
Abstract
Objective
To prioritize genes that were pleiotropically or potentially causally associated with central corneal thickness (CCT).
Methods
We applied the summary data-based Mendelian randomization (SMR) method integrating summarized data of genome-wide association study (GWAS) on CCT and expression quantitative trait loci (eQTL) data to identify genes that were pleiotropically associated with CCT. We performed separate SMR analysis using CAGE eQTL data and GTEx eQTL data. SMR analyses were done for participants of European and East Asian ancestries, separately.
Results
We identified multiple genes showing pleiotropic association with CCT in the participants of European ancestry. CLIC3 (ILMN_1796423; PSMR = 4.15 × 10− 12), PTGDS (ILMN_1664464; PSMR = 6.88 × 10− 9) and C9orf142 (ILMN_1761138; PSMR = 8.09 × 10− 9) were the top three genes using the CAGE eQTL data, and RP11-458F8.4 (ENSG00000273142.1; PSMR = 5.89 × 10− 9), LCNL1 (ENSG00000214402.6; PSMR = 5.67 × 10− 8), and PTGDS (ENSG00000107317.7; PSMR = 1.92 × 10− 7) were the top three genes using the GTEx eQTL data. No genes showed significantly pleiotropic association with CCT in the participants of East Asian ancestry after correction for multiple testing.
Conclusion
We identified several genes pleiotropically associated with CCT, some of which represented novel genes influencing CCT. Our findings provided important leads to a better understanding of the genetic factors influencing CCT, and revealed potential therapeutic targets for the treatment of primary open-angle glaucoma and keratoconus.
Similar content being viewed by others


Introduction
The cornea is a highly collagenous and transparent tissue through which light reaches the interior structures of the eye. Previous studies highlighted the importance of central corneal thickness (CCT) in relation to several ocular and non-ocular conditions. For example, decrease in CCT is significantly associated with intraocular pressure (IOP) [1]. Thinner CCT has been demonstrated as an important feature of keratoconus and a risk factor for primary open-angle glaucoma (POAG) in patients with ocular hypertension [2,3,4,5,6,7]. Keratoconus is the leading cause of corneal transplants worldwide, with an estimated prevalence of 0.14% [8], while POAG is the most common cause of irreversible blindness worldwide, accounting for approximately 70% of all the cases of glaucoma [9].
Epidemiologic studies have shown that CCT differs among ethnic groups, with Europeans having higher CCT values than Africans, and Asians showing a larger variation in CCT [10]. CCT is highly heritable, with an estimated heritability ranging from 68 to 95% [11,12,13]. Previous genome-wide association studies (GWAS) identified a number of CCT-associated loci in Europeans and Asians, such as genetic loci in ZNF469, FOXO1, LRRK1 and IBTK [14,15,16,17,18,19,20]. Recent genetic studies revealed additional novel loci associated with CCT, some of which conferred relatively high risks of keratoconus and POAG, highlighting the potential involvement of CCT-associated genes underlying the pathogenesis of keratoconus and POAG [21, 22].
Mendelian randomization (MR) uses genetic variants as the proxy to randomization and is a promising tool to explore pleiotropic/potentially causal effect of an exposure (e.g., gene expression) on the outcome (e.g., CCT) [23]. MR could reduce confounding and reverse causation that are commonly encountered in traditional association studies, and has been successful in identifying gene expression probes or DNA methylation loci that are pleiotropically/potentially causally associated with various phenotypes, such as neuropathologies of Alzheimer’s disease and severity of COVID-19 [24, 25].
In this paper, we applied the summary data-based MR (SMR) method integrating summarized GWAS data for CCT and cis- eQTL (expression quantitative trait loci) data to prioritize genes that are pleiotropically/potentially causally associated with CCT.
Methods
Data sources
eQTL data
In the SMR analysis, cis-eQTL genetic variants were used as the instrumental variables (IVs) for gene expression. We performed SMR analysis using gene expression in blood due to unavailability of eQTL data for the eye. Specifically, we used the CAGE eQTL summarized data for blood [26], which included 2765 participants, and the V7 release of the GTEx eQTL summarized data for blood [27], which included 338 participants. The eQTL data can be downloaded at https://cnsgenomics.com/data/SMR/#eQTLsummarydata.
