
Abstract
Background
MicroRNAs (miRNAs), a class of small regulatory RNAs, have been proven to play important roles in plant growth, development and stress responses. Sweet potato (Ipomoea batatas L.) is an important food and industrial crop that ranks seventh in staple food production. However, the regulatory mechanism of miRNA-mediated abiotic stress response in sweet potato remains unclear.
Results
In this study, we employed deep sequencing to identify both conserved and novel miRNAs from salinity-exposed sweet potato cultivars and its untreated control. Twelve small non-coding RNA libraries from NaCl-free (CK) and NaCl-treated (Na150) sweet potato leaves and roots were constructed for salt-responsive miRNA identification in sweet potatoes. A total of 475 known miRNAs (belonging to 66 miRNA families) and 175 novel miRNAs were identified. Among them, 51 (22 known miRNAs and 29 novel miRNAs) were significantly up-regulated and 76 (61 known miRNAs and 15 novel miRNAs) were significantly down-regulated by salinity stress in sweet potato leaves; 13 (12 known miRNAs and 1 novel miRNAs) were significantly up-regulated and 9 (7 known miRNAs and 2 novel miRNAs) were significantly down-regulated in sweet potato roots. Furthermore, 636 target genes of 314 miRNAs were validated by degradome sequencing. Deep sequencing results confirmed by qRT-PCR experiments indicated that the expression of most miRNAs exhibit a negative correlation with the expression of their targets under salt stress.
Conclusions
This study provides insights into the regulatory mechanism of miRNA-mediated salt response and molecular breeding of sweet potatoes though miRNA manipulation.
Similar content being viewed by others


Background
Soil salinity, one of the major environmental factors, reduces the productivity of crops worldwide [1]. It is estimated that 50% of all arable land will be affected by salinity by 2050 [2], and approximately 20% of the irrigated soils worldwide are suffering salt stress [3]. Meanwhile, the crop production demands continue to increase with the rapidly growing population [4]. Therefore, to sustain or increase food supply, salt tolerance is an important agronomic trait to support crop plant growth and production in marginal and high-salinity soils [5].
MicroRNAs (miRNAs) are a class of endogenous small non-coding RNAs (sRNAs) that are 21–24 nt in length and negatively regulate gene expression at transcriptional and post-transcriptional levels [6,7,8]. In plants, the primary miRNA precursor is transcribed from DNA and then sequentially processed by Dicer-like 1 (DCL1) via two steps: firstly, into precursor-miRNAs (pre-miRNAs), and secondly, into miRNA/miRNA* duplex. The mature miRNA from one of the duplex strands incorporates into the RNA-induced silencing complex (RISC) and then guides the RISC to target mRNA, either cleaving the target with near perfect base pairing complementarity or repressing its translation with lower complementarity [9, 10]. Previous studies showed that miRNAs regulate diverse processes in plants, including leaf morphogenesis and polarity, root initiation and development, flower differentiation and development, stem and vascular development, phase switch from vegetative growth to reproductive growth and response to abiotic and biotic stresses [7, 11, 12].
Salinity stress affects the photosynthesis, signal transduction, protein synthesis and degradation processes in plants and inhibits crop yield dramatically. miRNAs are indispensable for plants to respond to salt stress and play an important regulatory role by regulating the expression of their target genes. For example, miR396 increases Arabidopsis salt tolerance through regulating its target gene GRF [13]; miR398 protects cell membrane structure against salt stress by targeting CSD1 and CSD2 [14]; The miR172c-NNC1 module modulates root plastic development in response to salt in soybean [15]; and the target gene of miR169c can control the stomatal opening and closure in maize leaves, thereby reducing water loss and resisting salt stress [16]. The decrease in miR414/miR408/miR164e leads to up-regulated helicase (OsABP, OsDSHCT and OsDBH) expression, thereby regulating gene recombination replication and repair to resist saline environment [17]. The overexpression of osa-miR393 negatively regulates rice salt tolerance [18]. The miR397-resistant transgenic Arabidopsis plant decreases its salt tolerance ability [19]. MiR319 and miR528 can enhance cotton plant tolerance to salt stress by down-regulating their target genes [20, 21]. Recently, deep sequencing data showed that several miRNAs are significantly altered under NaCl stress in various plants, such as rice [22], soybean [23] and Dunaliella salina [24]. Evidence suggests that miRNA plays an important regulatory role in plant salt stress tolerance.
