
Abstract
Background
The family of NAC proteins (NAM, ATAF1/2, and CUC2) represent a class of large plant-specific transcription factors. However, identification and functional surveys of NAC genes of tomato (Solanum lycopersicum) remain unstudied, despite the tomato genome being decoded for several years. This study aims to identify the NAC gene family and investigate their potential roles in responding to Al stress.
Results
Ninety-three NAC genes were identified and named in accordance with their chromosome location. Phylogenetic analysis found SlNACs are broadly distributed in 5 groups. Gene expression analysis showed that SlNACs had different expression levels in various tissues and at different fruit development stages. Cycloheximide treatment and qRT-PCR analysis indicated that SlNACs may aid regulation of tomato in response to Al stress, 19 of which were significantly up- or down-regulated in roots of tomato following Al stress.
Conclusion
This work establishes a knowledge base for further studies on biological functions of SlNACs in tomato and will aid in improving agricultural traits of tomato in the future.
Similar content being viewed by others


Background
Aluminum (Al) is the most abundant metal element in the earth’s crust. Although it is nontoxic when it exists in oxides or hydroxides in neutral and alkaline conditions, the solubility of Al increases dramatically when soil pH is lower than 5.5, and solubilized Al is highly toxic to most plant species [1]. However, nearly 30% of arable lands and 50% of potentially arable lands are estimated to be acidic [2]. Therefore, Al toxicity is well recognized as one of the major edaphic factors threatening food security worldwide [1]. To survive the acidic Al toxic environment, plants have developed complicated coping mechanisms, which are largely controlled by transcriptional regulation in response to Al stress [3].
Al-induced changes in gene expression occur within hours of exposure in the root apex of some plant species, suggesting that transcriptional regulation is vital for plants to adapt to the stress [4,5,6]. Plant transcription factors (TFs) are central regulators that direct transcription via binding to special nucleotide sequences in response to developmental cues and environmental stresses [7]. Since the first report on an Arabidopsis mutant hypersensitive to both low pH and Al, STOP1 (Sensitive to proton rhizotoxicity 1) and its homologous genes from other plant species have been well-documented as a very important TF regulating several critical processes involved in Al tolerance [8]. In addition, several other TFs have also been characterized and implicated in Al tolerance. However, the majority are demonstrated to play minor roles in regulation of the expression of genes involved in organic acid anion secretion [8]. For example, whilst AtALMT1 (Al-activated malate transporter 1) expression was predominantly controlled by STOP1, CAMTA2 (CALMODULIN-BINDING TRANSCRIPTION ACTIVATOR2) and WRKY46 had a positive and negative role, respectively, in regulating AtALMT1 expression under Al stress [9, 10]. Although ART1 (Al resistance transcription factor 1) is a master TF controlling the expression of Al-tolerance genes including OsFRDL4 in rice, WRKY22 was recently reported to bind to the promoter of OsFRDL4 and regulate its expression [11]. However, other TFs in Al tolerance remain to be characterized.
As an important class of TFs, NAC, which is a descendent of 3 proteins of NAM (No apical meristem), ATAF 1/2 (Arabidopsis transcription activator factor 1/2) and CUC2 (Cup shaped cotyledon) [12], is a class of plant specific TFs and constitute one of the largest TF families in plants [13]. Typically, NAC TFs have a conserved NAM domain at the N-terminus and a diverse transcription regulatory region at the C-terminus [14]. It has been shown that NAC TFs have a crucial position not only in plant development and growth, but also in stress responses [15, 16].
Recently, several lines of evidence suggest the implication of NAC TFs in response to Al stress in plants. For instance, 25 NAC genes were found to be differentially expressed among different rice genotypes in response to Al stress and most of these NAC genes belong to the NAM subfamily [17]. We previously identified a NAC transcription factor gene up-regulated by Al stress in the root apex of rice bean [4]. Further functional characterization of this rice bean NAC gene showed that it could regulate WAK1 (Wall-associated protein kinases) expression and cell wall pectin metabolism when ectopically overexpressed in Arabidopsis [18]. SOG1 (SUPPRESSOR OF GAMMA RESPONSE1) is a NAC protein that acts as a central DNA damage response component [19]. Interestingly, SOG1 loss-of-function mutant displayed better root growth in comparison with wild-type plants during long-term exposure to low dosage Al [19]. However, sog1 mutant became extremely sensitive to Al when higher Al concentrations were applied in the growth medium [20]. Although these results suggest a complexity of responses of Arabidopsis plants to Al-induced DNA damage, it provided solid evidence that a NAC protein, SOG1, is involved in the Arabidopsis response to Al stress.
Tomato (Solanum lycopersicum) ranks fourth among the leading world vegetables in production. It is a rich source of nutrients and a model plant for fleshy fruit development [21]. However, with a continuously expanding scale of cultivation of tomato, they have suffered serious damage in recent years, not only caused by abiotic stresses like drought or temperature stress but also various pathogens and pests, such as fungi, insects and nematodes [22]. Unfortunately, few studies have focused on the response of tomato to Al stress. In a previous study, we characterized root organic acid anions secretion from tomato roots [23]; however, the underlying molecular basis is unknown. In the preset study, we aimed to provide a comprehensive view of the NAC gene family in tomato and to identify members involved in the response to Al stress.