GWAS data for corneal thickness
The GWAS summarized data for CCT were provided by a recent genome-wide association meta-analysis of CCT [21]. The results were based on meta-analyses of 1000 genomes phase 1 imputed GWASs on CCT, with a total of 19 cohorts from the International Glaucoma Genetics consortium (IGGC). Specifically, the meta-analysis for participants of European ancestry included 14 cohorts with a sample of size of 17,803, and the meta-analysis for participants of East Asian ancestry included 5 cohorts with a sample size of 8107. All participating studies assumed an additive genetic model, adjusting for age, sex and at least the first five principal components. The GWAS summarized data can be downloaded at https://datashare.is.ed.ac.uk/handle/10283/2976.
SMR analysis
We conducted the SMR analysis with cis-eQTL as the IV, gene expression as the exposure, and CCT as the outcome. The analysis was done using the method as implemented in the software SMR. Detailed information regarding the SMR method was reported in a previous publication [28]. In brief, SMR applies the principles of MR to jointly analyze GWAS and eQTL summary statistics in order to test for pleiotropic association between gene expression and a trait due to a shared and potentially causal variant at a locus. In SMR, the top cis-eQTL was used to estimate the effect of gene expression on the outcome. SMR analysis relies on the validity of the genetic variants as IVs to obtain consistent estimates. A genetic variant has to satisfy three assumptions to be a valid IV: 1) it is associated with the risk factor (gene expression); 2) it is not associated with any confounder of the risk factor-outcome association; and 3) it is conditionally independent of the outcome given the risk factor and confounders. It was found that if the P-value in a linear regression of the eQTL analysis for each variant is less than 1 × 10− 5, then weak instrument bias was small [29]. In the SMR analysis, we adopted the default threshold of PeQTL = 5 × 10− 8 to select the top associated cis-eQTL (with a default window size of 2000 Kb) for the SMR analysis, minimizing the risk of weak IV. By default, we removed SNPs with allele frequency difference > 0.2 between any pairwise data sets, including the LD reference sample data, the eQTL summary data and the GWAS summary data. We also conducted the heterogeneity in dependent instruments (HEIDI) test to evaluate the existence of linkage in the observed association. Rejection of the null hypothesis (i.e., PHEIDI < 0.05) indicates that the observed association could be due to two distinct genetic variants in high linkage disequilibrium with each other. In conducting the HEIDI test, we adopted the default settings: removing SNPs in very strong linkage disequilibrium [LD, r2 > 0.9] with the top associated eQTL, and SNPs in low LD or not in LD [r2 < 0.05] with the top associated eQTL; PeQTL < 1.5 × 10− 3; number of cis-SNPs≥3 for a HEIDI as HEIDI test loses power if cis-SNPs< 3; and maximum eQTLs in a HEIDI test = 20. We used false discovery rate (FDR) to adjust for multiple testing.
Data curation and statistical/bioinformatical analysis was performed using R version 4.0.3 (https://www.r-project.org/), PLINK 1.9 (https://www.cog-genomics.org/plink/1.9/) and SMR (https://cnsgenomics.com/software/smr/).
Results
Basic information of the summarized data
The number of participants of the CAGE eQTL data is much larger than that of the GTEx eQTL data, so is the number of eligible probes. The sample size of the GWAS data for the European ancestry is much large than that for the East Asian ancestry, so is the number of eligible genetic variants. The detailed information was shown in Table 1.
SMR analysis in the European population
In participants with European ancestry, we identified multiple genes showing pleiotropic association with CCT after correction for multiple testing using FDR (Table 2). Specifically, using the CAGE eQTL data, our SMR analysis identified 12 genes that were pleiotropically/potentially causally associated with CCT, with CLIC3 (ILMN_1796423; PSMR = 4.15 × 10− 12), PTGDS (ILMN_1664464; PSMR = 6.88 × 10− 9) and C9orf142 (ILMN_1761138; PSMR = 8.09 × 10− 9) being the top three genes (Fig. 1).

