone modifications: a review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol Res Pract. 2017;213(11):1329–39.
Tachiwana H, Osakabe A, Shiga T, Miya Y, Kimura H, Kagawa W, et al. Structures of human nucleosomes containing major histone H3 variants. Acta Crystallogr D Biol Crystallogr. 2011;67(6):578–83. https://doi.org/10.1107/S0907444911014818.
Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51. https://doi.org/10.1038/nrc1779.
Holliday R. DNA methylation and epigenetic defects in carcinogenesis. MUTAT RES-FUND MOL M. 1987;181(2):215–7. https://doi.org/10.1016/0027-5107(87)90098-4.
Bowman GD, Poirier MG. Post-translational modifications of histones that influence nucleosome dynamics. Chem Rev. 2014;115(6):2274–95. https://doi.org/10.1021/cr500350x.
Nadal S, Raj R, Mohammed S, Davis BG. Synthetic post-translational modification of histones. Curr Opin Chem Biol. 2018;45:35–47. https://doi.org/10.1016/j.cbpa.2018.02.004.
Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25(1):1–4. https://doi.org/10.1016/j.molcel.2006.12.010.
Niu Y, Bai J, Zheng S. The regulation and function of histone methylation. J Plant Biol. 2018;61(6):347–57. https://doi.org/10.1007/s12374-018-0176-6.
Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6(1):73–7. https://doi.org/10.1038/ncb1076.
Wood A, Shilatifard A. Posttranslational modifications of histones by methylation. Adv Protein Chem. 2004;67:201–22. https://doi.org/10.1016/S0065-3233(04)67008-2.
Liu C, Lu F, Cui X, Cao X. Histone methylation in higher plants. Annu Rev Plant Biol. 2010;61(1):395–420. https://doi.org/10.1146/annurev.arplant.043008.091939.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329(5992):689–93. https://doi.org/10.1126/science.1192002.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. https://doi.org/10.1016/j.cell.2007.02.005.
Biterge B, Schneider R. Histone variants: key players of chromatin. Cell Tissue Res. 2014;356(3):457–66. https://doi.org/10.1007/s00441-014-1862-4.
Antunez-Sanchez J, Naish M, Ramirez-Prado JS, Ohno S, Huang Y, Dawson A, et al. A new role for histone demethylases in the maintenance of plant genome integrity. Elife. 2020;9:e58533. https://doi.org/10.7554/eLife.58533.
Tsukada YI, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–6. https://doi.org/10.1038/nature04433.
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–53. https://doi.org/10.1016/j.cell.2004.12.012.
Chen Q, Chen X, Wang Q, Zhang F, Lou Z, Zhang Q, et al. Structural basis of a histone H3 lysine 4 demethylase required for stem elongation in rice. PLoS Genet. 2013;9(1):e1003239. https://doi.org/10.1371/journal.pgen.1003239.
Lu F, Li G, Cui X, Liu C, Wang XJ, Cao X. Comparative analysis of JmjC domain-containing proteins reveals the potential histone demethylases in Arabidopsis and rice. J Integr Plant Biol. 2008;50(7):886–96. https://doi.org/10.1111/j.1744-7909.2008.00692.x.
Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, et al. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 2007;128(6):1063–76. https://doi.org/10.1016/j.cell.2007.02.003.
Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128(6):1077–88. https://doi.org/10.1016/j.cell.2007.02.017.
Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, et al. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128(5):889–900. https://doi.org/10.1016/j.cell.2007.02.013.
Lee JH, Tate CM, You JS, Skalnik DG. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem. 2007;282(18):13419–28. https://doi.org/10.1074/jbc.M609809200.
Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U.S.A. 2007;104(49):19226–31.
Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. The putative oncogene GASC1 demethylates tri-and dimethylated lysine 9 on histone H3. Nature. 2006;442(7100):307–11. https://doi.org/10.1038/nature04837.
Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–27. https://doi.org/10.1038/nrg1945.
Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125(3):467–81. https://doi.org/10.1016/j.cell.2006.03.028.