Results
Genome-wide identification and phylogenetic analysis of the NAC gene family in tomato
In our study, BLAST and HMM searches were performed to broadly identify tomato NAC family using the NAC protein sequences in Arabidopsis and rice as queries. All of the putative proteins fulfilled the criteria of NAC proteins as described in previous research [7, 24]. As a result, 93 putative NAC proteins were identified in the S. lycopersicum genome, which were designated as SlNAC1-SlNAC93 based on their locations on the chromosomes (Table S1). The number of amino acid residues of the predicted SlNACs ranged from 108 to 1029, and their molecular mass varied from 12.28 to 117.0 kDa (Table S1). To probe the phylogenetic relationships among these 93 SlNACs, a phylogenetic tree was constructed by combining SlNACs with Arabidopsis NAC proteins (AtNACs). Because sequence lengths varied dramatically, phylogenetic tree was constructed based on maximum likelihood algorithm following [7]. The results indicated that the NAC family could be divided into 5 subfamilies (Group I, Group IIa, Group IIb, Group IIIa, and Group IIIb) (Fig. 1). Group III was the largest with 39 SlNACs and 2 subgroups (IIIa and IIIb) followed by groups II with 34 proteins and 2 subgroups (IIa and IIb) and Group I including 20 NACs was a species-specific subgroups of tomato (Fig. 1). These results suggest that these NACs may have crucial roles in the evolution of the tomato genome.

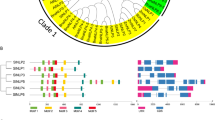
Phylogenetic analysis of tomato (Solanum lycopersicum) NACs (SlNACs). Phylogenetic analysis of NACs from tomato and Arabidopsis using the complete protein sequences. The Neighbor-joining (NJ) tree was constructed using MUSCLE and MEGA 7.0 software with the pairwise deletion option and 1000 bootstrap replicates were used to assess tree reliability. NACs from each plant species have colored labels. NACs of different plant species fell in 5 separate subfamilies as Group I, Group IIa, Group IIb, Group IIIa and Group IIIb
Gene structure and protein motif analysis of SlNAC genes
During the evolution of multigene families, the diversification of gene structure is responsible for evolving gene new function to adapt to the change of the living environments [25, 26]. To understand the structural diversity of SlNAC genes, intron/exon organization and conserved motifs were analyzed as described in previous research [13, 14]. Gene structure analysis showed that among these 93 SlNAC genes, 14 had no intron, and the others had at least one intron. Most of SlNAC members in the same subfamily displayed similar exon-intron structure (Fig. 2). Interestingly, most numbers in group I had only one exon (Fig. 2). This may be because that they are a specific class of NACs of tomato.

The exon-intron structure of SlNAC genes in accordance to the phylogenetic relationship. The unrooted phylogenetic tree was constructed with 1000 bootstrap based on the full length sequences of SlNACs. Exon-intron structure analysis of SlNAC genes was performed by using the online tool GSDS. Lengths of exons and introns of each SlNAC gene were exhibited proportionally
To further detect potential conserved motifs of SlNAC proteins (SlNACs), we also analyzed the putative motifs using the MEME program as described in previous research [7, 26]. As a result, 20 divergent motifs were identified in SlNACs, which were successively named as motifs 1–20 (Fig. 3). As expected, the closely-related members in the phylogenetic tree generally had mutual motif compositions and only minor differences were observed at subgroup levels (Fig. 3), indicating that there might have functional similarities among the SlNAC proteins within the same subgroup. This is consistent with a previous study showing that Solanaceae plants have specific NAC transcription factors [27]. Collectively, these results suggest that SlNACs possessing similar gene structures and motifs were clustered in the same subgroup and might have similar functions in the evolution of tomato.

Conserved motifs of SlNAC proteins in accordance to the phylogenetic relationship. The conserved motifs in the SlNAC proteins were identified by MEME. Grey lines represent the non-conserved sequences, and each motif is indicated by a colored box numbered at the bottom. The length of motifs in each protein was displayed proportionally
Chromosomal distribution and synteny analysis of SlNAC genes
To examine the chromosomal distribution of the SlNACs, the genomic sequence of each SlNAC was utilized to search against the tomato genome database with BLAST software. Physical map positions demonstrated that all of the 93 SlNAC genes could be mapped on 12 chromosomes in increasing order from short arm to long arm telomere (Fig. 4). Although each chromosome encompasses some SlNAC genes, the distribution is uneven (Fig. 4). The gene density per Chr (chromosome) ranged from 2.15% (2 SlNAC genes on Chr 09) to 16.13% (15 SlNAC genes on Chr 02), and relatively low numbers of SlNAC genes were observed in some chromosomes, such Chrs 01 and 12 (Fig. 1).

Schematic representations for the distribution and duplication of 93 SlNAC genes. Black lines represent the chromosomal location of SlNAC genes, and the red lines indicate duplicated SlNAC gene pairs. The chromosome number is indicated on the left side of each chromosome
Furthermore, we also investigated tandem repeats and segmental duplication events of the SlNAC genes to explore the mechanism underlying the expansion of the SlNAC gene family. In this study, multiple potential pairs linked each of at least 5 tandem repeats and 17 chromosomal segmental duplications were identified (Fig. 4), such as the large sections of Chrs 02 and 07 and Chrs 06 and 08. A previous report has demonstrated that the relatively recent (> 50 million years ago) genome-wide duplication (GWD) has caused a transition of 7 ancestral chromosomes to 12 chromosomes in the tomato [21]. Consistently, we found that there were at least 34 SlNAC genes involved in the GWD segment (Fig. 4). These results suggest that some SlNACs were possibly produced by gene duplication and the segmental duplication events, which might play a major driving force for SlNAC evolution in tomato.
Tissue specific expression patterns of SlNACs
To further explore the expression patterns of the putative SlNAC genes, we analyzed their expression profiles in different tissues and development stages of a cultivar Heinz cultivar and wild species S. pimpinellifolium using public RNA-seq data [20]. It showed that 96.8% and 94.6.3% of SlNACs were expressed in at least one tissue (stage) of Heinz and S. pimpinellifolium, respectively (Fig. 5). Twenty-one genes (SlNAC001, SlNAC003, SlNAC024, SlNAC025, SlNAC035, SlNAC037, SlNAC039, SlNAC040, SlNAC043, SlNAC044, SlNAC047, SlNAC055, SlNAC063, SlNAC064, SlNAC078, SlNAC081, SlNAC082, SlNAC083, SlNAC084, SlNAC090, and SlNAC093) were constitutively expressed in all the stages analyzed in the Heinz cultivar, whereas the transcripts of 11 genes (SlNAC012, SlNAC014, SlNAC021, SlNAC023, SlNAC029, SlNAC034, SlNAC052, SlNAC057, SlNAC061, SlNAC086, and SlNAC092) were hardly detectable. Among these genes, SlNAC082 had the highest expression level in both the Heinz cultivar and wild species S. pimpinellifolium (Fig. 5).