Sweet potato (Ipomoea batatas L.) is an important food and industrial crop [25]. It is a valuable food source that contains various nutrients, including high starch content, complex carbohydrates, dietary fibre, vitamins and anthocyanins. Salt stress adversely influences sweet potato growth, fresh weight, health-promoting compounds and antioxidant activity. As a source of bio-energy, sweet potato is mainly planted on marginal land; hence, improving its salt tolerance is important to maintain productivity [26, 27]. Until now, a series of salt-tolerance-associated genes, such as IbOr, IbNFU1, IbP5CR, IbMas, IbSIMT1, IbMIPS1 and IbZFP1, has already been well characterised in sweet potatoes [26, 28,29,30,31,32,33,34,35]. However, very little work has been done on sweet potato miRNAs, and miRNA-related salt stress in sweet potato has not been investigated.
In the present study, we employed high-throughput sequencing technology and bioinformatic analysis to identify conserved and novel salt-responsive miRNAs by constructing small RNA libraries from NaCl-free (CK) and NaCl-treated (Na150) sweet potato roots and leaves. The expression profiles of the miRNAs between the roots and the leaves were investigated. We also predicted the targets of miRNAs and further investigated their network by GO and KEGG analyses using sweet potato transcriptome and degradome data. This study contributed in elucidating the potential miRNA-mediated regulatory mechanism of salt stress response in sweet potatoes. The specific miRNAs in sweet potatoes can be used to breed salt-tolerant plants that can grow on marginal lands.
Results
Transcriptome sequencing analysis
The transcriptome sequencing of sweet potato (Xu32) was performed by Illumina high-throughput sequencing. Approximately 90,528,282, 109,002,500, 82,914,414 and 90,852,454 raw reads were obtained from the SRC, SLC, SRN and SLN libraries, respectively, and the total number of bases was approximately 56.01Gb (Additional file 1: Table S1). Through the Trinity de novo assembly method, a total of 27,712 non-redundant genes and 41,879 transcripts were obtained. The N50 length of these transcripts was 1338 bp, with an average length of 924 bp and a total of 25,967 transcripts over 500 bp (63%) (Additional file 2: Table S2 and Additional file 3: Table S3). All unigenes were searched in the Swiss-Prot, Nr, Pfam, KEGG, KOG and GO databases, and a total of 14,671, 22,756, 17,289, 8516, 19,462 and 12,959 were detected, respectively (Additional file 4: Table S4).
Sequencing, annotation and sequence characterisation of small RNA
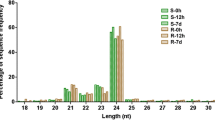
To explore the regulatory mechanisms of miRNAs in response to salt stress in sweet potato, we established four different sample groups, namely, NaCl-treated (Na150) group of roots (SRN) and leaves (SLN) and the NaCl-free group of roots (SRC) and leaves (SLC). Every sample group contains three biological replicates. As listed in Additional file 5: Table S5, a total of 12 sRNA libraries were established and subjected to high-throughput sequencing. Using Illumina’s Solexa sequencing platform, raw data were obtained as follows: 16,855,549 (SLC26), 20,085,489 (SLC28), 19,050,745 (SLC32), 17,881,455 (SLN18), 21,223,113 (SLN20), 17,902,270 (SLN22), 20,477,313 (SRC25), 20,464,252 (SRC27), 17,965,687 (SRC29), 16,221,626 (SRN17), 18,904,028 (SRN19) and 22,740,499 (SRN21). After filtering low-quality data and 3′ joint contamination data and sequences with length less than 18 nt or greater than 25 nt and excluding non-coding RNAs (e.g. rRNAs, tRNAs, snRNAs and snoRNAs), the remaining unannotated data were used for miRNA identification. The size distributions of the 12 small RNA libraries are presented in Fig. 1. The distribution patterns of the most redundant sRNA peaks at 21, 22 and 24 nt are shown in Fig. 1a. However, in the distribution pattern of non-redundant sRNA length, 24 nt sRNAs are the most abundant category (average of 33.7%) followed by 22 and 21 nt (Fig. 1b).