Yamane K, Toumazou C, Tsukada YI, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125(3):483–95. https://doi.org/10.1016/j.cell.2006.03.027.
Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science. 2007;318(5849):444–7. https://doi.org/10.1126/science.1145801.
Jones MA, Morohashi K, Grotewold E, Harmer SL. Arabidopsis JMJD5/JMJ30 acts independently of LUX ARRHYTHMO within the plant circadian clock to enable temperature compensation. Front Plant Sci. 2019;10:57. https://doi.org/10.3389/fpls.2019.00057.
Zhang J, Feng J, Liu W, Ren Z, Zhao J, Pei X, et al. Characterization and stress response of the JmjC domain-containing histone demethylase gene family in the Allotetraploid cotton species Gossypium hirsutum. Plants. 2020;9(11):1617. https://doi.org/10.3390/plants9111617.
Qian Y, Chen C, Jiang L, Zhang J, Ren Q. Genome-wide identification, classification and expression analysis of the JmjC domain-containing histone demethylase gene family in maize. BMC Genomics. 2019;20(1):1–6.
Han Y, Li X, Cheng L, Liu Y, Wang H, Ke D, et al. Genome-wide analysis of soybean JmjC domain-containing proteins suggests evolutionary conservation following whole-genome duplication. Front Plant Sci. 2016;7:1800.
Dong Y, Lu J, Liu J, Jalal A, Wang C. Genome-wide identification and functional analysis of JmjC domain-containing genes in flower development of Rosa chinensis. Plant Mol Biol. 2020;102(4):417–30. https://doi.org/10.1007/s11103-019-00955-2.
Lu F, Cui X, Zhang S, Liu C, Cao X. JMJ14 is an H3K4 demethylase regulating flowering time in Arabidopsis. Cell Res. 2010;20(3):387–90. https://doi.org/10.1038/cr.2010.27.
Yang H, Mo H, Fan D, Cao Y, Cui S, Ma L. Overexpression of a histone H3K4 demethylase, JMJ15, accelerates flowering time in Arabidopsis. Plant Cell Rep. 2012;31(7):1297–308. https://doi.org/10.1007/s00299-012-1249-5.
Yang H, Han Z, Cao Y, Fan D, Li H, Mo H, et al. A companion cell–dominant and developmentally regulated H3K4 demethylase controls flowering time in Arabidopsis via the repression of FLC expression. PLoS Genet. 2012;8(4):e1002664. https://doi.org/10.1371/journal.pgen.1002664.
Lu F, Cui X, Zhang S, Jenuwein T, Cao X. Arabidopsis REF6 is a histone H3 lysine 27 demethylase. Nat Genet. 2011;43(7):715–9. https://doi.org/10.1038/ng.854.
Crevillén P, Yang H, Cui X, Greeff C, Trick M, Qiu Q, et al. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state. Nature. 2014;515(7528):587–90. https://doi.org/10.1038/nature13722.
Gan ES, Xu Y, Wong JY, Goh JG, Sun B, Wee WY, et al. Jumonji demethylases moderate precocious flowering at elevated temperature via regulation of FLC in Arabidopsis. Nat Commun. 2014;5(1):1–3. https://doi.org/10.1038/ncomms6098.
Cho JN, Ryu JY, Jeong YM, Park J, Song JJ, Amasino RM, et al. Control of seed germination by light-induced histone arginine demethylation activity. Dev Cell. 2012;22(4):736–48. https://doi.org/10.1016/j.devcel.2012.01.024.
Huang S, Zhang A, Jin JB, Zhao B, Wang TJ, Wu Y, et al. Arabidopsis histone H3K4 demethylase JMJ 17 functions in dehydration stress response. New Phytol. 2019;223(3):1372–87. https://doi.org/10.1111/nph.15874.
Shen Y. Conde e Silva N, Audonnet L, Servet C, Wei W, Zhou DX. Over-expression of histone H3K4 demethylase gene JMJ15 enhances salt tolerance in Arabidopsis. Front. Plant Sci. 2014;5:290.