Temporal and tissue-specific expression patterns of 93 SlNAC genes. a Expression profile of SlNAC genes in cultivated tomato cultivar Heniz. b Expression profile of SlNAC genes in wild species S. pimpinellifolium. Expression data were processed with Log2 normalization. The colour scale represents relative expression levels. DPA, days post anthesis
When the expression levels of SlNACs in various tested organs were compared between the Heinz cultivar and S. pimpinellifolium, 45 showed similar expression patterns in both genotypes of tomato, with 11 genes barely expressed in all tested organs. Conversely, 39 genes showed significant differential expression patterns in the two tomato genotypes (Fig. 5). Notably, the expression of eleven genes was restricted to the leaf (SlNAC073) and root (SlNAC007, SlNAC013, SlNAC017, SlNAC041, SlNAC042, SlNAC050, SlNAC051, SlNAC068, SlNAC075, and SlNAC091) in Heniz cultivar, whilst only one gene was noted in the root (SlNAC050) in S. pimpinellifolium. Furthermore, in the Heniz tomato cultivar, expression of three SlNAC genes (SlNAC015, SlNAC032, and SlNAC076) was hardly detectable in young tomato fruits (1 cm-, 2 cm-, and 3 cm-fruit), whereas a distinct expression pattern was detected in the breaker fruits (Fig. 5a). In S. pimpinellifolium, expression of five SlNAC genes (SlNAC003, SlNAC013, SlNAC028, SlNAC059, and SlNAC078) in young fruits (10 DPA and 20 DPA) was higher than that in breaker fruits (30 DPA) (Fig. 5b). This suggests that the SlNACs are regulated in a tissue-specific manner in tomato.
Expression profiles of SlNAC genes in response to Al stress
Following an extensive analysis of SlNAC gene family in tomato, we next attempted to investigate the potential implication of SlNACs in responding to Al stress. The inhibition of root elongation was the primary visible symptom of Al toxicity and the relative root elongation is widely used to indicate Al toxicity or Al tolerance. Our preliminary experiment indicated that the relative root elongation was about 60% when 5 uM Al was applied for 6 h (Fig. S1), suggesting that 5 uM of Al and 6 h of exposure is suitable for investigating the effects of Al on tomato roots. To this end, the gene expression profiles of SlNACs in a tomato cultivar Ailsa Craig were examined using transcriptome analysis. As shown in Table S2, a total of 6 samples were subjected to RNA-Seq and generated about 6.77Gb data for each sample on average. The average genome mapping rate is 87.50% and the average gene mapping rate was 76.22%. Next, clean reads were mapped to the reference genome after merging novel coding transcripts with reference transcripts, and RNA-Seq by Expectation Maximization tool, which was utilized to calculate gene expression levels of both gene and transcript [28]. The number of genes and transcripts of each sample is shown in Table S3. Based on the gene expression level, a total of 1620 up-regulated and 789 down-regulated differentially expressed genes (DEGs) were identified (Fig. S2). The gene lists are shown in Tables S4 and S5 for up- and down-regulated DEGs. Finally, 19 out of 93 SlNACs were found to have differential expression patterns after 6-h of exposure to 10 μM Al (Table S6). Among 19 Al-responsive SlNAC genes, 7 were found to have relatively high expression levels than others (Fig. 6a). The reliability of the RNA-Seq data was further verified by qRT-PCR analysis which was validated on 15 selected SlNAC genes. As shown in Fig. 6b, all of these 15 selected SlNAC genes exhibited similar expression patterns to that obtained by RNA-Seq. The Pearson correlation analysis showed a good correlation (R2 = 0.7514) between RNA-Seq data and qRT-PCR results (Fig. 6b). These results suggest that the RNA-Seq data accurately mirrored the transcriptional changes induced by Al stress.

Expression profiles of the SlNAC genes under Al stress. a Hierachical clustering of expression profiles of SlNAC genes during Al stress. b Correlation of gene expression levels between RNA-Seq data and qRT-PCR analysis. Fifteen SlNACs potentially responding to Al were selected and subjected to qRT-PCR analysis using the same RNA as for RNA-Seq. Both x- and y-axes are shown in Log2 scale
Expression of selected SlNACs under Al and CHX
The rapid induction of SlNAC gene expression in response to Al stress led us to question whether these SlNAC TFs were early genes or late genes involved in Al tolerance in tomato. To verify this, a protein translation inhibitor, CHX, was applied before Al stress. It can be assumed that de novo protein synthesis is not required for early-gene expression activation, and thus cannot be repressed by CHX. We choose 7 among 19 Al-responsive SlNACs because they have higher expression levels. Intriguingly, we found that the expression of all 7 tested SlNAC TFs was substantially induced by CHX even in the absence of Al (Fig. 7), implying that there may be a transcriptional repressor which blocks the transcriptional activation of SlNAC TFs in the absence of Al, and Al stress might cause the degradation of the repressor. To exclude the possibility that the up-regulation of these 7 SlNACs was caused by the toxic effects of CHX, we analyzed other SlNACs expression under CHX. We found that CHX treatment could both up-regulate and down-regulate the expression of SlNAC genes. For example, the expression of SlNAC056 was repressed by CHX (Fig. S3). In addition, we identified three FRD3-like genes in our RNA-Seq data, and found that the ability of Al to induce the expression of three FRD3-like genes was abolished by CHX (Fig. S4). These results suggest that these SlNAC TFs represent early genes involved in the Al stress response in tomato root apex.