Length distribution and abundance of small RNAs in 12 sRNA libraries in sweet potato. a Size distribution of redundant sequences. b Size distribution of unique sequences
Identification of known miRNAs in sweet potato
To identify the known miRNAs in sweet potato, we screened the sequences that matched the sweet potato transcriptome sequences in the 12 small RNA libraries and then aligned these sequences with the matured miRNA sequences in miRBase 21.0 database. Finally, 444 pre-miRNAs corresponding to 475 known unique mature miRNAs were identified as homologues of known miRNAs from the other 42 plants (Additional file 6: Table S6). Approximately 33.89% of these sweet potato miRNAs can be found in Solanaceae plants, such as Solanum tuberosum (111), Solanum lycopersicum (19) and Nicotiana tabacum (31) (Additional file 7: Figure S1). Among these known miRNAs, 53 and 42 were specifically expressed in sweet potato leaves and roots, respectively, and 380 were co-expressed in the roots and leaves (Fig. 2a). The length of known miRNAs ranges from 18 nt to 25 nt, with 21 nt miRNAs being the most abundant (41.47%) (Fig. 3a). In the first nucleotide selection, uracil accounted for 53.19, 49.49, 55.17 and 54.55% in miRNAs of 20 nt to 23 nt in length, respectively (Fig. 3b and Fig. 3c). Among these known miRNAs, 379 belong to 66 families (Additional file 6: Table S6 and Fig. 4), whereas the families of the other 96 miRNAs are unknown. The three largest families were miR156 (30 miRNA members), miRNA159 (24 members) and miR166 (23 members), whereas only 22 families contained only 1 member (Fig. 4). Among these 66 miRNA families, 55, 58, 54 and 49 families were expressed in the SRN, SRC, SLN and SLC library, respectively (Additional file 8: Figure S2).

Distribution of miRNAs in sweet potato roots and leaves. a Distribution of known miRNAs in sweet potato roots and leaves. b Distribution of novel miRNAs in sweet potato roots and leaves

Analyses of the length distribution and nucleotide base bias of known miRNAs. a Size distribution of known miRNA. b Percentage of first nucleotide bias in known miRNAs. c Known miRNA nucleotide bias at first position

Distribution of conserved miRNAs in miRNA family
High-throughput sequencing detects the type of miRNA and the abundance of different miRNAs [36]. The expression abundance of some miRNA families in the four libraries varied (Additional file 8: Figure S2). Eleven families (miR166, miR156, miR168, miR2111, miR2275, miR398_2, miR858, miR6426, miR827_5, miR827_4 and miR8674) were up-regulated in the roots and leaves of sweet potato under salt stress condition, and 15 families (miR169_1, miR164, miR390, miR408, miR397, etc.) were down-regulated (Additional file 8: Figure S2). A total of 24 miRNA families exhibited opposite expression patterns in sweet potato leaves and roots under salt stress condition. For example, miR162_1 and miR398 were down-regulated in the roots but up-regulated in the leaves, whereas miRNA159 and miRNA160 were up-regulated in the roots but down-regulated in the leaves. miR166, miR168, miR2118, miR397, miR156, miR398, miR396, miR167_1, miR159 and miR408 were highly expressed in all four libraries. The different abundance of miRNAs indicated that they played different roles under salt stress.
Identification of novel miRNAs in sweet potato
According to the annotation standards of novel miRNA [37] and after normalising the expression level, miRNAs with a copy number of less than 10 were eliminated in all samples. Finally, 175 novel miRNAs were predicted from 157 pre-miRNAs with a length of 21–24 nt (Additional file 9: Table S7 and Additional file 10: Figure S3A), which was in line with the size of miRNA fragments generated by AGO1 protein cleavage. Among these novel miRNAs, 4 were specifically expressed in the leaves, 7 were specifically expressed in the roots, and 163 were co-expressed in the roots and leaves (Fig. 2b). Among the novel miRNAs, the first nucleotide of 5′ was A (adenine) (33.91%) and U (uracil) (35.63%) (Additional file 10: Figure S3B and S3C). These pre-miRNAs range in length from 59 nt to 259 nt with an average length of 138 nt, which is consistent with the general length of pre-miRNAs. The CG percentages (CG%) of these novel pre-miRNAs range from 12.20 to 61.20%, and their minimal folding free energy index (MFEI) ranges from 0.90 to 2.50 with an average of 1.29. The secondary structure of the precursor of the novel miRNA was obtained using Mfold software, and the structure of the representative miRNA is listed in Additional file 11: Figure S4.
