
Abstract
Background
Fertility is an important economic trait in the production of meat goat, and follicular development plays an important role in fertility. Although many mRNAs and microRNAs (miRNAs) have been found to play critical roles in ovarian biological processes, the interaction between mRNAs and miRNAs in follicular development is not yet completely understood. In addition, less attention has been given to the study of single follicle (dominant or atretic follicle) in goats. This study aimed to identify mRNAs, miRNAs, and signaling pathways as well as their interaction networks in the ovarian follicles (large follicles and small follicles) of uniparous and multiple Chuanzhong black goats at estrus phase using RNA-sequencing (RNA-seq) technique.
Results
The results showed that there was a significant difference in the number of large follicles between uniparous and multiple goats (P < 0.05), but no difference in the number of small follicles was observed (P > 0.05). For the small follicles of uniparous and multiple goats at estrus phase, 289 differentially expressed mRNAs (DEmRNAs) and 16 DEmiRNAs were identified; and for the large follicles, 195 DEmRNAs and 7 DEmiRNAs were identified. The functional enrichment analysis showed that DE genes in small follicles were significantly enriched in ovarian steroidogenesis and steroid hormone biosynthesis, while in large follicles were significantly enriched in ABC transporters and steroid hormone biosynthesis. The results of quantitative real-time polymerase chain reaction were consistent with those of RNA-seq. Analysis of the mRNA-miRNA interaction network suggested that CD36 (miR-122, miR-200a, miR-141), TNFAIP6 (miR-141, miR-200a, miR-182), CYP11A1 (miR-122), SERPINA5 (miR-1, miR-206, miR-133a-3p, miR-133b), and PTGFR (miR-182, miR-122) might be related to fertility, but requires further research on follicular somatic cells.
Conclusions
This study was used for the first time to reveal the DEmRNAs and DEmiRNAs as well as their interaction in the follicles of uniparous and multiple goats at estrus phase using RNA-seq technology. Our findings provide new clues to uncover the molecular mechanisms and signaling networks of goat reproduction that could be potentially used to increase ovulation rate and kidding rate in goat.
Similar content being viewed by others


Background
Ovulation rate is a key factor affecting the kidding rate, which is one of the most important economic traits for goat production [1,2,3]. However, the genetic mechanism of kidding rate associated with ovulation rate is poorly understood, which largely limits the improvement of kidding rate through genetic selection. The major function of the ovary is to produce oocytes for fertilization and secrete steroid hormones for regulating follicular development during the estrus cycle in goat [4]. Oocytes develop and mature in the ovarian follicle, and acquire their developmental competence in follicle through tight bidirectional communication with follicular somatic cells [5, 6]. The follicular somatic cells include epithelial-like granulosa cells (GC), mesenchymal-like theca cells (TC) and cumulus cells (CC), with each type secrets specific regulation factors [7, 8]. GC and TC face each other across a basement membrane. CC are the differentiated GC, which are tightly connected and metabolically coupled with an oocyte via gap junctions and form the cumulus oocyte complex (COC) [9]. The pre-antral follicle is filled with a fluid that is rich in proteins, steroids, and lipids, coming from the blood and secretory activity of follicular somatic cells [10, 11]. The microenvironment of the oocyte has a crucial impact on the acquisition of oocyte developmental competence and possesses molecular factors that are predictive of oocyte developmental potential. However, only 1% of follicles reach ovulation, more than 99% of follicles undergo atresia in mammals [12,13,14,15,16]. In goat, the number of small follicles [S, diameter (d) < 3 mm] far exceeding the number of mid-follicles (d > 3 mm) is a mechanism to regulate the number of oocytes ovulated and to contribute to the timing of ovulation. Hence, it is important to study the contribution of ovarian follicular compartments (follicular fluid, oocyte, CC, GC, and TC) of small follicles and large follicles to the regulation of ovulation number and the timing of ovulation.
In mammals, studies on follicles have mainly been conducted in mice [17], human [18], pig [19], bovine [20], rat [21], and sheep [22]. These studies revealed the effects of GCs and theca cells on follicular development, follicular atresia, and luteal development, and further demonstrated the mechanism of genes and signaling pathways. However, little is known about goat follicles. In goat, previous studies have identified the key genes involved in the regulation of ovulation rate and kidding rate by transcriptome analysis of goat ovaries, as well as the signaling pathways that affect ovulation and fertility [1, 4, 23,24,25,26,27,28,29]. Studies on litter size of goats have shown that PDGFRB, MARCH1, KDM6A, CSN1S1, SIRT3, KITLG, GHR, ATBF1, INHA, GNRH1, and GDF9 might be candidate genes for goat reproductive traits [26, 30,31,32,33,34,35,36,37,38,39]. Growth hormones and members of the insulin-like growth factor (IGF) system (IGF-I and IGF-II) may play a key role in follicular development and atresia [2, 40], and the genes FER1 L4 and SRD5A2 may be associated with the high fecundity of goats [41]. In addition, many studies have suggested that microRNAs (miRNAs) influence ovarian biological processes in goat, and several differentially expressed miRNAs (DEmiRNAs), such as miR-21, miR-99a, miRNA-143, let-7f, miR-493, and miR-200b have been identified and comparatively analyzed in the ovaries of prolific and non-prolifc goats [1, 29, 42]. However, the major genes and miRNAs related to ovulation rate and litter size have not yet been identified in goats through transcriptome sequencing of the ovary as a whole. Hence, since the follicle is a unique microenvironment within which the oocyte can develop and mature into a fertilizable gamete, it is important to individually study single follicles to explore factors that affect ovulation rate and kidding rate in goats.
Chuanzhong (CZ) black goat is an excellent local goat resource in China. The resources are abundant in China as well as Southeast Asia, and play an important role in herbivorous livestock [43]. After long-term natural selection and artificial cultivation, CZ black goat has gradually formed local meat goat breeds with high genetic stability [44]. However, low fecundity remains a key bottleneck limiting the development of goat industry.
To better understand the role and importance of follicles in kidding rate, we performed transcriptome profiling of small follicles (S, d < 3 mm) and large follicles (L, d > 10 mm) from uniparous and multiple CZ black goats at the estrus phase to identify DEmRNAs and DEmiRNAs, respectively. Furthermore, the interaction networks of DEmRNAs and DEmiRNAs were constructed, and Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were carried out for DEmRNAs and target genes of DEmiRNAs. In addition, we explored the role of ovarian follicular mRNAs and miRNAs in goat reproduction. Collectively, our findings provide a theoretical basis for improving ovulation and kidding rates in the future.
Results
Comparison of follicles between uniparous and multiple CZ black goats
The follicles around the large follicles were sacrificed during follicle separation, including small follicles (d < 3 mm) and mid-follicles (3 < d < 10 mm). Sometimes the nearby large follicle (d > 10 mm) had to be sacrificed, too. Unfortunately, some small or large follicles were broken up during follicle separation. Finally, eight to fifteen small follicles were isolated from each goat, one to two large follicles were isolated from each uniparous goat, and one to three large follicles each multiple goat. Among the separated follicles used for sequencing, the large follicles (d > 10 mm) were larger in size than the small follicles (d < 3 mm) (Fig. 1a). After separation of follicles, the number of follicles in uniparous and multiple goats was counted and analyzed (Table 1). Multiple goats showed a greater number of follicles than uniparous goats (P < 0.05). Furthermore, there was a significant difference in the number of large follicles between uniparous and multiple goats (P < 0.05), but no difference in the number of small follicles was observed (P > 0.05).

Images of ovaries and follicles. a Large follicles (black arrows) and small follicles (yellow arrows) in the ovaries before follicle separation. b Large follicles after separation. c Small follicles after separation
Transcriptome sequencing analysis and mapping
We collected twelve large follicles from four uniparous and four multiple goats, and nine small follicular pools from four uniparous and five multiple goats. The large follicle with few or no blood vessels on the surface were not used to RNA-seq because they considered to be atretic follicles [45, 46]. Eight to ten small follicles were pooled from each goat for a replicate, and only one single large follicle with clear follicular fluid and abundant blood vessels on the surface from each goat for a replicate (Fig. 1b, c). Next, we sequenced the RNA libraries of these seventeen follicular samples (eight large follicles and nine small follicular pools). Clean read counts, mapped ration, and Q30 of sequencing data are displayed in Table 2. The data showed that the sequencing results met the requirements and could be used for further analysis. The expression of 21,343 mRNAs and 436 miRNAs was detected by RNA-sequencing (RNA-seq). Finally, samples were divided into four groups for further analysis: uniparous-small follicles vs multiple-small follicles (Uni-S vs Mul-S) and uniparous-large follicles vs multiple-large follicles (Uni-L vs Mul-L). Unsupervised hierarchical clustering of mRNAs and miRNAs showed that each group clustered together, despite inter-individual variation (Figure S1).
