
Abstract
Background
Not all cells in a given individual are identical in their genomic makeup. Mosaicism describes such a phenomenon where a mixture of genotypic states in certain genomic segments exists within the same individual. Mosaicism is a prevalent and impactful class of non-integer state copy number variation (CNV). Mosaicism implies that certain cell types or subset of cells contain a CNV in a segment of the genome while other cells in the same individual do not. Several studies have investigated the impact of mosaicism in single patients or small cohorts but no comprehensive scan of mosaic CNVs has been undertaken to accurately detect such variants and interpret their impact on human health and disease.
Results
We developed a tool called Montage to improve the accuracy of detection of mosaic copy number variants in a high throughput fashion. Montage directly interfaces with ParseCNV2 algorithm to establish disease phenotype genome-wide association and determine which genomic ranges had more or less than expected frequency of mosaic events. We screened for mosaic events in over 350,000 samples using 1% allele frequency as the detection limit. Additionally, we uncovered disease associations of multiple phenotypes with mosaic CNVs at several genomic loci. We additionally investigated the allele imbalance observations genome-wide to define non-diploid and non-integer copy number states.
Conclusions
Our novel algorithm presents an efficient tool with fast computational runtime and high levels of accuracy of mosaic CNV detection. A curated mosaic CNV callset of 3716 events in 2269 samples is presented with comparability to previous reports and disease phenotype associations. The new algorithm can be freely accessed via: https://github.com/CAG-CNV/MONTAGE.
Similar content being viewed by others

Background
Mosaicism is non-integer CNV resulting from a mixture of deleted and diploid or duplicated and diploid cells. Mosaic CNV creation mechanisms include: chromosome nondisjunction, anaphase lag, and endoreplication. Mosaicism was first studied in fruit flies by Alfred Sturtevant and Curt Stern demonstrating mitotic recombination. “Somatic mosaicism” terminology was used by C. W. Cotterman in his seminal paper about antigenic variation [1]. Mosaic CNV detection is important in clinical settings for accurate assessment and estimate of disease recurrence risk [2,3,4,5].
Almost all CNV detection algorithms interpret a splitting of genotypes into allelic imbalance to mean duplication. However, splitting of genotypes into allelic imbalance clusters means duplication only when paired with a gain in intensity, otherwise the event is actually a mosaic deletion. In this way, mosaicism is often incorrectly classified or missed entirely by conventional CNV detection algorithms.
Mosaicism calls from publically accessible programs, such as R-GADA-MAD, BAFSegmentation, Mocha, and triPOD have provided insight into the prevalence and impact of mosaic CNVs [6,7,8]. However, we were motivated to create a computationally efficient and easy to perform algorithm, particularly at the scale needed for large projects. The goal of the algorithm was to deliver results in a high-throughput way with few spurious calls and to successfully detect mosaic events multiple megabases in size. Previous studies have been limited by small sample size to achieve accurate frequency estimates ([9, 10], Bonnefond et al. 2013, Schick et al. 2013, [11, 12], and Rodriguez-Santiago et al. 2010).
Mosaic Alteration Detection (MAD) is among the most commonly used computational tool to identify mosaic events using both B-allele frequency (BAF) and log R ratio (LRR) values from SNP array data. The MAD method first performs a segmentation procedure using the GADA algorithm (Pique-Regi R, Caceres A, Gonzalez JR. BMC Bioinformatics, 2010), and then searches for aberrant segments (mosaic regions) with B-deviations (Bdev) different from zero. Bdev occurs in mosaic regions when the locus has a mixture of genotypes from normal and mosaic tissue. The detected mosaic regions are further classified as copy-loss, copy-gain, or copy-neutral events based on the alteration of the LRR from baseline. The MAD method is implemented as a package in R. and the program is available at GitHub (https://github.com/isglobal-brge/MAD). The state of mosaic alterations is determined based on log2-ratio segment values together with the percentage of normal heterozygous (BAF ∼ 0.5) and homozygous (BAF ∼ 0 or 1) probes. While MAD has been successfully applied in several mosaicism projects (Forsberg et al. 2012, 2014 [11];), the program installation remains difficult requiring many undocumented dependencies including the R package devtools rendering it difficult to operate.