Song Y, Ji D, Li S, Wang P, Li Q, Xiang F. The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS One. 2012;7(7):e41274. https://doi.org/10.1371/journal.pone.0041274.
Shen Y, Wu X, Liu D, Song S, Liu D, Wang H. Cold-dependent alternative splicing of a Jumonji C domain-containing gene MtJMJC5 in Medicago truncatula. Biochem Biophys Res Commun. 2016;474(2):271–6. https://doi.org/10.1016/j.bbrc.2016.04.062.
Khuri S, Bakker FT, Dunwell JM. Phylogeny, function, and evolution of the cupins, a structurally conserved, functionally diverse superfamily of proteins. Mol Biol Evol. 2001;18(4):593–605. https://doi.org/10.1093/oxfordjournals.molbev.a003840.
Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125(4):691–702. https://doi.org/10.1016/j.cell.2006.04.024.
Huang Y, Chen D, Liu C, Shen W, Ruan Y. Evolution and conservation of JmjC domain proteins in the green lineage. Mol Genet Genomics. 2016;291(1):33–49. https://doi.org/10.1007/s00438-015-1089-4.
Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4(1):1–21.
Mehan MR, Freimer NB, Ophoff RA. A genome-wide survey of segmental duplications that mediate common human genetic variation of chromosomal architecture. Hum Genomics. 2004;1(5):1–0.
Lei L, Zhou SL, Ma H, Zhang LS. Expansion and diversification of the SET domain gene family following whole-genome duplications in Populus trichocarpa. BMC Evol Biol. 2012;12(1):1–7.
Chen S, Lin X, Zhang D, Li Q, Zhao X, Chen S. Genome-wide analysis of NAC gene family in Betula pendula. Forests. 2019;10(9):741. https://doi.org/10.3390/f10090741.
Le Hir H, Nott A, Moore MJ. How introns influence and enhance eukaryotic gene expression. Trends Biochem Sci. 2003;28(4):215–20. https://doi.org/10.1016/S0968-0004(03)00052-5.
Beck EH, Heim R, Hansen J. Plant resistance to cold stress: mechanisms and environmental signals triggering frost hardening and dehardening. J Biosci. 2004;29(4):449–59. https://doi.org/10.1007/BF02712118.
Zhao W, Shafiq S, Berr A, Shen WH. Genome-wide gene expression profiling to investigate molecular phenotypes of Arabidopsis mutants deprived in distinct histone methyltransferases and demethylases. Genomics data. 2015;4:143–5. https://doi.org/10.1016/j.gdata.2015.04.006.
Zong W, Zhong X, You J, Xiong L. Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol Biol. 2013;81(1):175–88. https://doi.org/10.1007/s11103-012-9990-2.
Zhang S, Zhou B, Kang Y, Cui X, Liu A, Deleris A, et al. C-terminal domains of histone demethylase JMJ14 interact with a pair of NAC transcription factors to mediate specific chromatin association. Cell Discov. 2015;1(1):1–3.
Yang W, Jiang D, Jiang J, He Y. A plant-specific histone H3 lysine 4 demethylase represses the floral transition in Arabidopsis. Plant J. 2010;62(4):663–73. https://doi.org/10.1111/j.1365-313X.2010.04182.x.
Yokoo T, Saito H, Yoshitake Y, Xu Q, Asami T, Tsukiyama T, et al. Se14, encoding a JmjC domain-containing protein, plays key roles in long-day suppression of rice flowering through the demethylation of H3K4me3 of RFT1. PLoS One. 2014;9(4):e96064. https://doi.org/10.1371/journal.pone.0096064.
Song T, Zhang Q, Wang H, Han J, Xu Z, Yan S, et al. OsJMJ703, a rice histone demethylase gene, plays key roles in plant development and responds to drought stress. Plant Physiol Biochem. 2018;132:183–8. https://doi.org/10.1016/j.plaphy.2018.09.007.
Li Y, Mukherjee I, Thum KE, Tanurdzic M, Katari MS, Obertello M, et al. The histone methyltransferase SDG8 mediates the epigenetic modification of light and carbon responsive genes in plants. Genome Biol. 2015;16(1):1–5.