Identification of DEmRNAs and DEmiRNAs
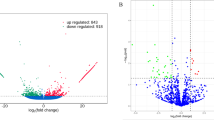
DEmRNAs and DEmiRNAs were initially identified by P < 0.05 (Additional files 1 and 2). For small follicles, a total of 289 DEmRNAs (131 upregulated and 158 downregulated in multiple goats) and seven DEmiRNAs (seven downregulated in multiple goats) were identified in Uni-S vs Mul-S (Fig. 2a and Table 3a). For large follicles, a total of 195 DEmRNAs (120 upregulated and 75 downregulated in multiple goats) and 16 DEmiRNAs (4 upregulated and 12 downregulated in multiple goats) were identified in Uni-L vs Mul-L (Fig. 2b and Table 3b). For better analysis, the fragments per kilobase million (FPKM) values > 1 of at least three samples per group was used to further quantify the mRNA expression levels. We identified 119 and 37 DEmRNAs from Uni-S vs Mul-S and Uni-L vs Mul-L, respectively, and the top 10 DEmRNAs and DEmiRNAs are shown in Table 3. The venn diagrams of shared DEmRNAs and DEmiRNAs are shown in Fig. 2c, d. Two shared DEmRNAs (AMDHD1 and LOC102190765) and five shared DEmiRNAs (miR-141, miR451-5p, miR-122, miR-182, and miR-206) were identified in both Uni-S vs Mul-S and Uni-L vs Mul-L.

RNA-seq data of DEmRNA expression in large and small follicles from uniparous and multiple goats. a Unsupervised clustering analysis showing the expression profiles of DEmRNAs between Uni-S and Mul-S groups. b Unsupervised clustering analysis showing the expression profiles of DEmRNAs between Uni-L and Mul-L groups. c Venn diagrams demonstrating the distribution of shared DEmRNAs in Uni-S vs Mul-S and Uni-L vs Mul-L groups. d Venn diagrams demonstrating the distribution of shared DEmiRNAs in Uni-S vs Mul-S and Uni-L vs Mul-L groups, respectively. e Top 20 KEGG pathways of DEmRNAs in Uni-S vs Mul-S. f Top 20 KEGG pathways of DEmRNAs in Uni-L vs Mul-L.
Functional annotation of DEmRNAs
DEmRNAs were enriched in biological process, cellular component, and molecular function categories by GO analysis (http://www.geneontology.org/). GO terms with P < 0.05 were considered significantly enriched in DEmRNAs. For small follicles, 455 GO terms were significantly enriched, including cell periphery, plasma membrane, steroid biosynthetic process, steroid hydroxylase activity, and receptor binding between the Uni-S and Mul-S groups (Table 4, Additional file 3). For large follicles, 322 GO terms were significantly enriched, including cell periphery, plasma membrane, animal organ development, embryo development, and anion channel activity (Table 4, Additional file 3).
DEmRNAs were also plotted to KEGG reference pathways (https://www.kegg.jp/kegg/pathway.html). The significantly enriched KEGG pathways (P < 0.05) were listed in Additional file 4. Of these KEGG pathways, ovarian steroidogenesis, cortisol synthesis and secretion, cytokine-cytokine receptor interaction, steroid hormone biosynthesis, and metabolism of xenobiotics by cytochrome P450 were significantly enriched between the Uni-S and Mul-S groups (Fig. 2e), and ABC transporters, retinol metabolism, steroid hormone biosynthesis, drug metabolism-cytochrome P450, and metabolism of xenobiotics by cytochrome P450 were significantly enriched between the Uni-L and Mul-L groups (Fig. 2f).
DEmRNA-DEmiRNA interaction network
For small follicles (Uni-S vs Mul-S), the miRNA–mRNA target prediction analyses identified 76 miRNA–mRNA target pairs, including only seven significant miRNAs, chi-miR-200a (degree = 20, degree means the number of DEmiRNAs’target genes), chi-miR-141 (degree = 20), chi-miR-182 (degree = 16), chi-miR-206 (degree = 10), chi-miR-122 (degree = 7), chi-miR-184 (degree = 2) and chi-miR-145-5p (degree = 1) (Fig. 3a). For large follicles (Uni-L vs Mul-L), a total of 153 possible significant miRNA–mRNA interaction pairs were obtained; chi-miR-141 (degree = 17), chi-miR-182 (degree = 13), chi-miR-122 (degree = 12) and chi-miR-154b-3p (degree = 12) were the top DEmiRNAs that had most target genes (Fig. 3b).

The original and selected DEmRNA-DEmiRNA interaction network of Uni-S vs Mul-S and Uni-L vs Mul-L. a The original DEmRNA-DEmiRNA interaction network of Uni-S vs Mul-S. b The original DEmRNA-DEmiRNA interaction network of Uni-L vs Mul-L. c The selected DEmRNA-DEmiRNA interaction network of Uni-S vs Mul-S. DEmRNAs were screened according to FPKM > 1 of at least three samples per group. d The selected DEmRNA-DEmiRNA interaction network of Uni-L vs Mul-L. DEmRNAs were screened according to FPKM > 1 of at least three samples per group. Ellipses and triangle represent DEmRNAs and DEmiRNAs, respectively. Red and blue colored nodes represent upregulation and downregulation, respectively
In order to further narrow the scope of genes and obtain more meaningful candidate genes, DEmRNAs were screened according to FPKM > 1 of at least three samples per group. Then, 19 pairs and 19 pairs of DEmRNAs-DEmiRNAs were obtained from Uni-S vs Mul-S and Uni-L vs Mul-L, respectively (Fig. 3c, d). Among them, in Uni-S vs Mul-S, TNFAIP6 (degree = 3) was upregulated in multiple goats; CD36 (degree = 3), BTK (degree = 2) and AKAP4 (degree = 2) were downregulated in multiple goats. In Uni-L vs Mul-L, SERPINA5 (degree = 5) was upregulated in multiple goats; ENSCHIG00000017462 (degree = 6) and PTGFR (degree = 4) were downregulated in multiple goats.
Validation by quantitative real-time polymerase chain reaction (qRT-PCR)
A total of 6 DEmRNAs and 5 DEmiRNAs were selected for verification by qRT-PCR. Based on the RNA-seq results, 3BHSD and STAR expression were upregulated and LEPR expression was downregulated in small follicles of multiple goat, and CCL21, RARRES1, and DPT expression were downregulated in large follicles of multiple goat. Notably, genes 3BHSD and STAR are both involved in the four significant pathways (ovarian steroidogenesis, cushing’s syndrome, cortisol synthesis and secretion and aldosterone synthesis and secretion), LEPR gene is involved in the two significant pathways (cytokine-cytokine receptor interaction and neuroactive ligand-receptor interaction). Five DEmiRNAs were downregulated in multiple large follicles. Based on the qRT-PCR results, expression of these 6 DEmRNAs and 5 DEmiRNAs were consistent with that in the RNA-seq results (Fig. 4).