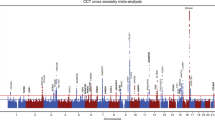
Prioritizing gene around CLIC3, PTGDS and C9orf142 in pleiotropic association with CCT in the participants of European ancestry using CAGE eQTL data. Top plot, grey dots represent the -log10(P values) for SNPs from the GWAS of CCT, and rhombuses represent the -log10(P values) for probes from the SMR test with solid rhombuses indicating that the probes pass HEIDI test and hollow rhombuses indicating that the probes do not pass the HEIDI test. Middle plot, eQTL results. Bottom plot, location of genes tagged by the probes. CCT central corneal thickness, GWAS genome-wide association studies, SMR summary data-based Mendelian randomization, HEIDI heterogeneity in dependent instruments, eQTL expression quantitative trait loci
Using the GTEx eQTL data, our SMR analysis identified six genes that were pleiotropically/potentially causally associated with CCT, with RP11-458F8.4 (ENSG00000273142.1; PSMR = 5.89 × 10− 9; Fig. 2), LCNL1 (ENSG00000214402.6; PSMR = 5.67 × 10− 8; Fig. 3), and PTGDS (ENSG00000107317.7; PSMR = 1.92 × 10− 7; Fig. 3) being the top three genes. It should be noted that the gene PTGDS was the top gene showing significant pleiotropic association in both SMR analyses.

Prioritizing gene around RP11-458F8.4 in pleiotropic association with CCT in the participants of European ancestry using GTEx eQTL data. Top plot, grey dots represent the -log10(P values) for SNPs from the GWAS of CCT, and rhombuses represent the -log10(P values) for probes from the SMR test with hollow rhombuses indicating that the probes do not pass the HEIDI test. Middle plot, eQTL results. Bottom plot, location of genes tagged by the probes. CCT central corneal thickness, GWAS genome-wide association studies, SMR summary data-based Mendelian randomization, HEIDI heterogeneity in dependent instruments, eQTL expression quantitative trait loci

Prioritizing gene around LCNL1 and PTGDS in pleiotropic association with CCT in the participants of European ancestry using CAGE eQTL data. Top plot, grey dots represent the -log10(P values) for SNPs from the GWAS of CCT, and rhombuses represent the -log10(P values) for probes from the SMR test with solid rhombuses indicating that the probes pass HEIDI test and hollow rhombuses indicating that the probes do not pass the HEIDI test. Middle plot, eQTL results. Bottom plot, location of genes tagged by the probes. CCT central corneal thickness, GWAS genome-wide association studies, SMR summary data-based Mendelian randomization, HEIDI heterogeneity in dependent instruments, eQTL expression quantitative trait loci
SMR analysis in the east Asian population
In participants of East Asian ancestry, we identified no genes showing significant pleiotropic association with CCT after correction for multiple testing using FDR (Table 3). Specifically, using the CAGE eQTL data, we found that two genes, PASK (ILMN_1754858, PSMR = 7.98 × 10− 5; ILMN_1667022, PSMR = 8.24 × 10− 5) and HIATL1 (ILMN_1703229, PSMR = 4.30 × 10− 4; ILMN_1737964, PSMR = 5.84 × 10− 4), each of which was tagged by two probes, were among the top hits in the SMR analysis. However, none of the genes survived multiple comparison. Using the GTEx eQTL data, we found that several genes overlapped with the top genes in the SMR analysis using CAGE eQTL data, including MAP 7D1, HIATL1, POLA2 and ACADM. Again, none of the genes survived multiple comparison.
Discussion
In the present study, we integrated summarized data of GWAS on CCT and eQTL data in the MR analysis to explore putative genes that showed pleiotropic/potentially causal association with CCT. In the participants of European ancestry, we identified multiple genes showing significantly pleiotropic association with CCT, some of which represented novel genes associated with CCT. Our findings provided important leads to a better understanding of the genetic factors influencing CCT, and revealed potential therapeutic targets for the treatment of POAG and keratoconus.