Effects of a protein translation inhibitor, cycloheximide (CHX), on the expression of SlNAC genes. Three-day-old seedlings (cv. AC) were subjected to 1/5 strength Hoagland nutrient solution (10 μM Pi; pH 5.0) containing 0 or 5 μM CHX for 1 h, and then the seedlings were transferred to the same nutrient solution containing 0 or 10 μM Al for 6 h. Data are means ± SD (n = 3)
Discussion
In this present study, we systemically analyzed the NAC gene family in tomato, and identified a total of 93 SlNAC genes (Table S1). Numerous studies have shown that NAC TFs are widely distributed in different plant species and have potential roles in regulating plant development, growth and stress responses [15]. This family seemed to be one of the largest TFs up till now. There were 117 NAC genes in Arabidopsis [29], 151 in rice [30], 79 in grape [24], 180 in apple [13], 152 in maize [31], 71 in chickpea [32], 96 in cassava [26], 87 in sesame [14], 185 in Asian pears [7], and 80 in tartary buckwheat [33]. These data suggest that NAC genes have extensively expanded with their evolution. Therefore, phylogeny-based functional prediction is useful for functional characterization of SlNACs. We further divided the SlNAC gene family into 5 distinct subgroups based on the molecular phylogenetic analysis (Fig. 1). SlNACs and AtNACs from groups IIa, IIb, IIIa and IIIb showed that these genes were not only homologous but might even be evolved from a common ancestor. However, NACs from group I indicated that tomato NAC genes had a different ancestor from Arabidopsis (Fig. 1). Here, we provided the nomenclature of this gene family according to their chromosomal position (Table S1). As a very important vegetable and model crop for fleshy fruit development, functional genomics has become more and more popular, and studies on functional characterization of NAC genes in tomato have increased in recent years [34]. However, nomenclature on NAC genes is confusing in published studies. For example, a recent characterized tomato SlNAP2 involved in leaf senescence and crop yield has actually been previously reported as SlNAC35 [35, 36]. Tomato No-ripening (NOR) is NAC protein functioning as a positive regulator of fruit ripening [37]. Gao et al. (2018) identified a new NAC transcription factor named NOR-like 1, which is involved in tomato fruit ripening [38]. However, NOR-like1 is the same as SlNAC4 already characterized by Zhu et al. (2014) [39]. As summarized in Table S7, we have listed all the names of reported SlNACs and their corresponding names presented in this study. It might be useful for standardizing the naming of NAC gene in tomato for future study as well as to eliminate the naming confusion of previous studies.
It has been shown that a GWD event happened in tomato about 83–123 Myr in prior to divergence with grape [21] and this gene duplication had crucial roles in the expansion, rearrangement and functional variation of NAC genes [7]. In this study, there are 22 SlNAC gene pairs were found to be associated with gene duplication, including 17 GWD duplicate pairs and 5 duplicate pairs. These duplications appeared in all five groups; however, group I had only 1 pair of duplication (Fig. 4). These results suggest that both GWD and tandem duplications contributed greatly to the expansion of the SlNAC gene family in tomato. Furthermore, MEME showed that groups IIa, IIb, IIIa and IIIb had main similar motifs (Motif 1–Motif 7) with minor changes and exchanges. In contrast, members of group I have evolved additional motifs (Motifs 17 and 18) (Fig. 3). Gene structure analysis also illustrated no intronic regions in mostly members of group I (Fig. 2). Therefore, SlNAC genes from groups I could have functions specific to Solanum species and the members from this subgroup in Arabidopsis might have been lost during the evolution. Alternatively, members were independently evolved in Solanum species.
Generally, gene expression patterns are able to provide essential cues for gene function. Therefore, we determined the expression levels of the 93 SlNAC genes in leaf, root, flower, and fruit tissues using RNA-Seq data downloaded from the TFGD database. As shown in Fig. 5, a high and/or preferential expression of 45 SlNACs was detected, which displayed tissue- and development-specific expression patterns in leaf, root, flower, and breaker fruit. These genes may have important roles in growth and development of tomato and their precise functions still remains to be elucidated in further investigations. Furthermore, the expression patterns of some SlNACs differed in different tissues and development stages, suggesting that the SlNAC TFs may have diverse functions. In addition, we also found that 9 genes (SlNAC25, SlNAC35, SlNAC37, SlNAC43, SlNAC47, SlNAC81, SlNAC82, SlNAC83, and SlNAC93) were highly expressed in all the examined tissues (Fig. 5), suggesting that they may be involved in specific housekeeping activity in the growth and development of tomato. In future, more research will be needed to examine the precise functions of the SlNAC genes in tomato.
We identified 19 SlNAC genes that responded quickly to Al stress in the root apex of tomato, of which 7 were expressed in abundance in the root apex (Fig. 6). Interestingly, these 7 SlNAC genes belong to IIb (SlNAC033, SlNAC063, SlNAC064, and SlNAC084) and IIIb (SlNAC019, SlNAC031, and SlNAC065) subgroups (Fig. 1). Similarly, most of Al-responsive NAC genes in rice belong to NAM subgroup i.e. IIb subgroup [17]. A few members from subgroup IIb have been well characterized with respect to fruit development and responses to biotic and abiotic stresses in tomato. For example, both SlNAC033 and SlNAC064 have been demonstrated to be implicated in tomato fruit development possibly via regulating the expression of genes in association with ethylene biosynthesis and cell wall metabolism [38, 39]. SlNAC063, previously known as JA2L, has proven to be necessary for jasmonic acid-mediated stomatal movement during pathogenesis [40]. By contrast, much less is known of the function of NAC genes from subgroup IIIb. In Arabidopsis, CUC1 and CUC2 belong to this subgroup functioning redundantly to regulate shoot apical meristem formation [41]. In tomato, the GOBLET gene, which encodes a NAC transcription factor (assigned as SlNAC062 in this study), directs the leaflet boundaries in compound leaves of tomato [42]. The involvement of three subgroup IIIb SlNACs in Al stress suggests that Al might affect root apex meristem development. Alternatively, the function of NAC genes from this subgroup is not limited to development. However, their functions in Al stress tolerance need to be examined in the future.