Verification of differently expressed genes and miRNAs by qRT-PCR. a The expression level of six genes were validated by qRT-PCR and compared with the results of RNA-seq. 3BHSD, STAR and LEPR were selected from Uni-S vs Mul-S, and CCL21, RARRES1 and DPT were selected from Uni-L vs Mul-L. b The expression level of five miRNAs were validated by qRT-PCR and compared with the results of RNA-seq. These five DEmiRNAs were selected from Uni-L vs Mul-L. Data were presented as expression values of genes and miRNAs in Uni-S vs Mul-S and Uni-L vs Mul-L. For qRT-PCR data, mRNA expression was normalized to β-actin in the same cDNA sample, and miRNA expression was normalized to U6
Discussion
Exploring the genetic mechanism associated with ovulation rate is important to improve kidding rate, which is fundamental to goat production. The greater number of large follicles that stimulate ovulation is believed to be the primary reason for higher ovulation rate [39]. Although several mRNAs and miRNAs have been found to play critical roles in ovarian biological processes, the majority of the genes and miRNAs related to ovulation rate and kidding rate have not been identified in goats. In addition, the interaction between mRNAs and miRNAs in follicular development is not yet completely understood. Hence, we compared DEmRNAs and DEmiRNAs from different size follicles between uniparous and multiple CZ black goats at estrus phase using RNA-seq. The results showed that the number of large follicles in multiple goats was significantly higher than that in uniparous goats (P < 0.05), while no difference in the number of small follicles was observed between uniparous and multiple goats, verifying that the greater number of large follicles was related to higher ovulation rate [39]. Based on the RNA-seq data, we identified 119 and 37 DEmRNAs by comparing Uni-S with Mul-S and Uni-L with Mul-L, respectively (FPKM> 1 of at least three samples per group). These DEmRNAs were found to be involved in various ovarian development-related pathways, such as ovarian steroidogenesis, steroid hormone biosynthesis, and metabolism of xenobiotics by cytochrome P450, etc. (Fig. 2e, f). Approximately 37% of the DEgenes in the Uni-S vs Mul-S group and 41% in the Uni-L vs Mul-L group were reported to be associated with mammalian reproduction, such as TNFAIP6, MMP9, INSL3, LEPR, 3BHSD, LHCGR, ARL4C, CD36, CYP11A1, AMDHD1, SPOCK2, AMDHD1, MFAP5, CCL21, PTGFR, and SERPINA5 [23, 47,48,49,50,51,52,53,54,55,56,57,58,59,60]. Of these genes, TNFAIP6, CYP11A1, CD36, PTGFR, and SERPINA5 were found to be associated with the ovulation rate. In addition, TNFAIP6, CYP11A1 and CD36 were found to be differentially expressed when comparing Uni-S vs Mul-S, while PTGFR and SERPINA5 were found to be differentially expressed when comparing Uni-L vs Mul-L.
TNFAIP6 is a secretory protein of the hyaluronan-binding protein family that played a role in CC stabilization and expansion, and it was upregulated in bovine GCs during ovulation [47, 61,62,63]. TNFAIP6-deficient female mice were sterile [44]. The present study reported that TNFAIP6 expression was 8-fold higher in Mul-S than in Uni-S, suggesting a possible role of TNFAIP6 in CC expansion in the small follicle of multiple goats during the estrus phase. CYP11A1 played a key role in the regulation of steroid-producing pathways in GCs [64]. The first step in steroid biosynthesis was the conversion of cholesterol into pregnenolone through the action of CYP11A1 in the mitochondria, and then pregnenolone acted as a substrate for progesterone synthesis through the mediation of 3b-HSD expression [65, 66]. In this study, CYP11A1 expression was found to be upregulated in the small follicle of multiple goats, which was consistent with previous reports [67]; this observation demonstrates that CYP11A1 might play multiple roles in goat ovarian development. CD36 was a multifunctional receptor-binding autocrine growth factor that could regulate angiogenesis, cell growth, and adhesion. The expression of CD36 was found to be follicle-type dependent with the greatest expression in atretic follicles, and the lowest expresssion in healthy follicles [68,69,70,71]. CD36 knockdown has been shown to increase proliferation and expression of survival and angiogenic markers in GCs [70]. In this study, CD36 expression was shown to be downregulated in multiple goats and it was associated with specific hematopoietic cell lineages, suggesting that the low expression of CD36 expression in multiple individuals may promote follicular maturation by stimulating GCs proliferation or angiogenesis, which then increases the kidding rate. Overall, TNFAIP6 and CYP11A1 played a positive role in the regulation of ovulation, while CD36 contributed to follicular atresia. Accordingly, TNFAIP6 and CYP11A1 expression was upregulated in the small follicles of multiple goats, while CD36 expression was downregulated, indicating that there were more small follicles that could grow into dominant follicles in multiple goats. Thus, these genes might play a key role in the ovulation rate or kidding rate in goats.
PTGFR was a regulatory factor in follicular development that affected mammalian reproductive pathways [72,73,74]. In the GCs of periovulatory follicles of mice, the expression of PTGFR was shown to be drastically reduced [75].. Our results showed that PTGFR expression was downregulated in the large follicle from multiple goats, suggesting that the greater reduction of PTGFR expression observed in multiple goats may contribute to a the higher ovulation rate. Previous studies have reported that SERPINA5, a protease inhibitor, was expressed in the reproductive tract of adult mice and in the GCs of bovine follicles and that was highly expressed in bovine healthy follicles [60, 76]. SERPINA5 expression was shown to be downregulated in ovarian cancer (OC) [77,78,79]. Here, we found that SERPINA5 expression was upregulated in the large follicles of multiple goats, suggesting that SERPINA5 may affect follicular development and the kidding rate in goats. Taken together, downregulation of PTGFR and upregulation of SERPINA5 in large follicles may represent useful strategies for increasing the ovulation rate in multiple goats.
We identified 17 and 16 DEmiRNAs in the Uni-S vs Mul-S and Uni-L vs Mul-L groups, respectively. The miRNAs, miR-200a, miR-451-5p, miR-141, miR-182, miR-206, and miR-122 were highly expressed in the small follicles of multiple goats, while miR-1, miR-206, miR-133a-3p, miR-133b, miR-182, miR-215-5p, miR-122, and miR-451-5p were highly expressed in the large follicles of multiple goats. However, only miR-200a has been previously reported to be highly expressed in goat ovaries [1], demonstrating that the the independence of the expression of miRNAs in whole ovary, small follicles, and large follicles. Of these 14 highly expressed DEmiRNAs, miR-200a, miR-141, miR-1, miR-206, miR-133b, miR-133a-3p, miR-182, and miR-122 have been reported to play important roles in basic reproductive activities. miR-200a was frequently overexpressed and is closely related to the migratory, proliferative, and invasive abilities of OC cells [80,81,82]. miR-141 was shown to be significantly upregulated in OC cell lines and advanced metastatic OC [83, 84], and can inhibit GC apoptosis by targeting DAPK1 through the MAPK signaling pathway, leading to the development of polycystic ovary syndrome (PCOS) [85]. In this study, miR-200a and miR-141 expression were upregulated in the small follicles of uniparous goats, suggesting that these miRNAs might affect normal GC development, thereby affecting follicular development. miR-206 and miR-1 were potential tumor suppressors that have been shown to be downregulated in OC tissues, to inhibit c-Met expression, and to regulate cell proliferation, migration, and invasion [86, 87]. And miR-206 has also been shown to induce apoptosis [88, 89]. Here, miR-206 and miR-1 expression was downregulated in the large follicles of multiple goats, which may be related to oocyte maturation and ovulation in goats. Members of the miR-133 family (miR-133a-3p and miR-133b) have been shown to be involved in the regulation of various cellular processes, such as cell proliferation, apoptosis, migration, and invasion [90, 91]. FOXL2 was a conserved, early-acting gene in vertebrate ovarian development, and it played an important role in GC proliferation and oocyte maturation [68, 69]. Recently, FOXL2 expression has been reported to be regulated by miR-133b. miR-133b can bind to the FOXL2–3′UTR in GCs to inhibit the expression of the downstream genes, STAR and CYP19A1, which simultaneously promoted estrogen secretion in GCs [92]. miR-133b has been shown to be upregulated more than 30-fold in metaphase I oocytes after IGF-1 treatment, and it may play important roles in oocyte growth and maturation by regulating the expression of its potential target gene TAGLN2 [93].
Comparing Uni-S with Mul-S and Uni-L with Mul-L, miR-182 and miR-122 were found to be differentially expressed in both groups. miR-182 was shown to be upregulated in the follicular fluid of patients with PCOS [94], and its expression was significantly increased in OC cell lines [95]. miR-122 inhibited epithelial mesenchymal transition by regulating P4HA1 expression in OC cells [43]. In addition, miR-122 regulated LHCGR expression, which was crucial for mediating LH action in growing follicles via modulating LRBP levels during FSH-induced follicle growth [96]. miR-122 has been shown to increase LHR mRNA levels by modulating the expression of LRBP through the regulation of SREBP activation, which was crucial for regulating key reproductive processes, such as ovulation and CL function [97]. In this study, both miR-182 and miR-122 were upregulated in uniparous goats, suggesting that they may affect the ovulation rate by affecting follicular growth in uniparous goats.