We found that PTGDS (prostaglandin D2 synthase) showed significantly pleiotropic association with CCT in the participants of European ancestry using both CAGE and GTEx eQTL data. PTGDS encodes the glutathione-independent prostaglandin D2 synthase which catalyzes the conversion of prostaglandin H2 (PGH2) to prostaglandin D2 (PGD2), an important marker for keratocytes [30, 31]. A genetic polymorphism in PTGDS (rs11145951) was reported to be associated with CCT in the European population (P = 9.20 × 10− 12) but not in the Asian population (P = 2.30 × 10− 2) [15]. The association of this polymorphism with CCT did not reach genome-wide significance in the Latino population (P = 1.15 × 10− 5), suggesting ethnic-specific effect of this genetic polymorphism on CCT [32]. PTGDS was found to be highly expressed in corneal endothelial cells (CECs) [33], ranked the among the top 50 most highly expressed genes in CECs [34]. When using a novel dual media culture approach for the in vitro expansion of primary human corneal endothelial cells (hCECs), the expression of PTGDS increased by 12.64 folds following exposure of cultivated hCECs from proliferative (M4) to maintenance (M5) medium [35]. Given that thinner CCT was an important feature for keratoconus and a potential risk factor for POAG, these findings, together with ours, demonstrated the important role of PTGDS in influencing CCT and highlighted the potential of this gene as a promising target for the prevention and treatment of keratoconus and POAG.
RP11-458F8.4 showed the most significantly pleiotropic association with CCT using GTEx eQTL data in the participants of European ancestry (Table 2). RP11-458F8.4 is a long intergenic noncoding RNA (lincRNA). LincRNAs exercise various tissue-specific functions such as remodeling chromatin and genome architecture, RNA stabilization and transcription regulation [36]. RP11-458F8.4 was reported to be associated with various types of malignant tumors. For example, it was upregulated in late-stage colon cancers, and its expression may be involved in the progression of colon cancer [37]. It was found to be a prognostic lincRNA in high-grade serous epithelial ovarian cancer [38], and was differently expressed in patients with breast cancer [39]. No studies have reported the association of this gene with CCT. As a result, further investigation is needed to elucidate the exact functions of this gene and examine its role in influencing CCT.
VKORC1L1 (vitamin K epoxide reductase complex subunit 1 like 1) was pleiotropically associated with CCT in the participants of European ancestry using CAGE eQTL data. VKORC1L1 can mediate vitamin K-dependent intracellular antioxidant functions [40]. A genetic variant in VKORC1L1 (rs11763147) was found to be associated with CCT in a combined meta-analysis of the European and the Asian samples (4.0 × 10− 9, 15]. Another genetic variant (rs10563220) located upstream of VKORC1L1 was found to be significantly associated with intraocular pressure in a GWAS of 8552 Chinese participants [41]. More studies are needed to elucidate the exact functions of VKORC1L1 in association with CCT, and explore whether/how it is involved in the pathogenesis of POAG and keratoconus.
RUNX2 (runt-related transcription factor 2) showed significantly pleiotropic association with CCT in the participants of European ancestry using the CAGE eQTL data (Table 2). RUNX2 is a member of the RUNX family of transcription factors and encodes a nuclear protein, a key transcription factor of osteoblast differentiation [42, 43]. A genetic variant in RUNX2 (rs13191376) was found to be associated with CCT in a cross-ancestry GWAS [21]. Another genetic variant in RUNX2 (rs1755056) was found to be significantly associated with IOP and weakly associated with POAG in a GWAS combing participants from the UK Biobank and published data from International Glaucoma Genetic Consortium [44]. In a transient transfection experiment using MG-63 cells and primary bovine corneal keratocytes, RUNX2 transcription factors affected the expression of several small leucine rich proteoglycans (SLRP) including mimecan, biglycan and keratocan [45], which were shown to be important for the development and maintenance of corneal transparency [46,47,48]. In another study using the rabbit corneal epithelial cell line RCE1(5 T5), RNAseq based transcriptome analysis showed that RUNX2 exhibited the highest expression in terminally differentiated corneal epithelial cells [49]. In breast cancer, it was found that RUNX2 functioned through the androgen receptor to regulate prolactin-induced protein (PIP) [50], a new biomarker for keratoconus [51]. These findings suggested that RUNX2 likely plays an important role in affecting CCT and the susceptibility of POAG and keratoconus.