It appears that Al-responsive SlNAC genes are early factors involved in Al stress responses as evidence by a protein translation inhibitor experiment. Generally, early genes are required for the transcription of secondary response or late genes. Here, we found that Al-induced expression of a tomato FRD3-like gene (Solyc01g087150) was almost completely blocked by CHX (Fig. S4), suggesting that this gene represents one of the late genes required for long-term responses (possibly citrate efflux) to Al stress in tomato. A similar expression regulation pattern has been reported previously in rice bean, in which VuSTOP1 expression could also be induced by CHX in the absence of Al stress, whereas Al-induced VuMATE1 expression was completely inhibited [43]. At present, the direct target gene of these SlNAC TFs remains unknown. A recent study showed that a rice bean (Vigna umbellata) NAC TF, VuNAR1 can activate the expressiong of cell wall related receptor kinase 1 (WAK1) gene that contributes to reduced pectin content in the cell wall and Al3 +binding capacity, thus enhancing Al tolerance. Since several lines of evidence suggest the role of NAC TFs in cell wall metabolism, it seems very likely that these SlNACs may also be involved in the Al stress response by regulating cell wall metabolism, which requires further investigation.
Conclusions
In this study, the first integrated analysis including gene identification, structure, chromosomal location, duplications, tissue and Al response expression patterns of the NAC gene family in tomato was carried out. A total of 93 SlNAC genes were identified, which will provide essential information for the functional characterization of SlNAC genes in tomato. Analysis of previously published RNA-Seq data indicates that SlNAC genes may participate in the development of tomato. Our RNA-Seq data showed that the expression levels of 19 SlNAC genes were significantly changed under Al stress. These data are helpful for further investigation of NAC gene-mediated physiological and molecular processes involved in Al stress, and provides new insight into NAC gene family and a basis for further exploration on its functional mechanisms in tomato.
Methods
Identification of the NAC family genes in tomato
The Hidden Markov Model (HMM) file corresponding to the NAC domain (PF02365) was download from the Pfam protein family database (http://pfam.xfam.org/) [44]. HMMER 3.2 was used to search against the NAC genes from the tomato genome database from Phytozome v12.1 (https://phytozome.jgi.doe.gov/pz/portal.html) [45, 46]. All candidate genes that may contain NAC domain based on HMMER results were further examined by confirming the existence of the NAC core sequences using PFAM and the SMART program (http://smart.embl-heidelberg.de/smart/batch.pl) [47]. Length of sequences, protein molecular weights, transmembrane domains and subcellular location of identified tomato NAC proteins were obtained by using tools from ExPasy website (https://www.expasy.org/).
Phylogenetic analysis of the NAC gene family members
The NAC domain sequences of 93 identified tomato NACs and 110 Arabidopsis NACs from PlantTFDB 4.0 4http://planttfdb.cbi.pku.edu.cn/) [48] were used to create multiple protein sequence alignments using ClustalW in MEGA 7.0 (https://www.megasoftware.net/) [49] with default parameters. The alignment results were used to construct a phylogenetic tree using the neighbor-joining method with 1000 bootstrap replicates. The phylogenetic tree was displayed with the online tools iTOL v4 (https://itol.embl.de/) [50].
Gene structure and conserved motif analysis
The exon-intron distribution of each tomato NAC genes (SlNACs) was analyzed by comparing predicted coding sequences with their corresponding genomic sequences acording to Gene Structure Display Server 2.0 (http://gsds.cbi.pku.edu.cn/) [51]. Conserved motifs of tomato NAC protein sequences were investigated using the online software MEME5.0.4 (http://meme-suite.org/tools/meme) [52] with the following motif parameters: number of repetitions (any), maximum number of motif (20), and the optimum width of each motif, (between 6 and 100 residues).
Chromosomal distribution and gene duplication analysis
All SlNACs were mapped to 12 tomato chromosomes based on physical location information from the database of tomato genome using TBtools program (https://github.com/CJ-Chen/TBtools) [53]. Multiple Collinearity Scan Toolkit (MCScanX) with the default parameters was used to analyze the tandem repeats and segmental duplication events of SlNAC gene family in the tomato genome (http://chibba.pgml.uga.edu/mcscan2/) [54].
Tissue-specific expression analysis
To investigate the expression patterns of putative SlNACs genes in different tissues of development stages of tomato, in silico analysis of RNA-seq data [20] from Tomato Functional Genomics Database (TFGD, http://ted.bti.cornell.edu/cgi-bin/TFGD/digital/home.cgi) were carried out. Different tissues in cultivated tomato (Solanum lycopersicum cv. Heinz) including leaves, roots, flower buds, fully opened flowers, 1 cm, 2 cm, 3 cm, mature green, breaker, and breaker+ 10 fruits were selected as described previously [55], In the wild species (Solanum pimpinellifolium), ten tissues and organs, which included leaves, whole root, hypocotyl, cotyledons, flower buds, 10 days before anthesis or younger, flowers at anthesis, 10 days post anthesis (DPA) fruit, 20 DPA fruit and breaker stage ripening fruit, were selected for analysis. Digital gene expression analysis of the putative SlNACs was visualized using MultiExperiment Viewer (MeV) software [56].