Taken together, the genes CD36, TNFAIP6, CYP11A1, SERPINA5, and PTGFR were reported to be crucial for regulating the proliferation, migration, invasion and apoptosis of follicular somatic cells. The present study showed that TNFAIP6, CYP11A1, and CD36 were found to be differentially expressed when comparing Uni-S with Mul-S, and PTGFR and SERPINA5 were found to be differentially expressed when comparing Uni-L with Mul-L, suggesting that they may affect follicular development by affecting the growth of follicular somatic cells in goats. Furthermore, we predicted the target genes for DEmiRNAs and some of the targets showed high expression including CD36 (miR-122, miR-200a), TNFAIP6 (miR-200a, miR-182), CYP11A1 (miR-122), SERPINA5 (miR-1, miR-206, miR-133a-3p, and miR-133b), and PTGFR (miR-182 and miR-122); these expression regulation mechanisms may be related to ovulation and kidding rates.
Conclusions
Identifying the specific subset of genes and miRNAs involved in follicular development is essential for fully comprehending the cascade of events leading to ovulation and will likely contribute to an improved ability to control fertility. This study was the first to reveal the DEmRNAs and DEmiRNAs as well as their interaction in the follicles of uniparous and multiple goats at the estrus phase using RNA-seq technology. Numerous DEmiRNAs were more highly expressed in the small follicular libraries of multiple goats (miR-200a, miR-451-5p, miR-141, miR-182, miR-206, and miR-122), and while other DEmiRNAs were more highly expressed in the large follicular libraries of multiple goats (miR-1, miR-206, miR-133a-3p, miR-133b, miR-182, miR-215-5p, miR-122 and miR-451-5p). The higher expression of TNFAIP6, CYP11A1 and CD36 in the small follicles of multiple goats, and the higher expression of PTGFR and SERPINA5 in the large follicles of multiple goats may play a critical role in goat prolificacy. Our findings provide a basic foundation for elucidating the regulatory mechanisms of mRNAs and miRNAs in CZ black goats and a unique source for exploring the corresponding targets of the miRNAs in the future.
Methods
Animals and sample preparation
In the present study, CZ goats were obtained from the South China Agriculture University, Guangdong, China. A total of eleven healthy female goats of the same age (about 3.5–4.5 years old) with more than three litters were raised under natural light conditions with free access to food and water. Five goats were uniparous with only one kid per birth, and six goats were multiple with an average of 2.8 kids per birth. Mul: the six goats had three litters whose kidding ≥2. Uni: the five goats had three litters whose kidding = 1.
In order to achieve estrus synchronization, each goat was injected intramuscularly with 0.1 mg chloroprostenol. Eighteen days later, male goats (vasectomy and ligation of vas deferens) were used to confirm whether the estrous was synchronized. At 24 h after estrus (in the middle of estrus), all goats were weighed and slaughtered at a local slaughterhouse. The intact ovaries were rapidly collected and washed with 75% alcohol thrice. Then they were soaked into phosphate buffered saline (PBS). The follicles were achieved by using micro-blades and tweezers under surgical dissecting microscope within 30 min. Isolated follicles from each ovarian were washed with PBS to eliminate debris, then froze in liquid nitrogen instantly and stored at − 80 °C for generating RNA libraries. The small follicles (S, d < 3 mm) or large antral follicles (L, d > 1 cm) were isolated from the ovarian stromal tissues with tweezers by immersing the ovary in PBS, and then placed in liquid nitrogen (Table 5). The small and large follicles were collected at the same time within 30 min.
RNA extraction and qualification
Total RNA was extracted from the whole ovarian follicle using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA quality was evaluated with the NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). RNA integrity was evaluated using 1% agarose gel. Purified RNA was stored at − 80 °C until further use. RNA with amount > 6 μg, concentration ≥ 200 ng/mL, 1.8 < OD260/280 < 2.2, and RNA integrity number (RIN) > 8.5 was used for the preparation of cDNA libraries.
mRNA sequencing and data processing
A total of 3 μg RNA per sample was used as input material for removing ribosomal RNA using the Ribo-Zero Magnetic kit (EpiCentre, Madison, WI, USA). RNA was then fragmented into 200–300 bp by ion interruption. The first cDNA strand was synthesized using 6-base random hexamer primers and reverse transcriptase, and the second cDNA strand was synthesized with dUTP instead of dTTP. The library was constructed and amplified according to the size of the fragments (300–400 bp) by PCR using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The hybrid library was uniformly diluted to 2 nM through proportionally mixing the libraries containing different index sequences and forming a single chain library using TruseqTM RNA sample prep kit (Illumina, San Diego, CA, USA). The libraries were sequenced by paired-end sequencing on the HiSeq 2500 sequencer (Illumina, San Diego, CA, USA).
By using base calling, raw data obtained from high-throughput RNA-seq were translated into raw FASTQ sequence data. Raw reads of FASTQ format were then processed with Perl scripts to assess the quality of data used for subsequent analysis. To obtain clean reads from RNA-seq data for mRNA analysis, low quality sequences, including adaptor sequences, sequences with quality score < 20, and sequences with N base rate of raw reads > 10% were removed using cutadapt (http://cufflinks.cbcb.umd.edu/). Next, low quality sequences, including adaptor sequences, sequences with quality score < 20, sequences with N base, and sequences less than 18 bp were removed using Fastx-Toolkit (http://hannonlab.cshl.edu/f astx toolkit/). After filtering the raw reads, clean reads were obtained. Statistical analysis was performed to evaluate its quantity and quality, including Q30 (the percentage of the number of bases with phared score > 30 in the original data to the total number of bases) statistics, data quantity statistics, base content statistics, etc.
After trimming the raw reads, the clean reads were mapped to the goat reference genome (GCF_001704415.1_ARS1) Ensembl V96 using Tophat2. The mismatch of default reads and reference genomic sequence was within 2, and the mapping ratio is generally higher than 70%.
Small RNA library construction, sequencing, and data processing
Following extraction and purification, about 2 μg of total RNA per sample was used to construct the small RNA library using the TruSeq Small RNA Sample Prep Kit (Illumina, San Diego CA, USA). All libraries for high-throughput sequencing of miRNA were amplified using PCR by adding the sequencing connector and the index part. Next, the 18–36 nucleotide RNA was purified using 6% Novex TBE PAGE gel (1.0 mm, 10 well) and quantified using the Agilent 2100 Bioanalyzer. Single-stranded cDNA template was subjected to bridge PCR followed by Illumina single-end sequencing on the HiSeq 2500 sequencer (Illumina, San Diego, CA, USA).
Raw reads were processed with the script developed by consisting of index trimming, read alignment, and read counting. To obtain clean reads, raw reads were further filtered according to the following rules: (1) Removing low quality reads containing more than one low quality (Q-value ≤20) base or containing unknown nucleotides (N); (2) Removing reads without 3′ adapters; (3) Removing reads containing 5′ adapters; (4) Removing reads containing 3′ and 5′ adapters but no small RNA fragment between them; (5) Removing reads containing poly A in the small RNA fragment; and (6) Removing reads shorter than 18 nt (not including adapters). The resected clean reads were mapped to the goat reference genome (GCF_001704415.1_ARS1) Ensembl V96 using miRDeep2, in which the mapper.pl program invokes Bowtie for the alignment between the de-repeat sequence and the reference genome sequence. The de-repeat sequences were aligned to the mature miRNA and precursor miRNA sequences of the species in the miRBase (http://www.mirbase.org/) [98], and the detected miRNA was annotated. Using mireap to analyze unannotated sequences of information, a new miRNA prediction analysis was carried out. According to the number of mature miRNA sequences of this species, the read count values of miRNA were calculated.
Identification of DEmRNAs and DEmiRNAs
For RNA-seq analysis, the criteria for measuring mRNA and miRNA expression levels were FPKM and CPM (CPM = C/N × 1,000,000; C is the total number of reads mapped onto the gene, and N is the total number of mapped reads) values, respectively. Differential expression in each group was identified using DESeq version 1.18.0 with R package, and DEmRNAs and DEmiRNAs were identified with the cut-off criteria of |log2FoldChange| > 1 and P-value < 0.05.