It’s important to be aware that inherited alleles at some loci may appear to affect the probability of somatic mutations, and at other loci they may constitute objects of positive or negative clonal selection. Several specific mosaic CNVs are strongly associated with future risk of hematological malignancies [8]. Loh et al. evaluated blood-derived DNA from 151,202 UK Biobank participants genotyped with Affymetrix arrays, finding 8342 mosaic CNVs ranging 50 kb–249 Mb by using phase-based programming (false discovery rate ≈7.5%). Mosaic deletions were observed more frequently in males while mosaic duplications were observed more frequently in elderly and male samples and copy number neutral loss of heterozygosity (CNN-LOH) affected the sexes equally.
In addition to the large number of samples from the Center for Applied Genomics at Children’s Hospital of Philadelphia biobank presented here (Supplementary Table 1), we also explored the pediatric and adolescent age range (Supplementary Fig. 1), providing further insights into early mosaic detection possibilities. Taken together, our results reveal clonal expansions with a wide range of effects on human health.
Implementation
Due to current limitations and difficulties in available mosaic genomic events callers, we have developed a mosaic calling tool programmed in Perl and freely available through our GitHub webpage https://github.com/CAG-CNV/MONTAGE. We wrote the Perl code to be flexible in terms of column order and column inclusion in sample based input files. The minimum column requirement for input files is SNP Name, BAF, and LRR. SNP Name with associated chromosome and base pair position can be specified separately or combined in the input. Sorting by chromosome and position (if not done already) is the first step. This is the most run time consuming step at 25 s. If sorting is detected to be done already, only the mosaic CNV detection portion of the code runs taking 10 s per sample (Table 1).
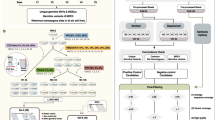
Since Windows computers are needed to run GenomeStudio to load idats and export BAF/LRR signal files, we remove any Windows carriage returns. We remove SNPs that failed to UCSC Liftover or low call rate SNPs marked with Position REMOVE. If not sorted, sort by chromosome and position (for linear runtime). Remove position 0 SNPs. A bash awk statement embedded in the Perl code efficiently performs a sliding window of 1 MB with 1 MB increments to roughly assess potential regions of mosaicism. The algorithm monitors position modulus window, if diff< 0 then report window: CHIP REGION AB ABLow ABHigh AAorBB AvgLRR BAF_SD(0.1–0.9). To define deletion, we take q3 + 1.5 interquartile range (iqr) as BAF standard deviation (SD) threshold and q1–1.5 iqr as LRR average threshold. To define duplication, we take q3 + 1.5 iqr as BAF SD threshold and q3 + 1.5 iqr as LRR average threshold. PennCNV script clean_cnv merges fragmented mosaic CNVs in neighboring genomic intervals. We record the first and last base pair position of mosaic evidence in these intervals to provide specific breakpoints (Fig. 2).
Methods used include programming in Bash, Perl, and R.
Efficient and minimal dependency coding allows for rapid ease of deployment of the software.
The BAF ranges used are tallied in the following intervals: (0–0.1) (0.1–0.4) (0.4–0.6) (0.6–0.9) (0.9–1). We use average LRR for each 1 Mb window in comparison with genome-wide average LRR. This presents the key command in the script. We note that our current version of MONTAGE, runs at 35 s per sample in a sample independent manner vs. 10 s if the input BAF/LRR signal file is sorted by chromosome and position.
Data preprocessing
We dynamically assess the column header to determine the presence and order of SNP Name, Chromosome, Position, B Allele Freq, and Log R Ratio column data in the user provided input files. These inputs may be generated by exporting text files from graphical user interface on Windows: Illumina GenomeStudio or Affymetrix Genotyping Workbench. Alternatively, these files can be generated by exporting text files from command line on Linux: Illumina iaap or Affymetrix apt. The flexibility to include only SNP Name, B Allele Freq, and Log R Ratio column data is allowed to minimize the disk space footprint of input data files, provided a separate map file linking SNP Name to chromosome and position. SNP microarray, whole exome sequencing and whole genome sequencing data are all supported input data types based on normalization of read depth to generate LRR signal and clustering of allele depth to generate BAF.