Differential expression analysis of miRNAs in sweet potato under salt stress condition
To find the salt-stress-responsive miRNAs in sweet potato (Xu32), the expression levels of all miRNAs were normalised and analysed. As shown in Fig. 5a, 33 and 28 miRNAs were specifically expressed in the control and under the salt stress condition, respectively. In the leaves, 41 and 50 miRNAs were specifically expressed in NaCl-free (SLC) and NaCl (150 mM) (SLN) groups, respectively; in the roots, 36 and 69 miRNAs were specifically expressed in CK (SRC) and NaCl (150 mM) (SRN) groups, respectively. In addition, 10, 11, 5 and 13 miRNAs were specifically expressed in SLN, SLC, SRN and SRC libraries, respectively (Fig. 5a). These results implied that the specifically expressed miRNAs under the control condition may play a negative role in salt response, whereas the specifically expressed miRNAs under salt condition may play a positive role in the salt response in sweet potato roots and/or leaves.

Distribution of miRNAs in CK and NaCl (150 mM) groups in sweet potato roots and leaves. a Distribution of total miRNAs in CK and NaCl (150 mM) groups in sweet potato roots and leaves. b Differentially expressed miRNAs in sweet potato roots and leaves were identified through two comparisons (SRC-VS-SRN and SLC-VS-SLN). MiRNAs with significant differences in sweet potato roots and leaves are shown in Additional file 12: Table S8
A total of 148 miRNAs (101 known miRNAs and 47 novel miRNAs) were significantly (P < 0.05 and |log2(fold change)| > 1) up- or down-regulated in sweet potato under salt stress (Additional file 12: Table S8, Fig. 5b). In the leaves, 50 miRNAs (21 known miRNAs and 29 novel miRNAs) were up-regulated, whereas 76 miRNAs (61 known miRNAs and 15 novel miRNAs) were down-regulated; in the roots, 12 miRNAs (11 known miRNAs and 1 novel miRNA) were up-regulated, whereas 9 miRNAs (7 known miRNAs and 2 novel miRNAs) were down-regulated. Among all the differentially expressed miRNAs, only one miRNA (ath-mir164b-3p) was up-regulated both in the roots and leaves after salt treatment. Among these significant differentially expressed miRNAs in leaves, six miRNAs (nta-miR156a_R + 3, far-miR159_L + 2_1ss22T, mes-MIR319e-p5_2ss12GC19GA, ssl-miR171b_1ss21TA, nta-miR169q_1ss14CA, PC-3p-786500_26) were only detected in the SLC library, whereas four miRNAs (gma-miR156b_L + 2R-1, gma-MIR171a-p5_2ss12TA18AC, PC-3p-14376_1292, PC-5p-150993_155) were only detected in the SLN library (Additional file 12: Table S8). In the roots, lus-MIR169j-p3-2ss6TC21TG and tcc-miR530a_R + 1_1ss12CT were only detected in the SRN library (Additional file 12: Table S8), indicating that these miRNAs may function in a tissue-specific manner in sweet potato under salt stress.
Target gene prediction and annotation of miRNA
To identify the target genes of miRNAs, we performed the miRNA-degradome of sweet potato and obtained 21,372,881 raw reads and 13,953,861 unique reads from the NaCl-treated (Na150) group (DSN) and 16,764,899 raw reads and 5,659,818 unique reads from the NaCl-free (CK) group (DSC) (Table 1). A total of 6,599,927 and 11,157,651 reads (called Transcript Mapped reads) mapped to the sweet potato transcriptome database were retained in DSN and DSC. Finally, 23,926 and 24,656 reads containing miRNA-mediated cleavage sites were identified in DSN and DSC. These data along with the sweet potato miRNA dataset were analysed according to the CleaveLand pipeline process [38]. Finally, the miRNA–mRNA pairings were obtained, that is, the target genes of the miRNA were identified. On the basis of the degradome data, a total of 392 target genes for 258 miRNAs (235 known miRNAs, 23 novel miRNAs) were identified in DSN (Additional file 13: Table S9 and Fig. 6a), and 458 target genes for 270 miRNAs (239 known miRNAs, 31 novel miRNAs) were identified in DSC (Additional file 13: Table S9 and Fig. 6a). Venn diagram analysis showed that 214 target genes were found in the two degradomes, and 244 and 178 specific target genes were present in the DSC and DSN libraries, respectively (Fig. 6a). According to the relationship between the cut position of the original fragment and the abundance of the mRNA, the target genes can be divided into five categories [39] (Additional file 14: Table S10). In the DSC and DSN libraries, the numbers of cleaved transcripts were 49 and 40 in Category 0; 4 and 8 in Category 1; 162 and 129 in Category 2; 16 and 9 in Category 3; and 256 and 236 