The volcano plots of differentially expressed genes were generated using the ggplots2 package in R. All genes and samples were clustered using the heatmap package in R software. The Euclidean distance was calculated based on the expression level of the same gene in different samples and the expression patterns of different genes in the same sample. The complete linkage hierarchical clustering method, which uses the largest intercluster distance, was used for clustering. Shared DEmRNAs or DEmiRNAs were identified in both Uni-S vs Mul-S and Uni-L vs Mul-L groups.
Prediction of the target genes of the DEmiRNAs and construction of mRNA-miRNA interaction network
Systematic bioinformatic analysis was developed based on possible functional relationships between DEmiRNAs and DEmRNA. In this study, miRanDa was used to predict the target genes of DEmiRNAs using the 3’UTR mRNA sequence of the species as the target sequence. The parameters used to determine miRNA-target predicted interaction were the mapping score more than 140 and the free energy less than 1.0. Due to multiple target genes, data were re-filtered and the criterion was that the gene could only be retained in FPKM > 1 of at least three samples per group. Based on the re-filtered data, DEmRNA-DEmiRNA pairs in Uni-S vs Mul-S and Uni-L vs Mul-L were constructed and visualized using Cytoscape (v3.5.1) software.
Functional enrichment analysis
GO (http://geneontology.org/) and KEGG (http://www.kegg.jp/) pathway enrichment analysis were used to analyze DEmRNAs and target genes of DEmiRNAs. GO and KEGG pathway analysis of the differentially expressed and target genes was performed with the software DAVID (https://david.ncifcrf.gov). Degree of enrichment was measured by Rich factor, FDR, and the number of genes that were enriched in the pathway. Both GO terms and KEGG pathways were corrected. A P-value ≤0.05 was considered to be significantly enriched.
RNA preparation and qRT-PCR
First, total RNA was extracted from the ovarian follicles with the Total RNA Kit II (OMEGA, USA) for qRT-PCR analysis of DEmRNAs and DEmiRNAs. Second, the levels of DEmRNAs were measured using the PrimeScript® RT Reagent Kit with gDNA Eraser (TaKaRa, China) and qRT-PCR was performed using SYBR® Green PCR Supermix (Bio-Rad, USA). Each 20 μL reaction included 10 μL of SYBR® Green PCR Supermix, 1 μL of each divergent primer, 1 μL of cDNA, and 7 μL of RNase-free water. The cycling conditions included an initial single cycle (95 °C for 1 min), followed by 34 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 1 min. Melting-curve analysis was performed to verify the product identity. The levels of DEmiRNAs were then measured by qRT-PCR using miDETECT A Track™ miRNA qRT-PCR Starter kit (RiboBio, Guangzhou, China). Each 20 μL reaction included 10 μL of SYBR Green Mix, 0.5 μL of each divergent primer, 1 μL of cDNA, and 8 μL of RNase-free water. The cycling conditions included an initial single cycle (95 °C for 10 min), followed by 40 cycles of 95 °C for 5 s, 60 °C for 30 s, and 72 °C for 30 s. Melting-curve analysis was performed to verify the product identity.
According to the results of RNA-Seq, 6 DEmRNAs (FPKM > 1) and 5 DEmiRNAs (CPM > 1) were selected for validation by qRT-PCR. Primers for DEmRNAs were designed by Premier 5 and obtained from Sangon Biotech (Shanghai, China), and primers for DEmiRNAs were designed and obtained from RiboBio Company (Guangzhou, China). Information regarding the quantitative primers used in this experiment is listed in Table 6. β-actin and U6 were used as endogenous controls for mRNA and miRNA, respectively, and all reactions were performed in triplicate. Relative expression levels were calculated using the 2−ΔΔCt method. The correlation between qRT-PCR and RNA-seq results was calculated with Microsoft Excel 2019.
Statistical analysis
One-way analysis of variance was conducted using JMP 8.0 software (SAS Institute, Cary, NC). All results were expressed as means ± standard error, and P-values below 0.05 were considered to indicate statistically significant differences.
Availability of data and materials
We have submitted the sequencing data to NCBI SRA repository under the BioProject ID PRJNA579007 and PRJNA579194.
Abbreviations
- miRNAs:
-
MicroRNAs
- CZ black goat:
-
Chuanzhong black goat
- FPKM:
-
Fragments Per Kilobase of exon model per Million mapped reads
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- RNA-seq:
-
RNA sequencing
- qRT-PCR:
-
Quantitative real time polymerase chain reaction
- DEmiRNA:
-
Different expressed miRNA
- DEmRNA:
-
Different expressed mRNA
- GH:
-
Growth hormone
- GCs:
-
Granulosa cells
- CCs:
-
Cumulus cells
- TCs:
-
theca cells
- CL:
-
Corpus luteum
- OC:
-
Ovarian cancer
- PCOS:
-
Polycystic ovary syndrome
References
Zi XD, Lu JY, Ma L. Identification and comparative analysis of the ovarian microRNAs of prolific and non-prolific goats during the follicular phase using high-throughput sequencing. Sci Rep. 2017;7(1):1921.
Zi XD, Mu XK, Wang Y. Variation in sequences and mRNA expression levels of growth hormone (GH), insulin-like growth factor I (IGF-I) and II (IGF-II) genes between prolific Lezhi black goat and non-prolific Tibetan goat (Capra hircus). Gen Comp Endocrinol. 2013;187:1–5.
Cui HX, Zhao SM, Cheng ML, Guo L, Ye RQ, Liu WQ, Gao SZ. Cloning and expression levels of genes relating to the ovulation rate of the Yunling black goat. Biol Reprod. 2009;80(2):219–26.
Liu Y, Qi B, Xie J, Wu X, Ling Y, Cao X, Kong F, Xin J, Jiang X, Wu Q, Wang W, Li Q, Zhang S, Wu F, Zhang D, Wand R, Zhang X, Li W. Filtered reproductive long non-coding RNAs by genome-wide analyses of goat ovary at different estrus periods. BMC Genomics. 2018;19(1):866.
Zhang J, Xu Y, Liu H, Pan Z. MicroRNAs in ovarian follicular atresia and granulosa cell apoptosis. Reprod Biol Endocrinol. 2019;17(1):9.
Gougeon A. Regulation of ovarian follicular development in primates: facts and hypotheses. Endocr Rev. 1996;17(2):121–55.
Mermillod P, Dalbies-Tran R, Uzbekova S, Thelie A, Traverso JM, Perreau C, Papillier P, Monget P. Factors affecting oocyte quality: who is driving the follicle? Reprod Domest Anim. 2008;43(Suppl 2):393–400.
Buratini J, Price CA. Follicular somatic cell factors and follicle development. Reprod Fertil Dev. 2011;23(1):32–9.
Gilchrist RB, Lane M, Thompson JG. Oocyte-secreted factors: regulators of cumulus cell function and oocyte quality. Hum Reprod Update. 2008;14(2):159–77.
Fortune JE. Ovarian follicular growth and development in mammals. Biol Reprod. 1994;50(2):225–32.
Dumesic DA, Meldrum DR, Katz-Jaffe MG, Krisher RL, Schoolcraft WB. Oocyte environment: follicular fluid and cumulus cells are critical for oocyte health. Fertil Steril. 2015;103(2):303–16.
Wang X, Zou P, He Y, Meng K, Quan F, Zhang Y. Effect of luteinizing hormone on goat theca cell apoptosis and steroidogenesis through activation of the PI3K/AKT pathway. Anim Reprod Sci. 2018;190:108–18.
Zhang GM, Deng MT, Lei ZH, Wan YJ, Nie HT, Wang ZY, Fan YX, Wang F, Zhang YL. Effects of NRF1 on steroidogenesis and apoptosis in goat luteinized granulosa cells. Reproduction. 2017;154(2):111–22.
Han P, Xin H, Peng J, Hou J, Zhang L, Song Y, Li G, Cao B, An X. Identification and expression of X-linked inhibitor of apoptosis protein during follicular development in goat ovary. Theriogenology. 2017;98:30–5.