Mosaicism detection
Using an ultra-efficient awk bash command, we are able to run an optimal non-overlapping sliding window algorithm to determine BAF in the mosaic deletion indicative ranges of (0.1–0.4) and (0.6–0.9) as well as the standard deviation of these observations to determine clarity (lack of noise) in the signal observed in a given sample. We implemented a sliding window approach to assess these BAF intervals for allelic imbalances and strong deviations from expected values. Average LRR values across the sliding window interval classify the mosaic CNV as a deletion or duplication relative to normal diploid copy number. High standard deviation of BAF (0.1,0.9) regions were prioritized (for those samples passing quality control with acceptably low genome-wide standard deviation of BAF).
Mosaicism breakpoint refinement
PennCNV (version 1.0.4) component script clean_cnv was used to combine segments in close proximity into one merged mosaic CNV call. Record the first and last base pair position of mosaic evidence in these intervals to provide specific breakpoints (Fig. 2).
Mosaicism algorithm differences
We do not perform phasing as done in MoCha to save on computational time. We do not require or use family information as done in triPOD. We focus our code in the extensible Perl and Bash programming languages as opposed to RGADA-MAD which is written in R. We use standard modern GitHub code tracking as opposed to BAFSegmentation which is on an institutional website.
Results
We assessed the performance of existing mosaic CNV detection algorithms (Table 1). We constructed a model reference of various levels of mosaicism (Fig. 1). We measured the allelic imbalance between proper heterozygous (AB) genotypes centered on 0.5 BAF versus those outside of this region (Fig. 2 and Supplementary Fig. 2). We evaluated mosaic events in 367,785 samples and found 3716 putative mosaic events in 2269 individuals with 2/3 of the raw mosaic calls being visually validated. In total, 187,096 mosaic CNV candidates were suggested by the first pass screening of our algorithm applied to approximately 350,000 SNP-array data sets. Next, we filtered out mosaic CNV candidates with overlapping homozygous deletion calls as detected by PennCNV, since the random noise in BAF for real homozygous deletions can give a false indication of aberrant BAF banding, leaving us with 126,020 mosaic CNV candidates in 43,781 samples. The MONTAGE algorithm no longer requires PennCNV homozygous deletion calls in order to minimize runtime using an approximation subroutine. There were 51,326 mosaic CNV candidates with at least one mosaic CNV candidate > = 3 Mb in genomic span. Finally, 19,090 mosaic CNV candidates had strictly one mosaic CNV candidate > = 3 Mb, suggesting high specificity of mosaic CNV detection in these samples (Fig. 3). Therefore, we set forth visualizing the underlying BAF and LRR profiles corresponding to these mosaic CNV candidates.

Example BAF and LRR Profiles Sampled from Various Levels of Mosaicism. Mosaic deletions have clearing of the 0.5 AB range and relatively equal banding of AAB and ABB range genotypes reflected by BAF. Less relative gain in LRR is observed in mosaic duplications, making them more difficult to detect with certainty. Data displayed is simulated to ensure consistent underlying data quality profiles for fair comparison of different mosaic copy number states

MONTAGE Algorithm Conceptual Flow Diagram. Code overview with key inputs and variables. First data is normalized to BAF and LRR values genome-wide for each sample. Then the number of deviating BAF values in a first pass sliding window are accounted to screen for potential mosaic events as an initial algorithmic step. The first quartile minus 1.5 interquartile range of the LRR paired with strong BAF deviation defines the calling threshold for mosaic deletion events. Fragmented windows meeting threshold are then merged together to form larger calls. The breakpoints are then refined based on the first and last specific base pair evidence of strongly deviating BAF within the merged window

MONTAGE Filtering to Arrive at Curated Callset. Filtering of the putative mosaic CNV calls and respective size of the curated callset at each step. PennCNV calls for homozygous deletions (copy number 0) on the same samples analyzed by MONTAGE were intersected with MONTAGE initial mosaic calls using bedtools software. Further visualization of BAF/LRR underlying potential mosaic CNV calls was conducted manually by a human expert reviewer (in the case only 1 mosaic call in the sample) or by DeepCNV algorithm (in the case 2 or more mosaic calls in the sample)
We verified ability to detect both high (Fig. 4) and low (Fig. 5) level mosaic CNV events with high sensitivity and specificity. We identified 273 putative mosaic CNV deletions in 76 out of 228 samples analyzed. Of those, 202 visually validated as true positive, confirming mosaic CNV deletions in 50 samples out of 228 samples. Median length of these CNVs was 8.5 Mb with average length of 24 Mb. The visually validated mosaic calls had at least 2 AB clearing ratio (equating to 15 AB(0.4–0.6 BAF), 16 low AB (0.1–0.4 BAF), and 16 high AB (0.6–0.9 BAF) observations in a 1 Mb genomic window. Approximately half of the mosaic calls had non-zero AB(0.4–0.6 BAF) signal indicating noise and or lower levels of mosaicism.