Plant material and growth conditions
Tomato (Solanum lycopersicum) cultivar Ailsa Craig (AC) (Horticulture Research International, Warwick, UK) was used in this study. Seeds were sterilized with 10% NaClO (v/v) for 15 min, then washed with sterilized water five times to remove the residual NaClO. Seeds were soaked in sterilized water overnight and then sown on agar plates containing 1/5 Hoagland nutrient solution (pH 5.5) consisting of KNO3 (1.0 mM), Ca (NO3)2 (1.0 mM), MgSO4 (0.4 mM) and (NH4)H2PO4 (0.2 mM), and the micronutrients NaFeEDTA (20 μM), H3BO3 (3.0 μM), MnCl2 (0.5 μM), CuSO4 (0.2 μM), ZnSO4 (0.4 μM) and (NH4)6Mo7O24 (1 μM), with 0.8% Agar (Sigma-Aldrich). Plates were kept in the dark at 4 °C for 2 d and then seeds were germinated in plant growth room with a daytime 16 h/24 °C and 8 h/22 °C night regime. After germination, uniform seedlings (until primary root length about 3–4 cm) were transferred to the 1/5 Hoagland nutrient solution (pH 5.0) with (NH4)H2PO4 concentration decreased to 10 μM either in the absence (−Al) or presence (+Al) of 5 μM Al for 6 h or 24 h in the same growth conditions. The tomato root tip (0-1 cm), basal root (1–2 cm) and leaves were collected for RNA extract and qRT-PCR analysis, root tips were used for further RNA-seq.
For the protein translation inhibitor cycloheximide (CHX), uniform AC seedlings (until primary root length about 3–4 cm) were pretreated with or without 10 μM CHX for 1 h, then transferred to 5 μM Al-containing solution or Al-free solution for 6 h (pH 5.0 with 10 μM (NH4)H2PO4). Root tips (0–1 cm) were collected for RNA-seq and qRT-PCR analysis.
RNA-Seq and quantitative RT-PCR (qRT-PCR) analysis
For RNA-Seq, RNA samples were extracted from both root tips (1 cm in length) treated with or without 5 μM Al for 6 h. RNA-seq was carried out on an Illumina HiSeq Platform (http://www.bgitechsolutions.cn). Three biological replicates were performed for each treatment.
For qRT-PCR analysis, one microgram of DNA-free RNA was transcribed into first strand cDNA by PrimeScriptTM RT Master Mix (TaKaRa). The qRT-PCR was carried out with the Roche LightCyler 480 instrument using SYBR Green chemistry (Toyobo). The reaction conditions were 40 cycles at 95 °C for 15 s, 60 °C for 10 s, and 72 °C for 15 s. The primer sequences used in this study are listed in Table S8. Expression data of target genes were normalized with expression of tomato GAPDH [35] and ACTIN [57], respectively, by the ΔΔCt method. Each reaction was performed with three repeats from different biological samples.
Statistical analysis
Student’s t-test was run in Microsoft Excel (v. 2016, Microsoft Corp., Redmond, WA, USA). Data are given as means ± standard deviation (SD) of three independent biological replicates. A p-value less than 0.05 (p < 0.05) was considered to be statistically significant.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. RNA-Seq data is available as accession number SRP227100–1005 in the NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra/SRP227103). Tomato (Solanum lycopersicum) cultivar Ailsa Craig (AC) used in this study is deposited in our Lab.
Abbreviations
- TF:
-
Transcription factor
- STOP1:
-
Sensitive to proton rhizotoxicity 1
- ALMT1:
-
Al-activated malate transporter 1
- CAMTA2:
-
CALMODULIN-BINDING TRANSCRIPTION ACTIVATOR2
- ART1:
-
Al resistance transcription factor 1
- NAM:
-
No apical meristem
- ATAF 1/2:
-
Arabidopsis transcription activator factor 1/2
- CUC2:
-
Cup shaped cotyledon
- WAK1:
-
Wall-associated protein kinases
- SOG1:
-
SUPPRESSOR OF GAMMA RESPONSE1
- HMM:
-
Hidden Markov model
- MCScanX:
-
Multiple Collinearity Scan Toolkit
- DPA:
-
Days post anthesis
- MeV:
-
MultiExperiment Viewer
- CHX:
-
Cycloheximide
References
Kochian LV. Cellular mechanisms of aluminum toxicity and resistance in plants. Annu Rev Plant Physiol Plant Mol Biol. 1995;46:237–60.
von Uexküll HR, Mutert E. Global extent, development and economic impact of acid soils. Plant Soil. 1995;171(1):1–15.
Delhaize E, Ma JF, Ryan PR. Transcriptional regulation of aluminium tolerance genes. Trend Plant Sci. 2012;17(6):341–8.
Fan W, Lou HQ, Gong YL, Liu MY, Wang ZQ, Yang JL, Zheng SJ. Identification of early Al-responsive genes in rice bean (Vigna umbellata) roots provides new clues to molecular mechanisms of Al toxicity and tolerance. Plant Cell Environ. 2014;37(7):1586–97.
Fan W, Xu JM, Wu P, Yang ZX, Lou HQ, Chen WW, Jin JF, Zheng SJ, Yang JL. Alleviation by abscisic acid of Al toxicity in rice bean is not associated with citrate efflux but depends on ABI5-mediated signal transduction pathways. J Integr Plant Biol. 2019;61(2):140–54.
Xu JM, Fan W, Jin JF, Lou HQ, Chen WW, Yang JL, Zheng SJ. Transcriptome analysis of Al-induced genes in buckwheat (Fagopyrum esculentum Moench) root apex: new insight into Al toxicity and resistance mechanisms in an al accumulating species. Front Plant Sci. 2012;8:1141.
Ahmad M, Yan X, Li J, Yang Q, Jamil W, Teng Y, Bai S. Genome wide identification and predicted functional analyses of NAC transcription factors in Asian pears. BMC Plant Biol. 2018;18(1):214.
Yang JL, Fan W, Zheng SJ. Mechanisms and regulation of aluminum-induced secretion of organic acid anions from plant roots. J Zhejiang Univ Sci B. 2019;20(6):513–27.
Ding ZJ, Yan JY, Xu XY, Li GX, Zheng SJ. WRKY46 functions as a transcriptional repressor of ALMT1, regulating aluminum-induced malate secretion in Arabidopsis. Plant J. 2013;76(5):825–35.