Yang D, Jiang T, Lin P, Chen H, Wang L, Wang N, Zhao F, Wang A, Jin Y. Knock-down of apoptosis inducing factor gene protects endoplasmic reticulum stress-mediated goat granulosa cell apoptosis. Theriogenology. 2017;88:89–97.
Yang D, Wang L, Lin P, Jiang T, Wang N, Zhao F, Chen H, Tang K, Zhou D, Wang A, Jin Y. An immortalized steroidogenic goat granulosa cell line as a model system to study the effect of the endoplasmic reticulum (ER)-stress response on steroidogenesis. J Reprod Dev. 2017;63(1):27–36.
Yang Y, Lin P, Chen F, Wang A, Lan X, Song Y, Jin Y. Luman recruiting factor regulates endoplasmic reticulum stress in mouse ovarian granulosa cell apoptosis. Theriogenology. 2013;79(4):633–9.
Zhang Y, Yan Z, Qin Q, Nisenblat V, Chang HM, Yu Y, Wang T, Lu C, Yang M, Yang S, Yao Y, Zhu X, Xia X, Dang Y, Ren Y, Yuan P, Li R, Liu P, Guo H, Han J, He H, Zhang K, Wang Y, Wu Y, Li M, Qiao J, Yan J, Yan L. Transcriptome landscape of human folliculogenesis reveals oocyte and granulosa cell interactions. Mol Cell. 2018;72(6):1021–34.
Zhang J, Liu Y, Yao W, Li Q, Liu H, Pan Z. Initiation of follicular atresia: gene networks during early atresia in pig ovaries. Reproduction. 2018;156(1):23–33.
Hatzirodos N, Irving-Rodgers HF, Hummitzsch K, Harland ML, Morris SE, Rodgers RJ. Transcriptome profiling of granulosa cells of bovine ovarian follicles during growth from small to large antral sizes. BMC Genomics. 2014;15:24.
Di Pietro M, Scotti L, Irusta G, Tesone M, Parborell F, Abramovich D. Local administration of platelet-derived growth factor B (PDGFB) improves follicular development and ovarian angiogenesis in a rat model of polycystic ovary syndrome. Mol Cell Endocrinol. 2016;433:47–55.
Huang Y, Yao XL, Meng JZ, Liu Y, Jiang XL, Chen JW, Li PF, Ren YS, Liu WZ, Yao JB, Folger JK, Smith GW, Ly LH. Intrafollicular expression and potential regulatory role of cocaine- and amphetamine-regulated transcript in the ovine ovary. Domest Anim Endocrinol. 2016;54:30–6.
Zi XD, Lu JY, Zhou H, Ma L, Xia W, Xiong XR, Lan DL, Wu XH. Comparative analysis of ovarian transcriptomes between prolific and non-prolific goat breeds via high-throughput sequencing. Reprod Domest Anim. 2018;53(2):344–51.
Ling Y, Xu L, Zhu L, Sui M, Zheng Q, Li W, Liu Y, Fang F, Zhang X. Identification and analysis of differentially expressed long non-coding RNAs between multiparous and uniparous goat (Capra hircus) ovaries. PLoS One. 2017;12(9):e183163.
Lai FN, Zhai HL, Cheng M, Ma JY, Cheng SF, Ge W, Zhang GL, Wang JJ, Zhang RQ, Wang X, Min LJ, Song JZ, Shen W. Whole-genome scanning for the litter size trait associated genes and SNPs under selection in dairy goat (Capra hircus). Sci Rep. 2016;6:38096.
An XP, Hou JX, Zhao HB, Li G, Bai L, Peng JY, YQ M, Song YX, Wang JG, Cao BY. Polymorphism identification in goat GNRH1 and GDF9 genes and their association analysis with litter size. Anim Genet. 2013;44(2):234–8.
Zi XD, Xu HW, Wang Y. Variation in sequences and mRNA expression levels of inhibin subunits alpha (INHA) and betaA (INHBA) genes between prolific and nonprolific goat breeds. Mol Reprod Dev. 2012;79(4):238.
Yang CX, Zi XD, Wang Y, Yang DQ, Ma L, Lu JY, Niu HR, Xiao X. Cloning and mRNA expression levels of GDF9, BMP15, and BMPR1B genes in prolific and non-prolific goat breeds. Mol Reprod Dev. 2012;79(1):2.
Ling YH, Ren CH, Guo XF, Xu LN, Huang YF, Luo JC, Zhang YH, Zhang XR, Zhang ZJ. Identification and characterization of microRNAs in the ovaries of multiple and uniparous goats (Capra hircus) during follicular phase. BMC Genomics. 2014;15:339.
Yang W, Yan H, Wang K, Cui Y, Zhou T, Xu H, Zhu H, Liu J, Lan X, Qu L, Pan C, Zhang E. Goat PDGFRB: unique mRNA expression profile in gonad and significant association between genetic variation and litter size. R Soc Open Sci. 2019;6(1):180805.
Kang Z, Jiang E, Wang K, Pan C, Chen H, Yan H, Zhu H, Liu J, Qu L, Lan X. Goat membrane associated ring-CH-type finger 1 (MARCH1) mRNA expression and association with litter size. Theriogenology. 2019;128:8–16.
Cui Y, Yan H, Wang K, Xu H, Zhang X, Zhu H, Liu J, Qu L, Lan X, Pan C. Insertion/deletion WITHin the KDM6A gene is significantly associated with litter size in goat. Front Genet. 2018;9:91.
Wang K, Yan H, Xu H, Yang Q, Zhang S, Pan C, Chen H, Zhu H, Liu J, Qu L, Lan X. A novel indel within goat casein alpha S1 gene is significantly associated with litter size. Gene. 2018;671:161–9.
Silpa MV, Naicy T, Aravindakshan TV, Radhika G, Boswell A, Mini M. Sirtuin3 (SIRT3) gene molecular characterization and SNP detection in prolific and low prolific goat breeds. Theriogenology. 2018;122:47–52.
An XP, Hou JX, Li G, Song YX, Wang JG, Chen QJ, Cui YH, Wang YF, Cao BY. Polymorphism identification in the goat KITLG gene and association analysis with litter size. Anim Genet. 2012;43(1):104–7.
Yang Q, Yan H, Li J, Xu H, Wang K, Zhu H, Chen H, Qu L, Lan X. A novel 14-bp duplicated deletion within goat GHR gene is significantly associated with growth traits and litter size. Anim Genet. 2017;48(4):499–500.
Wang K, Hui Y, Zhang S, Wang M, Yan H, Zhu H, Qu L, Lan X, Pan C. A deletion mutation within the ATBF1 gene is strongly associated with goat litter size. Anim Biotechnol. 2019:1–7.
Hou J, An X, Li G, Wang Y, Song Y, Cao B. Exploring polymorphisms and their effects on reproductive traits of the INHA and INHbetaA genes in three goat breeds. Anim Sci J. 2012;83(4):273–8.
Pramod RK, Sharma SK, Singhi A, Pan S, Mitra A. Differential ovarian morphometry and follicular expression of BMP15, GDF9 and BMPR1B influence the prolificacy in goat. Reprod Domest Anim. 2013;48(5):803–9.
Zhang C, Liu Y, Huang K, Zeng W, Xu D, Wen Q, Yang L. The association of two single nucleotide polymorphisms (SNPs) in growth hormone (GH) gene with litter size and superovulation response in goat-breeds. Genet Mol Biol. 2011;34(1):49–55.
Ling YH, Quan Q, Xiang H, Zhu L, Chu MX, Zhang XR, Han CY. Expression profiles of differentially expressed genes affecting fecundity in goat ovarian tissues. Genet Mol Res. 2015;14(4):18743–52.
An X, Song Y, Hou J, Li G, Zhao H, Wang J, Cao B. Identification and profiling of microRNAs in the ovaries of polytocous and monotocous goats during estrus. Theriogenology. 2016;85(4):769–80.
Li L, Zhang J, Zhu JQ, Gu S, Sun Q, Zhou GM, Fu CX, Li Q, Chen LY, Li DX, Liu SJ, Yang ZR. Genetic diversity of nine populations of the black goat (Capra hircus) in Sichuan. PR China Zoolog Sci. 2006;23(3):229–34.
Yu J, Jie W, Xingyan J. Genetic diversity of mtDNA D-loop of different black goat breeds in Sichuan Province. J Anhui Agric Sci. 2010;38(27):15052–4.