Higher Levels of Mosaicism Genome-wide LRR BAF plot representative for one Individual. Full deletion of chromosome 1p contrasted by duplication of 1q is shown along with mosaic deletions of high proportion of cells in the person’s sample on 2q, 8q, and 11p. We use alternating colors similar to a Manhattan plot for GWAS to represent the switch between chromosomes on a linear x axis

Lower Levels of Mosaicism Genome-wide LRR BAF plot representative for one Individual. Mosaic deletions of low proportion of cells in the person’s sample on 3p, 10q, 11q, and 14q
In order to validate the rest of the mosaic CNV candidates, we used a machine learning approach we developed called DeepCNV (in review). DeepCNV is based on a trained model of positive and negative mosaic CNV examples based on a human expert’s labeling. Using this model and images of LRR/BAF plots from PennCNV visualize_cnv which are standard and popularly used, probabilistic predictions of the mosaic CNV candidate being a true positive are output. This makes the prospect of visual validation much more tractable and reproducible with minimum generated bias.
We compared our observed mosaic CNV counts and frequencies to previous studies of mosaicism and found high concordance in genomic regions and their corresponding frequencies observed in populations (Fig. 6). In addition, when examined in the context of multiple disease phenotypes that these individuals harbored, several disease categories were associated with mosaic CNVs based on results generated using the ParseCNV software (Table 2). ParseCNV is a CNV GWAS tool [13]. Association p-values as low as 1E-39 were observed across phenotypes including: adhd, autism, autoimmune, cancer, congenital, healthy, and neurodegenerative [14,15,16]. Thirty three genomic loci were observed p < 5E-4 associated between mosaic events and human phenotypes. Interestingly, we observed potential protective association in healthy subjects and also may consider mosaic CNVs related to healthy subjects as negative controls [17, 18].

Genome-wide Count Mosaic CNVs Detected in Curated Callset in Comparison to Previous Studies. Blue line represents the current work from Montage mosaic CNV callset. Red line represents the Loh et al. mosaic CNV callset. Black line represents the other previously published mosaic CNV callsets. Previous includes: [9, 10], Bonnefond et al. 2013, Schick et al. 2013, [11, 12], and Rodriguez-Santiago et al. 2010. See Supplementary Dataset (glessner2020_hg19.txt, loh2018_hg19.txt, and prev_calls_hg19.txt)
We compared calling on the same sample data for 755 individuals using MONTAGE, MoCha, and RGADA-MAD (Fig. 7). Using a majority voting scheme, many more mosaic CNV calls overlap MONTAGE than MoCha or RGADA-MAD. If we assume samples selected by two or more callers as true positives, then we have 1748 + 194 + 61 + 418 = 2421. Apparently MONTAGE has the highest sensitivity: 1–61/2421 = 0.975, followed by MoCha 1–194/2421 = 0.920 and RGADA-MAD 1–1748/2488 = 0.280. To compute specificity using this similar majority-vote approach, we need to know the size of the background in the background, namely, the number of samples that are considered as negatives by all three callers, which is not well defined. Alternatively, we can look at accuracy (true positive rate), which is generally a trade-off for sensitivity. Based on the Venn diagram, MoCha has the largest number of samples called by itself (3316). If we assumed the samples called by only one caller are false positives, then the accuracy (true positive rate) of MONTAGE is (1748 + 194 + 418)/ (1748 + 194 + 418 + 1236) = 0.656 vs. MoCha (1748 + 61 + 418)/(1748 + 61 + 418 + 3316) = 0.402 vs. RGADA-MAD (194 + 61 + 418)/ (194 + 61 + 418 + 1130) = 0.373. This is a better tradeoff for MONTAGE. Namely, sensitivity of 0.975 at false positive rate 0.344 (MONTAGE) vs. sensitivity of 0.920 at false positive rate 0.598 (Mocha). We note that this estimate is contingent on assumptions. While we acknowledge that these assumptions are imperfect, this analysis gives good evidence that our FDR is well-controlled. (We also note that while we cannot completely rule out the possibility that our FDR is higher than we estimated, the key results of our paper are robust to higher FDRs than estimated; e.g., we would only expect a higher-than-estimated FDR to weaken GWAS associations and decrease effect sizes.)