Of the multiple genes showing significantly pleiotropic association with CCT in the participants of European ancestry, none was significant in the SMR analysis for the participants of the East Asian ancestry. And only RP11-458F8.4, which was pleiotropically associated with CCT in the participants of European ancestry using GTEx data, appeared among the top-hit genes in the SMR analysis for the participants of East Asian ancestry using GTEx data (Tables 2 and 3). These findings implied possible ethnicity-specific genetic mechanisms underlying CCT, leading to anatomic changes (e.g., cornea and optic nerves) [52,53,54] and differences in the susceptibility of CCT-related disorders/diseases. For example, the prevalence of POAG was reported to be high in Afro-Caribbeans and African Americans, intermediate in Latinos and Asians, and low in non-Hispanic whites [55]. Moreover, compared with Asians, Europeans have a higher rate of keratoconus incidence and an older age-of-onset of keratoconus [56, 57]. The findings of our study revealed genes harboring potential biomarkers for tailored screening and treatment of POAG and keratoconus in subjects of different ethnicities.
Our study has some limitations. The number of probes used in our SMR analysis was limited, especially in the SMR analysis of the participants of East Asian ancestry. Moreover, the sample size of eQTL analysis is limited, especially for the GTEx data, which may lead to reduced power in the eQTL analysis. As a result, we may have missed some important genes. Moreover, the sample size and the number of eligible genetic variants for GWAS in the participants of East Asian ancestry is limited, compared to GWAS in the participants of European ancestry, which may affect the power of our SMR analysis and contribute to the insignificant findings in the participants of East Asian ancestry. The HEIDI test was significant for some of the identified genes (Table 2-3). As results, the possibility of horizontal pleiotropy, i.e., the identified association might be due to two distinct genetic variants in high linkage disequilibrium with each other, could not be ruled out. We only used eQTL data in the blood due to the unavailability of eQTL data from the eye. Our findings need to be validated in the future when eQTL data from the eye are available. We used the same eQTL data in the SMR analysis for both European and Asian ancestry. It is possible that eQTL pattern could vary with ethnicity. Ethnicity-specific eQTL data with larger sample size are needed in future SMR research.
Conclusions
We identified several genes pleiotropically associated with CCT, some of which represented novel genes influencing CCT. Our findings provided important leads to a better understanding of the genetic factors influencing CCT, and revealed potential therapeutic targets for the treatment of POAG and keratoconus.
Availability of data and materials
All data generated or analyzed during this study are publicly available as specified in the methods section of this paper. Specifically, the eQTL data can be downloaded at https://cnsgenomics.com/data/SMR/#eQTLsummarydata, and the GWAS summarized data can be downloaded at https://datashare.is.ed.ac.uk/handle/10283/2976.
References
Cho P, Lam C. Factors affecting the central corneal thickness of Hong Kong-Chinese. Curr Eye Res. 1999;18(5):368–74.
Cohen EJ. Keratoconus and normal-tension glaucoma: a study of the possible association with abnormal biomechanical properties as measured by corneal hysteresis (An AOS Thesis). Trans Am Ophthalmol Soc. 2009;107:282–99.
Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, Johnson CA, et al. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120(6):714–20 discussion 829–730.
Herndon LW, Weizer JS, Stinnett SS. Central corneal thickness as a risk factor for advanced glaucoma damage. Arch Ophthalmol. 2004;122(1):17–21.
Kim JW, Chen PP. Central corneal pachymetry and visual field progression in patients with open-angle glaucoma. Ophthalmology. 2004;111(11):2126–32.