Brand A, de Jong WH. Qualitative and quantitative micromorphological investigations of the tertiary follicle population during the oestrous cycle in sheep. J Reprod Fertil. 1973;33(3):431–9.
Hay MR, Cran DG, Moor RM. Structural changes occurring during atresia in sheep ovarian follicles. Cell Tissue Res. 1976;169(4):515–29.
Lussier JG, Diouf MN, Levesque V, Sirois J, Ndiaye K. Gene expression profiling of upregulated mRNAs in granulosa cells of bovine ovulatory follicles following stimulation with hCG. Reprod Biol Endocrinol. 2017;15(1):88.
Kfir S, Basavaraja R, Wigoda N, Ben-Dor S, Orr I, Meidan R. Genomic profiling of bovine corpus luteum maturation. PLoS One. 2018;13(3):e194456.
Pelusi C, Fanelli F, Pariali M, Zanotti L, Gambineri A, Pasquali R. Parallel variations of insulin-like peptide 3 (INSL3) and antimullerian hormone (AMH) in women with the polycystic ovary syndrome according to menstrual cycle pattern. J Clin Endocrinol Metab. 2013;98(10):e1575–82.
Batista AM, Silva DM, Rego MJ, Silva FL, Silva EC, Beltrao EI, Gomes FM, Wischral A, Guerra MM. The expression and localization of leptin and its receptor in goat ovarian follicles. Anim Reprod Sci. 2013;141(3–4):142–7.
Convissar S, Winston NJ, Fierro MA, Scoccia H, Zamah AM, Stocco C. Sp1 regulates steroidogenic genes and LHCGR expression in primary human luteinized granulosa cells. J Steroid Biochem Mol Biol. 2019;190:183–92.
Wakinoue S, Chano T, Amano T, Isono T, Kimura F, Kushima R, Murakami T. ADP-ribosylation factor-like 4C predicts worse prognosis in endometriosis-associated ovarian cancers. Cancer Biomark. 2019;24(2):223–9.
Zhang N, Wang L, Luo G, Tang X, Ma L, Zheng Y, Liu S, PC A, Jiang Z. Arachidonic acid regulation of intracellular signaling pathways and target geneexpression in bovine ovarian granulosa cells. Animals (Basel). 2019;9(6):374.
Dentis JL, Schreiber NB, Burress AM, Spicer LJ. Effects of angiogenin on granulosa and theca cell function in cattle. Animal. 2017;11(5):811–9.
Angulo L, Guyader-Joly C, Auclair S, Hennequet-Antier C, Papillier P, Boussaha M, Fritz S, Hugot K, Moreews F, Ponsart C, Humblot P, Dalbies-Tran R. An integrated approach to bovine oocyte quality: from phenotype to genes. Reprod Fertil Dev. 2015; 28:1276-87.
Ren F, Wang D, Wang Y, Chen P, Guo C. SPOCK2 affects the biological behavior of endometrial cancer cells by regulation of MT1-MMP and MMP2. Reprod Sci. 2019;1933719119834341.
Leung CS, Yeung TL, Yip KP, Pradeep S, Balasubramanian L, Liu J, Wong KK, Mangala LS, Armaiz-Pena GN, Lopez-Berestein G, Sood AK, Birrer MJ, Mok SC. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat Commun. 2014;5:5092.
Ignacio R, Lee ES, Wilson AJ, Beeghly-Fadiel A, Whalen MM, Son DS. Chemokine network and overall survival in TP53 wild-type and mutant ovarian cancer. Immune Netw. 2018;18(4):e29.
Chang J, Frandsen S, D'Annibale-Tolhurst M, Palumbo N, Gadsby J. Prostaglandin (PTG) E and F receptors in the porcine corpus luteum; effect of tumor necrosis factor-alpha. Anim Reprod Sci. 2018;195:139–48.
Hayashi KG, Ushizawa K, Hosoe M, Takahashi T. Differential gene expression of serine protease inhibitors in bovine ovarian follicle: possible involvement in follicular growth and atresia. Reprod Biol Endocrinol. 2011;9:72.
Sayasith K, Bouchard N, Dore M, Sirois J. Regulation of bovine tumor necrosis factor-alpha-induced protein 6 in ovarian follicles during the ovulatory process and promoter activation in granulosa cells. Endocrinology. 2008;149(12):6213–25.
Fulop C, Szanto S, Mukhopadhyay D, Bardos T, Kamath RV, Rugg MS, Day AJ, Salustri A, Hascall VC, Glant TT, Mikecz K. Impaired cumulus mucification and female sterility in tumor necrosis factor-induced protein-6 deficient mice. Development. 2003;130(10):2253–61.
Assidi M, Dufort I, Ali A, Hamel M, Algriany O, Dielemann S, Sirard MA. Identification of potential markers of oocyte competence expressed in bovine cumulus cells matured with follicle-stimulating hormone and/or phorbol myristate acetate in vitro. Biol Reprod. 2008;79(2):209–22.
Bakhshalizadeh S, Amidi F, Shirazi R, Shabani NM. Vitamin D3 regulates steroidogenesis in granulosa cells through AMP-activated protein kinase (AMPK) activation in a mouse model of polycystic ovary syndrome. Cell Biochem Funct. 2018;36(4):183–93.
Manna PR, Stetson CL, Slominski AT, Pruitt K. Role of the steroidogenic acute regulatory protein in health and disease. Endocrine. 2016;51(1):7–21.
Liang D, Fan Z, Zou Y, Tan X, Wu Z, Jiao S, Li J, Zhang P, You F. Characteristics of Cyp11a during gonad differentiation of the olive flounder Paralichthys olivaceus. Int J Mol Sci. 2018;19(9):2641.
Wong QW, Sun MA, Lau SW, Parsania C, Zhou S, Zhong S, Ge W. Identification and characterization of a specific 13-miRNA expression signature during follicle activation in the zebrafish ovary. Biol Reprod. 2018;98(1):42–53.
Bertho S, Herpin A, Branthonne A, Jouanno E, Yano A, Nicol B, Muller T, Pannetier M, Pailhoux E, Miwa M, Yoshizaki G, Schartl M, Guiguen Y. The unusual rainbow trout sex determination gene hijacked the canonical vertebrate gonadal differentiation pathway. Proc Natl Acad Sci U S A. 2018;115(50):12781–6.
Loffler KA, Zarkower D, Koopman P. Etiology of ovarian failure in blepharophimosis ptosis epicanthus inversus syndrome: FOXL2 is a conserved, early-acting gene in vertebrate ovarian development. Endocrinology. 2003;144(7):3237–43.
Osz K, Ross M, Petrik J. The thrombospondin-1 receptor CD36 is an important mediator of ovarian angiogenesis and folliculogenesis. Reprod Biol Endocrinol. 2014;12:21.
Serke H, Bausenwein J, Hirrlinger J, Nowicki M, Vilser C, Jogschies P, Hmeidan FA, Blumenauer V, Spanel-Borowski K. Granulosa cell subtypes vary in response to oxidized low-density lipoprotein as regards specific lipoprotein receptors and antioxidant enzyme activity. J Clin Endocrinol Metab. 2010;95(7):3480–90.
Matzuk MM, Lamb DJ. Genetic dissection of mammalian fertility pathways. Nat Cell Biol. 2002;4(Suppl):s41–9.
Berisha B, Rodler D, Schams D, Sinowatz F, Pfaffl MW. Prostaglandins in superovulation induced bovine follicles during the preovulatory period and early corpus luteum. Front Endocrinol (Lausanne). 2019;10:467.
Shirasuna K, Akabane Y, Beindorff N, Nagai K, Sasaki M, Shimizu T, Bollwein H, Meidan R, Miyamoto A. Expression of prostaglandin F2alpha (PGF2alpha) receptor and its isoforms in the bovine corpus luteum during the estrous cycle and PGF2alpha-induced luteolysis. Domest Anim Endocrinol. 2012;43(3):227–38.
Lee-Thacker S, Choi Y, Taniuchi I, Takarada T, Yoneda Y, Ko C, Jo M. Core binding factor beta expression in ovarian granulosa cells is essential for female fertility. Endocrinology. 2018;159(5):2094–109.
Zhang X, Huang L, Wu T, Feng Y, Ding Y, Ye P, Yin Z. Transcriptomic analysis of ovaries from pigs with high and low litter size. PLoS One. 2015;10(10):e139514.