Tokizawa M, Kobayashi Y, Saito T, Kobayashi M, Iuchi S, Nomoto M, Tada Y, Yamamoto YY, Koyama H. Sensitive to proton rhizotoxicity1, calmodulin binding transcription activator2, and other transcription factors are involved in aluminum-activated malate transporter1 expression. Plant Physiol. 2015;167(3):991–1003.
Li GZ, Wang ZQ, Yokosho K, Ding B, Fan W, Gong QQ, Li GX, Wu YR, Yang JL, Ma JF, Zheng SJ. Transcription factor WRKY22 promotes aluminum tolerance via activation of OsFRDL4 expression and enhancement of citrate secretion in rice (Oryza sativa). New Phytol. 2018;219(1):149–62.
Olsen AN, Ernst HA, Leggio LL, Skriver K. NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005;10(2):79–87.
Su H, Zhang S, Yuan X, Chen C, Wang XF, Hao YJ. Genome-wide analysis and identification of stress-responsive genes of the NAM-ATAF1,2-CUC2 transcription factor family in apple. Plant Physiol Biochem. 2013;71:11–21.
Zhang H, Kang H, Su C, Qi Y, Liu X, Pu J. Genome-wide identification and expression profile analysis of the NAC transcription factor family during abiotic and biotic stress in woodland strawberry. PLoS One. 2018;13(6):e0197892.
Puranik S, Sahu PP, Srivastava PS, Prasad M. NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 2012;17(6):369–81.
Nuruzzaman M, Sharoni AM, Kikuchi S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front Microbiol. 2013;4(3):248.
Moreno-Alvarado M, García-Morales S, Trejo-Téllez LI, Hidalgo-Contreras JV, Gómez-Merino FC. Aluminum enhances growth and sugar concentration, alters macronutrient status and regulates the expression of NAC transcription factors in rice. Front Plant Sci. 2017;8:73.
Lou HQ, Fan W, Jin JF, Xu JM, Chen WW, Yang JL, Zheng SJ. A NAC-type transcription factor confers aluminium resistance by regulating cell wall-associated receptor kinase 1 and cell wall pectin. Plant Cell Environ. 2020;43(2):463–78.
Sjogren CA, Bolaris SC, Larsen PB. Aluminum-dependent terminal differentiation of the Arabidopsis root tip is mediated through an ATR-, ALT2-, and SOG1-regulated transcriptional response. Plant Cell. 2015;27(9):2501–15.
Chen P, Sjogren CA, Larsen PB, Schnittger A. A multi-level response to DNA damage induced by aluminium. Plant J. 2019;98(3):479–91.
Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485(7400):635–41.
Bai Y, Kissoudis C, Yan Z, Visser RGF, van der Linden G. Plant behaviour under combined stress: tomato responses to combined salinity and pathogen stress. Plant J. 2018;93(4):781–93.
Yang JL, Zhu XF, Peng YX, Zheng C, Ming F, Zheng SJ. Aluminum regulates oxalate secretion and plasma membrane H+-ATPase activity independently in tomato roots. Planta. 2011;234(2):281–91.
Wang N, Zheng Y, Xin H, Fang L, Li S. Comprehensive analysis of NAC domain transcription factor gene family in Vitis vinifera. Plant Cell Rep. 2013;32(1):61–75.
Shang H, Li W, Zou C, Yuan Y. Analyses of the NAC transcription factor gene family in Gossypium raimondii Ulbr.: chromosomal location, structure, phylogeny, and expression patterns. J Integr Plant Biol. 2013;55(7):663–76.
Hu W, Wei Y, Xia Z, Yan Y, Hou X, Zou M, Lu C, Wang W, Peng M. Genome-wide identification and expression analysis of the NAC transcription factor family in cassava. PLoS One. 2015;10(8):e0136993.
Rushton PJ, Bokowiec MT, Zhang H, Brannock JF, Chen X, Laudeman TW, Timko MP. Tobacco transcription factors: novel insights into transcriptional regulation in the Solanaceae. Plant Physiol. 2008;147(1):280–95.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Ooka H, Satoh K, Doi K, Nagata T, Otomo Y, Murakami K, Matsubara K, Osato N, Kawai J, Carninci P, Hayashizaki Y, Suzuki K, Kojima K, Takahara Y, Yamamoto K, Kikuchi S. Comprehensive analysis of NAC family genes in Oryza sativa and Arabidopsis thaliana. DNA Res. 2003;10(6):239–47.
Nuruzzaman M, Manimekalai R, Sharoni AM, Satoh K, Kondoh H, Ooka H, Kikuchi S. Genome-wide analysis of NAC transcription factor family in rice. Gene. 2010;465(1–2):30–44.
Shiriga K, Sharma R, Kumar K, Yadav SK, Hossain F, Thirunavukkarasu N. Genome-wide identification and expression pattern of drought-responsive members of the NAC family in maize. Meta Gene. 2014;2:407–17.
Ha CV, Esfahani MN, Watanabe Y, Tran UT, Sulieman S, Mochida K, Nguyen DV, Tran LS. Genome-wide identification and expression analysis of the CaNAC family members in chickpea during development, dehydration and ABA treatments. PLoS One. 2014;9(12):e114107.
Liu M, Ma Z, Sun W, Huang L, Wu Q, Tang Z, Bu T, Li C, Chen H. Genome-wide analysis of the NAC transcription factor family in Tartary buckwheat (Fagopyrum tataricum). BMC Genomics. 2019;20(1):113.
Tweneboah S, Oh SK. Biological roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in solanaceous crops. J Plant Biotechnol. 2017;44:1–11.
Ma XM, Zhang Y, Turečková V, Xue GP, Fernie AR, Mueller-Roeber B, Balazadeh S. The NAC transcription factor slnap2 regulates leaf senescence and fruit yield in tomato. Plant Physiol. 2018;177(3):1286–302.