Kim JM, Sasaki T, Ueda M, Sako K, Seki M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front Plant Sci. 2015;6:114.
Gautam A, Pandey P, Pandey AK. Proteomics in relation to abiotic stress tolerance in plants. In: Tripathi DK, Singh VP, Chauhan DK, Sharma S, Prasad SM, Dubey NK, et al., editors. Plant life under changing environment: Responses and management. Academic press, an imprint of Elsevier. 2020;513–41. https://doi.org/10.1016/B978-0-12-818204-8.00023-0.
Yang Z, Qiu Q, Chen W, Jia B, Chen X, Hu H, et al. Structure of the Arabidopsis JMJ14-H3K4me3 complex provides insight into the substrate specificity of KDM5 subfamily histone demethylases. Plant Cell. 2018;30(1):167–77. https://doi.org/10.1105/tpc.17.00666.
Insilico PS. Analysis of cis acting Regulatory Elements CAREs in Upstream Regions of Ascorbate Glutathione Pathway Genes from Oryza sativa [J]. Biochem Physiol Open Access. 2015;4(02). https://doi.org/10.4172/2168-9652.1000159.
Chunhui Wu, Caiyun Zheng, Gengsheng Ji, et al. Synergistic effects of HSE and LTR elements from hsp70 gene promoter of Ulva prolifera (Ulvophyceae, Chlorophyta) upon temperature induction1[J]. J Phycol. 2019;55(3). https://pubmed.ncbi.nlm.nih.gov/30843209/.
Bouyer D, Roudier F, Heese M, Andersen ED, Gey D, Nowack MK, et al. Polycomb repressive complex 2 controls the embryo-to-seedling phase transition. PLoS Genet. 2011;7(3):e1002014. https://doi.org/10.1371/journal.pgen.1002014.
Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34(suppl_1):D247–51.
Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, et al. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 2004;32(suppl_1):D142–4.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(suppl_2):W29–37.
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese- Scott C, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–9.
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s con- served domain database. Nucleic Acids Res. 2015;43(D1):D222–6. https://doi.org/10.1093/nar/gku1221.
Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95(11):5857–64. https://doi.org/10.1073/pnas.95.11.5857.
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–8. https://doi.org/10.1093/nar/gkg563.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202. https://doi.org/10.1016/j.molp.2020.06.009.
Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46(D1):D493–6. https://doi.org/10.1093/nar/gkx922.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–9. https://doi.org/10.1093/molbev/msy096.
Xie T, Chen C, Li C, Liu J, Liu C, He Y. Genome-wide investigation of WRKY gene family in pineapple: evolution and expression profiles during development and stress. BMC Genomics. 2018;19(1):1–8. https://doi.org/10.1186/s12864-018-4880-x.
Koch M, Al-Shehbaz IA, Mummenhoff K. Molecular systematics, evolution, and population biology in the mustard family (Brassicaceae). Ann Mo Bot Gard. 2003;1:151–71.
Rombauts S, Déhais P, Van Montagu M, Rouzé P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999;27(1):295–6. https://doi.org/10.1093/nar/27.1.295.
Yan S, Zhang D, Chen S, Chen S. Transcriptome sequencing analysis of birch (Betula platyphylla Sukaczev) under low-temperature stress. Forests. 2020;11(9):970. https://doi.org/10.3390/f11090970.
Shao F, Zhang L, Wilson IW, Qiu D. Transcriptomic analysis of Betula halophila in response to salt stress. Int J Mol Sci. 2018;19(11):3412. https://doi.org/10.3390/ijms19113412.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9(1):1–3. https://doi.org/10.1186/1471-2105-9-559.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. https://doi.org/10.1101/gr.1239303.
Klopfenstein DV, Zhang L, Pedersen BS, Ramírez F, Vesztrocy AW, Naldi A, et al. GOATOOLS: a Python library for gene ontology analyses. Sci Rep. 2018;8(1):1–7.
Acknowledgments
We thank the editors and reviewers for their work on promoting the manuscript.