in Category 4, respectively (Fig. 6b and Fig. 6c). In addition, on the basis of the sequencing results of the degradome, the same target gene corresponding to the different miRNAs may have different categories (Additional file 13: Table S9). For example, SPL13A (comp19729_c0), which is regulated by gma-mir156b_r-1, nta-mir156a_r + 1 and nta-mir156a_r + 3, belongs to Category 2 in the DSC library and Category 0 in the DSN library. In addition, conserved miRNAs can regulate multiple target genes. For example, ppe-MIR169i-p3_2ss1TC17GT regulates TOP1, GC4, HSP90, UBXN1, etc.; ath-miR156h_L + 1 regulates SPL13A, SBP1 and SPL12. A target gene was regulated by multiple conserved miRNAs. For example, AFB2 was co-regulated by stu-miR393-5p, stu-miR393-5p_R + 1_2ss21CT22CT and stu-miR393-5p_R-1_1ss21CT; DCL1 was regulated by stu-miR162a-3p and stu-miR162a-3p_R-2.

Target gene cleavage site analysis of miRNA. a Distribution of confirmed miRNA targets, separated by category in DSC library (left) and DSN library (right). b Distribution of target genes in two degradation group libraries. c Representative target plots (t-plot) depicting categories of the cleaved mRNAs confirmed by degradome sequencing, reflecting the reliability of the miRNA targets
Functional analysis of the target genes of significantly up- or down-regulated miRNA under salt stress
Although 148 miRNAs were significantly reduced or induced by salt stress, 66 miRNAs and 87 target genes generated 168 regulatory interactions according to the combined results of miRNA profiling, degradome sequencing and transcriptome profiling. Among 87 target genes, 56 genes are transcription factors (TFs), such as AP2, NAC, NFYA, GRF, SCL, SPL and TCP (Additional file 15: Table S11 and Additional file 16: Table S12), which are involved in the regulation of gene expression and signal transduction. The other miRNA target genes, including desaturase, peptidase, synthase, transferase and kinase-encoding genes (such as calcium-dependent protein kinase (nta-miR169q_L + 2R-2, comp15536_c0), omega-3 fatty acid desaturase (mes-MIR319e-p5_2ss12GC19GA, comp23770_c0) and Cu2+-exporting ATPase (stu-miR408b-3p, comp11674_c0)) were involved in plant growth and developmental adaptive responses. Nine target genes of significantly up- or down- regulated novel miRNAs have been successfully identified. Among them, only four target genes were annotated, namely, formin-like protein 20, small subunit ribosomal protein S19, translation initiation factor if-3 subunit 6 and mitochondrial import receptor subunit TOM40. These results indicated that miRNAs may play an important role in different biological processes under salt stress in sweet potato.
To further analyse the specific biological functions of sweet potato miRNAs under salt stress, we performed GO enrichment analysis on 87 target genes of 66 miRNAs in the regulatory interactions. The results indicated that the target genes were annotated into 39 GO terms (Fig. 7). Among them, the target genes of differentially expressed miRNAs have the largest proportion of cellular processes, followed by single-organism processes, metabolic process, biological regulation and regulation of biological processes. In the classification of cellular components, cells, cell parts and organelles (plasma membrane) have the top three number of genes (43, 43 and 38, respectively); in the molecular function classification, the two GO terms of binding and catalytic activity have the most target genes. In addition, we performed a significant analysis of these GO terms (P < 0.05, FDR < 0.05) (Additional file 17: Table S13). For the molecular function of target genes, the nucleic acid binding, DNA binding and nucleic acid binding TF activity of the GO terms were significantly enriched (Additional file 18: Figure S5A). For the cellular components, the target genes are mainly found in CCAAT-binding factor complex, RNA polymerase II TF complex and nuclear TF complex (Additional file 18: Figure S5B). For the biological processes, target genes are significantly enriched into transcription (DNA-templated), nucleic acid-templated transcriptions, RNA biosynthetic process and so on (Additional file 18: Figure S5C). Furthermore, we performed KEGG pathway analysis on these target genes. The result showed that 87 miRNA target genes participated in 11 metabolic pathways, including environmental adaptation, lipid metabolism, nucleotide metabolism, etc. (Additional file 18: Figure S5D).