Wang G, Zhang S, Ma X, Wang Y, Kong F, Meng Q. A stress-associated NAC transcription factor (SlNAC35) from tomato plays a positive role in biotic and abiotic stresses. Physiol Plantarum. 2016;158(1):45–64.
Moore S, Vrebalov J, Payton P, Giovannoni J. Use of genomics tools to isolate key ripening genes and analyse fruit maturation in tomato. J Exp Bot. 2002;53(377):2023–30.
Gao Y, Wei W, Zhao X, Tan X, Fan Z, Zhang Y, Jing Y, Meng L, Zhu B, Zhu H, Chen J, Jiang CZ, Grierson D, Luo Y, Fu DQ. A NAC transcription factor, NOR-like1, is a new positive regulator of tomato fruit ripening. Hortic Res. 2018;5:75.
Zhu M, Chen G, Zhou S, Tu Y, Wang Y, Dong T, Hu Z. A new tomato NAC (NAM/ATAF1/2/CUC2) transcription factor, SlNAC4, functions as a positive regulator of fruit ripening and carotenoid accumulation. Plant Cell Physiol. 2014;55(1):119–35.
Du M, Zhai Q, Deng L, Li S, Li H, Yan L, Huang Z, Wang B, Jiang H, Huang T, Li CB, Wei J, Kang L, Li J, Li C. Closely related NAC transcription factors of tomato differentially regulate stomatal closure and reopening during pathogen attack. Plant Cell. 2014;26(7):3167–84.
Cucinotta M, Manrique S, Cuesta C, Benkova E, Novak O, Colombo L. CUP-SHAPED COTYLEDON1 (CUC1) and CUC2 regulate cytokinin homeostasis to determine ovule number in Arabidopsis. J Exp Bot. 2018;69(21):5169–76.
Berger Y, Harpaz-Saad S, Brand A, Melnik H, Sirding N, Alvarez JP, Zinder M, Samach A, Eshed Y, Ori N. The NAC-domain transcription factor GOBLET specifies leaflet boundaries in compound tomato leaves. Development. 2009;136(5):823–32.
Fan W, Lou HQ, Gong YL, Liu MY, Cao MJ, Liu Y, Yang JL, Zheng SJ. Characterization of an inducible C2H2-type zinc finger transcription factor VuSTOP1 in rice bean (Vigna umbellata) reveals differential regulation between low pH and aluminum tolerance mechanisms. New Phytol. 2015;208(2):456–68.
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47(D1):D427–32.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37.
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(Database issue):D1178–86.
Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46(D1):D493–6.
Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, Gao G. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45(D1):D1040–5.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242–5.
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31(8):1296–7.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–8.
Chen C, Xia R, Chen H, He Y. TBtools, a Toolkit for Biologists integrating various biological data handling tools with a user-friendly interface. BioRxiv. 2018. https://doi.org/10.1101/289660.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, Kissinger JC, Paterson AH. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Yu J, Cheng Y, Feng K, Ruan M, Ye Q, Wang R, Li Z, Zhou G, Yao Z, Yang Y, Wan H. Genome-wide identification and expression profiling of tomato hsp20 gene family in response to biotic and abiotic stresses. Front Plant Sci. 2016;7:1215.
Howe E, Holton K, Nair S, Schlauch D, Sinha R, Quackenbush J. MeV: MultiExperiment Viewer. In: Ochs M, Casagrande J, Davuluri R, editors. Biomedical Informatics for Cancer Research. Boston: Springer; 2010. p. 267–77.
Song LX, Xu XC, Wang FN, Wang Y, Xia XJ, Shi K, Zhou YH, Zhou J, Yu JQ. Brassinosteroids act as a positive regulator for resistance against root-knot nematode involving RESPIRATORY BURST OXIDASE HOMOLOG-dependent activation of MAPKs in tomato. Plant Cell Environ. 2018;41(5):1113–25.
Funding
This work was supported financially by the National Natural Science Foundation of China (grant no. 31572193, 31760584), 111 project (grant no. B14027), the Chang Jiang Scholars Program (JLY) and the the project of yong and middle-aged talent of Yunnan province (No. 2019HB019). The design of this study and the manuscript writing were supported by 31572193, 31760584 and B14027. And the collection, analysis, and interpretation of data used in this study were supported by Chang Jiang Scholars Program and 2019HB019.
Author information
Authors and Affiliations
Contributions
YJL, FW and ZSJ conceived the study. JJF, WZQ, HQY, WJY, LPF, and XJM performed the experiments and carried out the analysis. YJL and FW designed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1
. Structural features of NAC family genes in tomato (Solanum lycopersicum). Table S2. Summary of RNA-Seq of tomato root tip in response to 0 or 10 μM Al for 6 h. Table S3. Statistics of genes and transcripts. Table S4. List of up-regulated differentially expressed genes (DEGs) in response to Al stress in tomato. Table S5. List of down-regulated differentially expressed genes (DEGs) in response to Al stress in tomato. Table S6. List of SlNAC genes whose expression was regulated by Al stress for 6 h. Table S7. List of published NAC genes in our naming system. Table S8. List of primers used in this study.
Additional file 2: Figure S1
: Effects of Al treatment on root growth of tomato seedlings. Figure S2. Volcano plot analysis of differentially expressed genes (DEGs) in roots of tomato seedlings under Al for 6 h. Figure S3. Effects of a protein translation inhibitor, cycloheximide (CHX), on the expression of SlNAC056 gene identified in our RNA-Seq data. Figure S4. Effects of a protein translation inhibitor, cycloheximide (CHX), on the expression of FRD3-like genes identified in our RNA-Seq data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, J.F., Wang, Z.Q., He, Q.Y. et al. Genome-wide identification and expression analysis of the NAC transcription factor family in tomato (Solanum lycopersicum) during aluminum stress. BMC Genomics 21, 288 (2020). https://doi.org/10.1186/s12864-020-6689-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6689-7