Naderan M, Shoar S, Rezagholizadeh F, Zolfaghari M, Naderan M. Characteristics and associations of keratoconus patients. Cont Lens Anterior Eye. 2015;38(3):199–205.
Song C, De Moraes CG, Forchheimer I, Prata TS, Ritch R, Liebmann JM. Risk calculation variability over time in ocular hypertensive subjects. J Glaucoma. 2014;23(1):1–4.
Hashemi H, Heydarian S, Hooshmand E, Saatchi M, Yekta A, Aghamirsalim M, et al. The Prevalence and Risk Factors for Keratoconus: A Systematic Review and Meta-Analysis. Cornea. 2020;39(2):263–70.
Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262–7.
Dimasi DP, Burdon KP, Craig JE. The genetics of central corneal thickness. Br J Ophthalmol. 2010;94(8):971–6.
Landers JA, Hewitt AW, Dimasi DP, Charlesworth JC, Straga T, Mills RA, et al. Heritability of central corneal thickness in nuclear families. Invest Ophthalmol Vis Sci. 2009;50(9):4087–90.
Toh T, Liew SH, MacKinnon JR, Hewitt AW, Poulsen JL, Spector TD, et al. Central corneal thickness is highly heritable: the twin eye studies. Invest Ophthalmol Vis Sci. 2005;46(10):3718–22.
Zheng Y, Ge J, Huang G, Zhang J, Liu B, Hur YM, et al. Heritability of central corneal thickness in Chinese: the Guangzhou Twin Eye Study. Invest Ophthalmol Vis Sci. 2008;49(10):4303–7.
Gao X, Gauderman WJ, Liu Y, Marjoram P, Torres M, Haritunians T, et al. A genome-wide association study of central corneal thickness in Latinos. Invest Ophthalmol Vis Sci. 2013;54(4):2435–43.
Lu Y, Vitart V, Burdon KP, Khor CC, Bykhovskaya Y, Mirshahi A, et al. Genome-wide association analyses identify multiple loci associated with central corneal thickness and keratoconus. Nat Genet. 2013;45(2):155–63.
Vitart V, Bencic G, Hayward C, Skunca Herman J, Huffman J, Campbell S, et al. New loci associated with central cornea thickness include COL5A1, AKAP13 and AVGR8. Hum Mol Genet. 2010;19(21):4304–11.
Cornes BK, Khor CC, Nongpiur ME, Xu L, Tay WT, Zheng Y, et al. Identification of four novel variants that influence central corneal thickness in multi-ethnic Asian populations. Hum Mol Genet. 2012;21(2):437–45.
Hoehn R, Zeller T, Verhoeven VJ, Grus F, Adler M, Wolfs RC, et al. Population-based meta-analysis in Caucasians confirms association with COL5A1 and ZNF469 but not COL8A2 with central corneal thickness. Hum Genet. 2012;131(11):1783–93.
Ulmer M, Li J, Yaspan BL, Ozel AB, Richards JE, Moroi SE, et al. Genome-wide analysis of central corneal thickness in primary open-angle glaucoma cases in the NEIGHBOR and GLAUGEN consortia. Invest Ophthalmol Vis Sci. 2012;53(8):4468–74.
Vithana EN, Aung T, Khor CC, Cornes BK, Tay WT, Sim X, et al. Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum Mol Genet. 2011;20(4):649–58.
Iglesias AI, Mishra A, Vitart V, Bykhovskaya Y, Hohn R, Springelkamp H, et al. Cross-ancestry genome-wide association analysis of corneal thickness strengthens link between complex and Mendelian eye diseases. Nat Commun. 2018;9(1):1864.
Choquet H, Melles RB, Yin J, Hoffmann TJ, Thai KK, Kvale MN, et al. A multiethnic genome-wide analysis of 44,039 individuals identifies 41 new loci associated with central corneal thickness. Commun Biol. 2020;3(1):301.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98.
Liu D, Wang Y, Jing H, Meng Q, Yang J. Mendelian randomization integrating GWAS and mQTL data identified novel pleiotropic DNA methylation loci for neuropathology of Alzheimer's disease. Neurobiol Aging. 2021;97:18–27.