GO analysis of significant differentially expressed miRNA target genes in response to salt stress. The y-axis represents the percent of a specific category of target genes in that main category. The x-axis represents the name of the GO sub-categories
qRT-PCR validation of miRNAs and their target genes
To verify the small RNA sequencing results, we randomly selected 10 miRNA–target pairs for qRT-PCR analysis (Fig. 8). The results showed that in the leaves, the expression of eight miRNA–target pairs (nta-miR156a_R + 3/SPL12/SL13A, stu-miR169a-5p_1ss20TA/NF-YA9/CRK1, stu-miR166b/ATHB-15, nta-miR168d_R + 1/AGO1, stu-miR396-5p/rpb-5, stu-miR8005a-p3_1ss4GA/EGD1 and vvi-miR319g/TCP2) displayed reverse expression pattern, indicating that miRNAs repressed their corresponding targets, and two miRNA–target pairs showed ‘unrelated miRNA–target pair in expression’. Similarly, in the roots, eight miRNA–target pairs showed a reverse expression pattern, and two miRNA–target pairs showed ‘unrelated miRNA–target pairs in expression’. Even previously studies described that miRNAs and targets might be involved in multiple expression patterns and their co-regulation can transcend the inverse of the expression [40, 41]. However, this ‘unrelated miRNA–target pairs in expression’ might be possibly caused by the false positive effect of the transcriptome itself, which was need to be further investigated carefully in the future.

qRT-PCR analysis of miRNAs and their targets under salt stress. The relative expression of the 10 randomly selected sweet potato miRNA–target pairs were analysed by qRT-PCR. The ARF transcript levels were used for normalisation. Error bars indicate standard deviation. qRT-PCR value represents an average of three biological samples ± SE. MiRNA and its target primers are listed in Additional file 19: Table S14
Discussion
High-throughput sequencing and identification of miRNAs in sweet potato
According to miRNA prediction criteria, 650 miRNAs, including 475 known miRNAs and 175 novel miRNAs, were obtained from 12 small RNA libraries. The use of multiple sRNA libraries can improve the reliability of novel miRNA prediction and provide a solid basis for further in-depth miRNA study in sweet potato. In our study, most of the known miRNA are 21 nt in length, and the novel miRNAs are mainly 21 and 24 nt in length (Fig. 3 and Additional file 10: Figure S3), which is consistent with the report of sRNA in sweet potato [42]. And the sRNA length distribution pattern in sweet potato is very similar to the typical pattern of sRNAs reported in Betula luminifera [43]. The difference in the length of small RNAs may be related to the species and the enzymes that process it. For example, the length of the small RNAs in plants processed by DCL1 and DCL2 is 21 and 24 nt, respectively [44]. The 21 or 22 nt miRNAs tend to function as silencing complexes with AGO proteins and cleave the target genes [45], whereas 24 nt miRNAs mediate DNA methylation by binding to AGO4 [46]. Among known miRNAs, most of the first base of 21 and 22 nt miRNA was U (uracil) biased at the 5′-end, and the first base 24 nt miRNA was A (adenine) biased. For 175 novel miRNAs, the first base of the 5′-end of miRNA was A (adenine), accounting for 33.91%, and U (uracil), accounting for 35.63%. Some studies have found that uracil (U) at the 5′-end can facilitate the miRNA and AGOl protein binding. The first base adenine (A) at the 5′-end of 24 nt sRNA is beneficial for binding to AGO4, which preferentially binds to siRNA [47,48,49]. The previous studies revealed that when MFEI was greater than 0.85, the sequence most likely originated from miRNAs [50]. In our study, the minimal folding free energy index (MFEI) of novel pre-miRNAs ranged from 0.90 to 2.50 with an average of 1.29, which was higher than that of other types of RNAs, such as tRNAs (0.64), rRNAs (0.59) and mRNAs (0.65) [51].