Funding
This research was financially supported by the Fundamental Research Fund for Central Universities (Grant No. 2572018AA27) and the National Natural Science Foundation of China (No. 31871220 and No. 31801444), National Nonprofit Institute Research Grant of the Chinese Academy of Forestry (No. CAFYBB2019ZY003), and Fundamental Research Funds for the Central Universities (No. 2572020DP01 and No. 2572017DA06).
Author information
Authors and Affiliations
Contributions
The conception of the presented idea, BWC, SA; performed research and analyzed data, BWC, XZ; Writing-original draft, BWC, SA; review & editing, YZ, MW. supervised and revised the manuscript LX, QZ. All the authors read and agreed with the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the People’s Republic of China and international authorities relevant guidelines and legislation, including the official website of the Committee on Publication Ethics (http://www.publicationethics.org/) and the European Association of Science Editors (EASE) and other institutions’ publishing ethics standards. All plants materials involved in this research are used for scientific research, which are allowed to be used and provided free of charge in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Basic information of JHDM genes identified in birch.
Additional file 2: Table S2
. Homolog similarity of JHDM genes between birch and Arabidopsis.
Additional file 3: Table S3
. The MEME motif anotation of JHDM members in birch.
Additional file 4: Table S4
. The distribution of functional domains in different JmjC subfamilies of birch.
Additional file 5: Table S5
. The classification of duplicate events in JHDM genes subfamily.
Additional file 6: Table S6
. Ka/Ks calculation of the paralog pairs of BpJMJ in birch.
Additional file 7: Table S7
. Expression levels (TPM) of JHDM genes in different birch tissues.
Additional file 8: Table S8
. The spearman correlation between the number of BpJMJ genes introns and their expression in different tissues.
Additional file 9: Table S9
. The spearman correlation between the number of BpJMJ genes cis-acting elements and their expression in different tissues.
Additional file 10: Table S10
. The quantitative distribution of cis-acting elements in the promoter region of BpJMJ gene family.
Additional file 11: Table S11
. Expression levels (TPM) of JHDM genes with cold stress.
Additional file 12: Table S12
. Expression levels (TPM) of JHDM genes with salt stress.
Additional file 13: Table S13
. JHDM gene distribution among Birch, Maize, Arabidopsis and Rice.
Additional file 14: Table S14
. The BpJMJ protein sequences of Arabidopsos, birch, rice and may involved in the phylogenetic tree.
Additional file 15: Table S15
. The Newick file obtained from MEGA software for constructing the phylogenetic tree.
Additional file 16: Table S16
. The primers for qRT-PCR used in this study.
Additional file 17: Figure S1
. The detailed sequences information of 20 motifs in BpJMJ proteins annotated from MEME website.
Additional file 18: Figure S2
. Protein sequence alignments of KDM5/JARID1 and KDM4/JHDM3 subfamily among birch, Arabidopsis, rice and may.
Additional file 19: Figure S3
. Protein sequence alignments of KDM3/JHDM2, JMJD6 and JmjC domain-only subfamily among birch, Arabidopsis, rice and may.
Additional file 20: Figure S4
. Distribution of cis-acting elements (hormone, light and stress) in 2kb promoter region of 21 BpJMJ genes.
Additional file 21: Figure S5
. The count of BpJMJ genes with different classification of cis-acting elements.
Additional file 22: Figure S6
. The expression of BpJMJ genes analyzed by transcriptome in response to salt treatment (200mM NaCl).
Additional file 23: Figure S7
. Different gene modules of birch under cold stress analyzed by WGCNA.
Additional file 24: Figure S8
. The co-expression network (network1) for the low-temperature response BpJMJ genes.
Additional file 25: Figure S9
. GO enrichment (MF) analysis of BpJMJ directly associated genes in co-expression network.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, B., Ali, S., Zhang, X. et al. Genome-wide identification, classification, and expression analysis of the JmjC domain-containing histone demethylase gene family in birch. BMC Genomics 22, 772 (2021). https://doi.org/10.1186/s12864-021-08063-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08063-6