Venn Diagram Callsets Mosaic Positive Samples. We compared calling on the same sample data for 755 individuals using MONTAGE, MoCha, and RGADA-MAD. Sensitivity of 0.975 at false positive rate 0.344 (MONTAGE) vs. sensitivity of 0.920 at false positive rate 0.598 (MoCha) vs. sensitivity of 0.280 at false positive rate 0.627 (RGADA-MAD) based on 755 Illumina Human610-Quadv1 samples analyzed by all algorithms. The MONTAGE algorithm provides a competitive tool to be used for mosaic CNV detection
63% of the mosaic events identified were found in males compared to the 50% male percentage in the input dataset Fisher’s exact test (2-Tail) p = 1.554e-11 (Supplementary Table 2). Mosaic duplications were observed more in males and older individuals. Mosaic CNN-LOH were found to affect the sexes equally. The sample race as determined by principal components analysis is provided in Supplementary Table 3.
Discussion
Mosaic CNVs of intermediate states between integer copy number variation are important genetic/genomic events in both clinical and research settings. However, detection of these mosaic events has been limited to incidental findings from CNV algorithms designed for integer discrete copy numbers and not the continuous nature of mosaic CNVs. For example, the tripod algorithm requires parents for calling mosaic events therefore is limited. Moreover, in a recent study using Mocha [8], 8342 mosaic chromosomal alterations (mCAs) were reported in 7585 individuals ranging in length from 50 kb–249 Mb. These mCAs were obtained from blood-derived DNA samples from 151,202 UK Biobank participants aged 40–70 years using new phase-based computational techniques (estimated false discovery rate, 6–9%). However, as 5522 of these mosaics have negative LRR values, they should be considered to be deletions. MAD was notably used in The Cancer Genome Atlas (TCGA) mosaic CNV analysis [19]. Exome sequencing (~ 8000 samples) was used to compare 22 different cancer phenotypes with more than 6000 controls using a case–control study design and demonstrate that mosaic protein truncating variants in these genes are also associated with solid-tumor cancers.
In light of shortage of high performance tools, we designed a new mosaic CNV detection tool aimed at providing high sensitivity and specificity mosaic CNV detection and fast runtime. In comparison, we show that in certain circumstances other algorithms miss critical mosaic events while overcalling other false events.
Others have shown that mosaic CNVs are enriched in males [8]. Our analysis concurs with this, showing that 63% of the mosaic events identified were found in males.
This mosaic CNV detection work has implications in cancer, cell free fetal DNA, and aging [20, 21]. Tumor-normal heterogeneity can appear similarly to germline mosaic CNV. Therefore, cancer phenotyping records are important in conditioning the assessment of supposed mosaic CNV callsets. Cell free fetal DNA is another application that such mosaic CNV detection and association presented here could be of utility. Prenatal testing could be enhanced by deconvolution of the maternal and child CNV genotype profile. Aging accumulating CNVs has been investigated previously [11, 12]. In older age cohorts which have had more exposure to potential environmental hazards inducing CNVs in subsets of cells is another important longitudinal consideration [22, 23].