Analysis of differential miRNAs in response to salt stress in sweet potato
Identification of differentially expressed miRNAs will provide a better understanding of the post-transcriptional regulation in sweet potato under salt stress. A total of 148 miRNAs (101 known miRNAs belonged to 24 miRNA families, 47 novel miRNAs) were significantly up- or down-regulated by salt stress treatment in sweet potato roots or leaves (P < 0.05) (Additional file 12: Table S8 and Fig. 5b). In the roots, miR169_1, miR171_1 and miR818 were significantly down-regulated under salt stress, whereas miR166, miR156, miR167_1 and miR159 were up-regulated. In the leaves, miR2111, miR8005 and miR827_5 were significantly up-regulated, whereas miR399, miR167–1, miR166, miR172, miR396, miR408, miR477, miR444, miR482, miR530 and miR2118 were significantly down-regulated. In the roots and leaves, the miR168 family was significantly up-regulated, whereas the miR169_1 family was significantly down-regulated. These results implied that down- or up-regulated miRNA may play negative or positive roles in salt response in sweet potato. Some miRNA members belonging to the same family showed opposite expression pattern after salt stress treatment in sweet potato roots and leaves. For example, in the roots, stu-MIR530-p3_2ss8GA20TG of the miR530 family was down-regulated, whereas tcc-miR530a_R + 1_1ss12CT was up-regulated. In the leaves, nta-miR156a_R + 3 was down-regulated, and gma-miR156b_L + 2R-1 was up-regulated. This result indicates that miRNAs in the same family may play different roles in response to salt stress. Arabidopsis miRNA168 and maize miRNA168 were previously reported to be induced by salt stress [52, 53], and maize miR167 and S. linnaeanum miR399b were down-regulated [52, 54]. This finding indicated that miRNAs, which show similar expression patterns in different species, may have similar salt stress response mechanisms. Although a large number of salt-stress-response-related miRNAs are conserved in plants, some miRNAs have different regulatory patterns in different species. For example, miR396 was significantly up-regulated in Arabidopsis under salt stress [53] but down-regulated in maize and cotton [52, 55]. The expression of miR172 is down-regulated in maize [52] but up-regulated in Arabidopsis [53]. Therefore, this species-specific miRNA regulation mechanism makes it necessary to analyse the miRNAs of specific plant species under salt stress.
Dissecting the roles of sweet potato miRNA in salinity response by combined analysis of sRNA and degradome sequencing
The target genes must be determined to elucidate the biological functions of miRNA. To further understand the regulation function of sweet potato miRNAs in salt stress response, we analysed the miRNAs and their target genes by miRNA expression profile, degradome and transcriptome. Sixty-six significant differentially expressed miRNAs and their 87 target genes were identified (Additional file 15: Table S11). These targets are TFs, hormone response genes, DNA/RNA binding proteins, protein-coding genes and enzymes. The general salt stress response pathway is: membrane receptors sense extracellular stress signals, signal transduction, transcriptional regulation and induction of salt-related gene expression and finally make physiological changes to cope with stress. Among them, salt stress signal transduction mainly includes ABA pathway, protein kinase pathway and SOS pathway. The potential regulatory network of miRNAs in sweet potato in salt stress response is shown in Fig. 9.

Potential regulatory network of miRNAs in the roots and leaves of sweet potato under salt stress
In sweet potato, nta-miR169q_L + 2R-2 targeted CRK1 and CPK32, and nta-miR169q_R-2_1ss1CT and nta-miR169q targeted CRK1. The calmodulin-dependent protein kinases (CDPK) cascade pathway is an important signalling pathway in the protein kinase pathway. When plants receive salt stress signals, the Ca2+-dependent signal transduction pathway can transmit signals through the synergistic effect of Ca2+ concentration as a second messenger and CDPK to induce TFs to play a role in enhancing plant salt tolerance [56]. AtCRK1 is involved in salt tolerance stress by altering the levels of MDA and proline in Arabidopsis [57]. CPK32 interacts with ABF4 and phosphorylates, and CPK32-overexpressing plants exhibit a highly sensitive phenotype for ABA and NaCl, suggesting that CPK32 is actively involved in ABA/stress signalling [58]. In the process of signal transduction, TFs receive upstream salt stress signals and regulate the expression of downstream salt-tolerant or salt-sensitive genes [59, 60]. In the present study, most of the targets of miRNAs were TF genes, including NACs, SPLs, AP2, TCP and NF-Y. NACs are the largest TF gene family in plants that play an important role in abiotic stress responses. OSNAC5 protein has been found to bind to the promoter region of OsLEA3, up-regulate the expression of stress-related genes and enhance rice stress tolerance [61]. In addition, GmNAC021 is highly expressed in drought-resistant soybeans and may play a role in soybean leaf growth and plant response to drought stress [62]. We found that ptc-miR164a_R + 3 was significantly down-regulated under salt stress and targets NAC021, suggesting that the decrease in ptc-miR164a_R + 3 may release the expression of its target NAC021 to regulate the salt stress adaption in sweet potato.