Liu D, Yang J, Feng B, Lu W, Zhao C, Li L. Mendelian randomization analysis identified genes pleiotropically associated with the risk and prognosis of COVID-19. J Infect. 2021;82(1):126-32. https://doi.org/10.1016/j.jinf.2020.11.03.
Lloyd-Jones LR, Holloway A, McRae A, Yang J, Small K, Zhao J, et al. The Genetic Architecture of Gene Expression in Peripheral Blood. Am J Hum Genet. 2017;100(2):228–37.
Consortium GT, Laboratory DA. Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida et al: Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–13.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–7.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65.
Chakravarti S, Wu F, Vij N, Roberts L, Joyce S. Microarray studies reveal macrophage-like function of stromal keratocytes in the cornea. Invest Ophthalmol Vis Sci. 2004;45(10):3475–84.
Dos Santos A, Balayan A, Funderburgh ML, Ngo J, Funderburgh JL, Deng SX. Differentiation Capacity of Human Mesenchymal Stem Cells into Keratocyte Lineage. Invest Ophthalmol Vis Sci. 2019;60(8):3013–23.
Gao X, Nannini DR, Corrao K, Torres M, Chen YI, Fan BJ, et al. International Glaucoma Genetics C, Taylor KD, Gauderman WJ et al: Genome-wide association study identifies WNT7B as a novel locus for central corneal thickness in Latinos. Hum Mol Genet. 2016;25(22):5035–45.
Sakai R, Kinouchi T, Kawamoto S, Dana MR, Hamamoto T, Tsuru T, et al. Construction of human corneal endothelial cDNA library and identification of novel active genes. Invest Ophthalmol Vis Sci. 2002;43(6):1749–56.
Gottsch JD, Seitzman GD, Margulies EH, Bowers AL, Michels AJ, Saha S, et al. Gene expression in donor corneal endothelium. Arch Ophthalmol. 2003;121(2):252–8.
Peh GS, Chng Z, Ang HP, Cheng TY, Adnan K, Seah XY, et al. Propagation of human corneal endothelial cells: a novel dual media approach. Cell Transplant. 2015;24(2):287–304.
Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol. 2018;19(3):143–57.
Cheng Y, Geng L, Wang K, Sun J, Xu W, Gong S, et al. Long Noncoding RNA Expression Signatures of Colon Cancer Based on the ceRNA Network and Their Prognostic Value. Dis Markers. 2019;2019:7636757.
Lin X, Spindler TJ, de Souza Fonseca MA, Corona RI, Seo JH, Dezem FS, et al. Super-Enhancer-Associated LncRNA UCA1 Interacts Directly with AMOT to Activate YAP Target Genes in Epithelial Ovarian Cancer. iScience. 2019;17:242–55.
Xiong H, Chen Z, Chen W, Li Q, Lin B, Jia Y. FKBP-related ncRNA-mRNA axis in breast cancer. Genomics. 2020;112(6):4595–607.
Westhofen P, Watzka M, Marinova M, Hass M, Kirfel G, Muller J, et al. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem. 2011;286(17):15085–94.
Huang L, Chen Y, Lin Y, Tam POS, Cheng Y, Shi Y, et al. Genome-wide analysis identified 17 new loci influencing intraocular pressure in Chinese population. Sci China Life Sci. 2019;62(2):153–64.
van Wijnen AJ, Stein GS, Gergen JP, Groner Y, Hiebert SW, Ito Y, et al. Nomenclature for Runt-related (RUNX) proteins. Oncogene. 2004;23(24):4209–10.
Bruderer M, Richards RG, Alini M, Stoddart MJ. Role and regulation of RUNX2 in osteogenesis. Eur Cell Mater. 2014;28:269–86.
MacGregor S, Ong JS, An J, Han X, Zhou T, Siggs OM, et al. Genome-wide association study of intraocular pressure uncovers new pathways to glaucoma. Nat Genet. 2018;50(8):1067–71.