Conclusions
Mosaic CNVs represent an important class of variation in clinical genetic diagnosis that are often missed. To successfully diagnose mosaic CNVs, it’s important to develop targeted detection tools and systematically apply them to large cohorts to truly understand its relevance and frequency of mosaic CNVs in the general population. Here we demonstrate the utility of our fast scalable tool, MONTAGE, specifically designed for mosaic CNV detection. We envision MONTAGE being an integral part to include for future mosaic CNV detection and analysis.
Availability and requirements
- Project name: MONTAGE.
- Project home page: https://github.com/CAG-CNV/MONTAGE
- Operating system(s): Platform independent.
- Programming language: bash, perl.
- Other requirements: NA.
- License: GNU GPL.
- Any restrictions to use by non-academics: license needed.
Availability of data and materials
https://github.com/CAG-CNV/MONTAGE
All data authorized for dbGaP submission have been deposited to dbGaP (accessions: phs000490.v1.p1, phs000607.v3.p2, phs000371.v1.p1, phs000490.v1.p1, phs001194.v2.p2, phs001194.v2.p2.c1, phs001661, and phs000233).
Abbreviations
- CNV:
-
Copy number variation
- BAF:
-
B-allele frequency
- LRR:
-
Log R ratio
- MAD:
-
Mosaic alteration detection
- MCA:
-
Mosaic chromosomal alteration
References
Cotterman CW. Somatic mosaicism for antigen A2. Acta Genet Stat Med. 1956;6:520–1.
Liu, Qian, et al. "Parental somatic mosaicism for CNV deletions–A need for more sensitive and precise detection methods in clinical diagnostics settings." Genomics (2020).
Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19(7):1263–75. https://doi.org/10.1093/hmg/ddq003 Epub 2010 Jan 6.
Luo M, Mulchandani S, Dubbs HA, Swarr D, Pyle L, Zackai EH, Spinner NB, Conlin LK. Detection of mutually exclusive mosaicism in a girl with genotype-phenotype discrepancies. Am J Med Genet A. 2015;167A(12):3091–5. https://doi.org/10.1002/ajmg.a.37261 Epub 2015 Jul 21.
Spinner NB, Conlin LK. Mosaicism and clinical genetics. Am J Med Genet C Semin Med Genet. 2014;166C(4):397–405. https://doi.org/10.1002/ajmg.c.31421 Epub 2014 Nov 25.
Baugher JD, Baugher BD, Shirley MD, Pevsner J. Sensitive and specific detection of mosaic chromosomal abnormalities using the parent-of-origin-based detection (POD) method. BMC Genomics. 2013 May 31;14:367. https://doi.org/10.1186/1471-2164-14-367.
González JR, Rodríguez-Santiago B, Cáceres A, Pique-Regi R, Rothman N, Chanock SJ, Armengol L, Pérez-Jurado LA. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011;12:166. https://doi.org/10.1186/1471-2105-12-166.
Loh PR, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA, Price AL. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018;559(7714):350–5. https://doi.org/10.1038/s41586-018-0321-x Epub 2018 Jul 11.
Machiela MJ, et al. Characterization of large structural genetic mosaicism in human autosomes. Am J Hum Genet. 2015;96:487–97 PubMed: 25748358.
Vattathil S, Scheet P. Extensive hidden genomic mosaicism revealed in normal tissue. Am J Hum Genet. 2016a;98:571–8 PubMed: 26942289.
Jacobs KB, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–8 PubMed: 22561519.
Laurie CC, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–50 PubMed: 22561516.
Glessner JT, Li J, Hakonarson H. ParseCNV integrative copy number variation association software with quality tracking. Nucleic Acids Res. 2013. https://doi.org/10.1093/nar/gks1346.
King DA, Jones WD, Crow YJ, Dominiczak AF, Foster NA, Gaunt TR, Harris J, Hellens SW, Homfray T, Innes J, Jones EA, Joss S, Kulkarni A, Mansour S, Morris AD, Parker MJ, Porteous DJ, Shihab HA, Smith BH, Tatton-Brown K, Tolmie JL, Trzaskowski M, Vasudevan PC, Wakeling E, Wright M, Plomin R, Timpson NJ, Hurles ME. Deciphering Developmental Disorders Study. Mosaic structural variation in children with developmental disorders. Hum Mol Genet. 2015;24(10):2733–45. https://doi.org/10.1093/hmg/ddv033 Epub 2015 Jan 29.
McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch J, Hall IA, Gage FH. Mosaic copy number variation in human neurons. Science. 2013a;342(6158):632–7.
Notini AJ, Craig JM, White SJ. Copy number variation and mosaicism. Cytogenet Genome Res. 2008;123(1–4):270–7. https://doi.org/10.1159/000184717 Epub 2009 Mar 11.
Collins RL et al. An open resource of structural variation for medical and population genetics. bioRxiv. n.d. 578674; doi: https://doi.org/https://doi.org/10.1101/578674.
Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease—clones picking up speed. Nat Rev Genet. 2017;18:128–42.
Artomov M, Rivas MA, Genovese G, Daly MJ. Mosaic mutations in blood DNA sequence are associated with solid tumor cancers. NPJ Genom Med. 2017;2:22. 2017 Jul 6. https://doi.org/10.1038/s41525-017-0025-4.
Ruttanajit T, Chanchamroen S, Cram DS, Sawakwongpra K, Suksalak W, Leng X, Fan J, Wang L, Yao Y, Quangkananurug W. Detection and quantitation of chromosomal mosaicism in human blastocysts using copy number variation sequencing. Prenat Diagn. 2016;36(2):154–62. https://doi.org/10.1002/pd.4759 Epub 2016 Jan 27.
Zhou B, Haney MS, Zhu X, Pattni R, Abyzov A, Urban AE. Detection and quantification of Mosaic genomic DNA variation in primary somatic tissues using ddPCR: analysis of Mosaic transposable-element insertions, copy-number variants, and single-nucleotide variants. Methods Mol Biol. 1768;2018:173–90. https://doi.org/10.1007/978-1-4939-7778-9_11.
Chronister WD, Burbulis IE, Wierman MB, Wolpert MJ, Haakenson MF, Smith ACB, Kleinman JE, Hyde TM, Weinberger DR, Bekiranov S, McConnell MJ. Neurons with complex karyotypes are rare in aged human neocortex. Cell Rep. 2019;26(4):825–835.e7. https://doi.org/10.1016/j.celrep.2018.12.107.
Cohen AS, Wilson SL, Trinh J, Ye XC. Detecting somatic mosaicism: considerations and clinical implications. Clin Genet. 2015;87(6):554–62. https://doi.org/10.1111/cge.12502 Epub 2014 Oct 7.
Acknowledgements
We thank the study participants who allowed for the use of genotyping, sequencing and disease phenotype data for this study, and to testers of the codes used in this study.
Funding
This work was supported by a sponsored research agreement from Aevi Genomic Medicine Inc. (HH) and by an Institutional Development Award from Children’s Hospital of Philadelphia (HH).
The funders had no role in this study including the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
JTG designed and wrote the software. JTG, JL, YL, MK, XC, ZW, PMAS, and HH wrote and drafted the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent authorizing the use of de-identified GWAS data was obtained from all subjects. The Institutional Review Board at The Children’s Hospital of Philadelphia approved the study. Written informed consent to participate in this study was obtained from the parents or legal guardians of any participant under the age of 16 with verbal assent from the participant. No administrative permissions and/or licenses were necessary to be acquired by our team to access the clinical/personal patient data used in our research.
The Institutional Review Board of The Children’s Hospital of Philadelphia approved this study. De-identified sample data was used to conduct this study.
Consent for publication
Not Applicable.
Competing interests
Dr. Hakonarson and CHOP own stock in Aevi Genomic Medicine (unrelated to this manuscript); otherwise, none declared.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Fig. 1.
Age Distribution of Studied Cohort. Supplementary Fig. 2. Modeling B-allele Frequency Standard Deviation for Mosaic Copy Number States. Supplementary Table 1. Samples Genotyped on Illumina SNP microarray platforms. Supplementary Table 2. Genotyping Sex. Supplementary Table 3. Genotyping Race.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Glessner, J.T., Chang, X., Liu, Y. et al. MONTAGE: a new tool for high-throughput detection of mosaic copy number variation. BMC Genomics 22, 133 (2021). https://doi.org/10.1186/s12864-021-07395-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07395-7