Yang H, Geiger M. Cell penetrating SERPINA5 (ProteinC inhibitor, PCI): more questions than answers. Semin Cell Dev Biol. 2017;62:187–93.
Jing Y, Jia D, Wong CM, Oi-Lin NI, Zhang Z, Liu L, Wang Q, Zhao F, Li J, Yao M, Wu X, He X. SERPINA5 inhibits tumor cell migration by modulating the fibronectin-integrin beta1 signaling pathway in hepatocellular carcinoma. Mol Oncol. 2014;8(2):366–77.
Bijsmans IT, Smits KM, de Graeff P, Wisman GB, van der Zee AG, Slangen BF, de Bruine AP, van Engeland M, Sieben NL, Van de Vijver KK. Loss of SerpinA5 protein expression is associated with advanced-stage serous ovarian tumors. Mod Pathol. 2011;24(3):463–70.
Suo HB, Zhang KC, Zhao J. miR-200a promotes cell invasion and migration of ovarian carcinoma by targeting PTEN. Eur Rev Med Pharmacol Sci. 2018;22(13):4080–9.
Zavesky L, Jandakova E, Weinberger V, Minar L, Hanzikova V, Duskova D, Drabkova LZ, Svobodova I, Horinek A. Ascites-derived extracellular micrornas as potential biomarkers for ovarian cancer. Reprod Sci. 2019;26(4):510–22.
Wang Y, Qiu C, Lu N, Liu Z, Jin C, Sun C, Bu H, Yu H, Dongol S, Kong B. FOXD1 is targeted by miR-30a-5p and miR-200a-5p and suppresses the proliferation of human ovarian carcinoma cells by promoting p21 expression in a p53-independent manner. Int J Oncol. 2018;52(6):2130–42.
Zhang M, Ren C, Xiao Y, Xia X, Fang X. Expression profile analysis of circular rnas in ovarian endometriosis by microarray and bioinformatics. Med Sci Monit. 2018;24:9240–50.
Mak CS, Yung MM, Hui LM, Leung LL, Liang R, Chen K, Liu SS, Qin Y, Leung TH, Lee KF, Chan KK, Ngan HY, Chan DW. MicroRNA-141 enhances anoikis resistance in metastatic progression of ovarian cancer through targeting KLF12/Sp1/survivin axis. Mol Cancer. 2017;16(1):11.
Li D, Xu D, Xu Y, Chen L, Li C, Dai X, Zhang L, Zheng L. MicroRNA-141-3p targets DAPK1 and inhibits apoptosis in rat ovarian granulosa cells. Cell Biochem Funct. 2017;35(4):197–201.
Dai C, Xie Y, Zhuang X, Yuan Z. MiR-206 inhibits epithelial ovarian cancer cells growth and invasion via blocking c-met/AKT/mTOR signaling pathway. Biomed Pharmacother. 2018;104:763–70.
Qu W, Chen X, Wang J, Lv J, Yan D. MicroRNA-1 inhibits ovarian cancer cell proliferation and migration through c-met pathway. Clin Chim Acta. 2017;473:237–44.
Ling C, Liu S, Wang Y, Zhang FC, DU Y. Role of miR-206/CDK4 in modulating the growth and chemotlerapy sensitivity of ovarian cancer cells. Nan Fang Yi Ke Da Xue Xue Bao. 2017;37(3):393–7.
Sheng N, Xu YZ, Xi QH, Jiang HY, Wang CY, Zhang Y, Ye Q. Overexpression of KIF2A is suppressed by miR-206 and associated with poor prognosis in ovarian Cancer. Cell Physiol Biochem. 2018;50(3):810–22.
Liu X, Li G. MicroRNA-133b inhibits proliferation and invasion of ovarian cancer cells through Akt and Erk1/2 inactivation by targeting epidermal growth factor receptor. Int J Clin Exp Pathol. 2015;8(9):10605–14.
Hao L, Li J, Tian Y, Wu J. Changes in the microrna profile of the mandible of ovariectomized mice. Cell Physiol Biochem. 2016;38(4):1267–87.
Dai A, Sun H, Fang T, Zhang Q, Wu S, Jiang Y, Ding L, Yan G, Hu Y. MicroRNA-133b stimulates ovarian estradiol synthesis by targeting Foxl2. FEBS Lett. 2013;587(15):2474–82.
Xiao G, Xia C, Yang J, Liu J, Du H, Kang X, Lin Y, Guan R, Yan P, Tang S. MiR-133b regulates the expression of the actin protein TAGLN2 during oocyte growth and maturation: a potential target for infertility therapy. PLoS One. 2014;9(6):e100751.
Naji M, Nekoonam S, Aleyasin A, Arefian E, Mahdian R, Azizi E, Shabani NM, Amidi F. Expression of miR-15a, miR-145, and miR-182 in granulosa-lutein cells, follicular fluid, and serum of women with polycystic ovary syndrome (PCOS). Arch Gynecol Obstet. 2018;297(1):221–31.
Wang A, Jin C, Li H, Qin Q, Li L. LncRNA ADAMTS9-AS2 regulates ovarian cancer progression by targeting miR-182-5p/FOXF2 signaling pathway. Int J Biol Macromol. 2018;120((Pt B):1705–13.
Duan Y, Dong Y, Dang R, Hu Z, Yang Y, Hu Y, Cheng J. MiR-122 inhibits epithelial mesenchymal transition by regulating P4HA1 in ovarian cancer cells. Cell Biol Int. 2018;42(11):1564–74.
Menon B, Gulappa T, Menon KM. Molecular regulation of LHCGR expression by miR-122 during follicle growth in the rat ovary. Mol Cell Endocrinol. 2017;442:81–9.
Griffiths-Jones S. The microRNA registry. Nucleic Acids Res. 2004;32(Database issue):D109–11.
Acknowledgements
We would like to thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript.
Funding
This research was supported by the Guangdong Provincial Department of Education Youth Innovative Talents Project (2017KQNCX014), Modern Agricultural Industrial Technology System of Guangdong Province (2019KJ127), the Guangdong Public Welfare Research and Capacity Building Project (2017B020201014, 2017A020208050), the Guangdong Provincial Promotion Project on Preservation and Utilization of Local Breed of Livestock and Poultry, and the Guangdong Special plan young top-notch talent (2015TQ01N843).
Author information
Authors and Affiliations
Contributions
XZ and TL contributed equally to this work. XZ, YL, GL and DL designed the experiments. XZ, TL, ZZ and ZL performed the experiment. XZ, TL, YG and BS carried out the data processing and analysis. XZ and TL were major contributors in writing the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments and sample collections from CZ black goats in the present study were performed in strict accordance with the Regulations of People’s Republic of China for the Administration of Laboratory Animals (2017 Revision, CLI.2.293192, State Council, China). All animal research protocols were approved by the Institutional Animal Care and Use Committees (IACUCs) of South China Agricultural University (Approval No. 2018-P002). All efforts were made to minimize animal suffering.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 Figure S1
, RNA-seq reveals distinct expression pattern of mRNAs and miRNAs among the four groups. (A) Unsupervised clustering analysis showing expression profiles of mRNAs between Uni-S and Mul-S groups. (B) Unsupervised clustering analysis showing expression profiles of mRNAs between Uni-L and Mul-L groups. (C) Unsupervised clustering analysis showing expression profiles of miRNAs between Uni-S and Mul-S groups. (D) Unsupervised clustering analysis showing expression profiles of miRNAs between Uni-L and Mul-L groups.
Additional file 2.
RNA-seq data of DEmRNA expression in large and small follicles from uniparous and multiple goats.
Additional file 3.
RNA-seq data of DEmiRNA expression in large and small follicles from uniparous and multiple goats.
Additional file 4.
The significant Go term of DEmRNAs in large and small follicles from uniparous and multiple goats.
Additional file 5.
The significant KEGG pathways of DEmRNAs in large and small follicles from uniparous and multiple goats.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zou, X., Lu, T., Zhao, Z. et al. Comprehensive analysis of mRNAs and miRNAs in the ovarian follicles of uniparous and multiple goats at estrus phase. BMC Genomics 21, 267 (2020). https://doi.org/10.1186/s12864-020-6671-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6671-4