Tasheva ES, Klocke B, Conrad GW. Analysis of transcriptional regulation of the small leucine rich proteoglycans. Mol Vis. 2004;10:758–72.
Plaas AH, West LA, Thonar EJ, Karcioglu ZA, Smith CJ, Klintworth GK, et al. Altered fine structures of corneal and skeletal keratan sulfate and chondroitin/dermatan sulfate in macular corneal dystrophy. J Biol Chem. 2001;276(43):39788–96.
Tanihara H, Inatani M, Koga T, Yano T, Kimura A. Proteoglycans in the eye. Cornea. 2002;21(7 Suppl):S62–9.
Podskochy A, Koulikovska M, Fagerholm P, van der Ploeg I. Biglycan gene expression in UVR-exposed rabbit corneas. Acta Ophthalmol Scand. 2004;82(2):200–4.
Ortiz-Melo MT, Garcia-Murillo MJ, Salazar-Rojas VM, Campos JE, Castro-Munozledo F. Transcriptional profiles along cell programming into corneal epithelial differentiation. Exp Eye Res. 2021;202:108302.
Naderi A. Prolactin-induced protein in breast cancer. Adv Exp Med Biol. 2015;846:189–200.
Sharif R, Bak-Nielsen S, Hjortdal J, Karamichos D. Pathogenesis of Keratoconus: The intriguing therapeutic potential of Prolactin-inducible protein. Prog Retin Eye Res. 2018;67:150–67.
Lee RY, Kao AA, Kasuga T, Vo BN, Cui QN, Chiu CS, et al. Ethnic variation in optic disc size by fundus photography. Curr Eye Res. 2013;38(11):1142–7.
Rao R, Dhrami-Gavazi E, Al-Aswad L, Ciarleglio A, Cioffi GA, Blumberg DM. Optic Nerve Head and Retinal Nerve Fiber Layer Differences Between Caribbean Black and African American Patients as Measured by Spectral Domain OCT. J Glaucoma. 2015;24(5):e43–6.
Chansangpetch S, Huang G, Coh P, Oldenburg C, Amoozgar B, He M, et al. Differences in Optic Nerve Head, Retinal Nerve Fiber Layer, and Ganglion Cell Complex Parameters Between Caucasian and Chinese Subjects. J Glaucoma. 2018;27(4):350–6.
Gedde SJ, Lind JT, Wright MM, Chen PP, Muir KW, Vinod K, et al. Primary Open-Angle Glaucoma Suspect Preferred Practice Pattern®. Ophthalmology. 2021;128(1):P151–92.
Georgiou T, Funnell CL, Cassels-Brown A, O'Conor R. Influence of ethnic origin on the incidence of keratoconus and associated atopic disease in Asians and white patients. Eye (Lond). 2004;18(4):379–83.
Pearson AR, Soneji B, Sarvananthan N, Sandford-Smith JH. Does ethnic origin influence the incidence or severity of keratoconus? Eye (Lond). 2000;14(Pt 4):625–8.
Acknowledgements
Not applicable.
Funding
The study was supported by Beijing Tianjin Hebei Basic Research Cooperation Project (J200006), Pharmaceutical Collaborative Innovation Research Project of Beijing Science and Technology Commission (L192062), and National Key Research and Development Project (SQ2018YFC200148). Dr. Jingyun Yang’s research was supported by NIH/NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG36042, U01AG61356 and 1RF1AG064312–01. Di Liu was supported by China Scholarship Council (CSC 201908110339).
Author information
Authors and Affiliations
Contributions
ZY, JY and WY designed and registered the study. DL and JY analyzed data and performed data interpretation. ZY, DL and JY wrote the initial draft, and JY and WY contributed writing to subsequent versions of the manuscript. All authors reviewed the study findings and read and approved the final version before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No potential conflicts of interest were disclosed by the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, Z., Yang, J., Liu, D. et al. Mendelian randomization analysis identified genes pleiotropically associated with central corneal thickness. BMC Genomics 22, 517 (2021). https://doi.org/10.1186/s12864-021-07860-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07860-3