The highly conserved miR156/SPL module was also identified in sweet potato. For example, significantly down-regulated sweet potato nta-miR156a_R + 3 and gma-miR156b_R-1 targeted to SPL12, SPL4 and SPL13A. qRT-PCR validation showed that nta-miR156a_R + 3/SPL13A target pairs presented a reverse expression pattern. Squamosa promoter binding protein-like (SPL) is a key regulator of plant abiotic stress tolerance. VpSBP16 and BpSPL9 enhance salt and drought stress tolerance in Arabidopsis and Betula platyphylla, respectively [63, 64], indicating that miR156 may also participate in the salt stress response in sweet potato. AP2 TFs have been reported to be regulated by miRNA172 and play an important role in the transcriptional regulation of biological processes involved in the growth and development and in the response to abiotic stress [65, 66]. In our study, four miRNAs (stu-miR172a-3p_R + 1, stu-miR172a-3p, stu-miR172d-3p_1ss21GT, stu-miR172a-3p_R-1) targeted to AP2/RAP2 were significantly down-regulated in leaves under salt stress, which may be responsible for the reduction of AP2/RAP2 degradation in sweet potato under salt stress. Tobacco transgenic plant overexpressing chickpea AP2 exhibited salt tolerance traits [67]. Overexpression of the sweet potato RAP2–12 gene in Arabidopsis increased its tolerance to salt and drought [68].
MiRNA319 targeted to a plant-specific TF named TCP, which plays an important role in plant growth and development and in the response to biotic/abiotic stress [60, 69]. In the present study, significantly down-regulated Vvi-miR319g targeted to TCP (TCP2 and TCP3) in sweet potato leaves under salt stress. qRT-PCR validation showed that vvi-miR319g/TCP2 target pairs presented a reverse expression pattern. In switchgrass (Panicum virgatum L.), 29 in 42 TCP proteins were related to salt treatment [70]. miR319-targeted TCP plays a role in the response of plants to bentgrass under salt stress [71]. Nuclear factor Y (NF-Y) is a ubiquitous TF in almost all eukaryotic organisms and has a high affinity for the CCAAT cassette for transcriptional regulation of a large number of genes. Our study found that most of the miRNAs in the miR169 family, which were significantly down-regulated under salt stress, targeted to NF-YA1, NF-YA2 and NF-YA9 in sweet potato leaves. qRT-PCR validation showed that nta-miR169q_L + 2R-2/NF-YA2 and stu-miR169a-5p_1ss20TA/NF-YA9/NF-YA2 target pairs presented a reverse expression pattern. This result is consistent with the miR169 expression in Spartina alterniflora leaves under salt stress [72]. In Arabidopsis, most members in the NF-Y gene family are regulated by miR169, and the salt tolerance of Arabidopsis plant was enhanced in PtNF-YA9-overexpressing plants [73].
ATHB-14/15 are members of the HD-ZIP III TF family. This study found that stu-miR166b, which was significantly down-regulated by salt stress, targeted to ATHB-14/15. Previous study found that STTM165/166 triggers significant degradation of all endogenous miRNA165/166 members, resulting in the increased expression of its target gene HD-ZIP III (PHB, PHV, REV, ATHB8 and ATHB15), which directly leads to sttm165/166 overexpression in Arabidopsis seedlings and salt tolerance enhancement [74]. At the same time, overexpression of sttm165/166 reduced the expression level of miR165/166d, disrupting miR165/166-mediated target inhibition, and the up-regulated expression of miR165/166 targets directly promoted the accumulation of ABI4 and BG1, thereby regulating ABA and the abiotic stress response and control of ABA homeostasis [75]. In the future, the target pair of stu-miR166b and ATHB-14/15 should be further studied to determine whether the salt stress signal is transmitted through the ABA signalling pathway in response of sweet potato to salt stress. Some other TF genes, such as GRF and SCL, were targeted by miRNA in sweet potato under salt stress. For instance, GRF2/3/6 were targeted by miR396, which were significantly down-regulated; In Arabidopsis, up-regulation of AtGRF1 and AtGRF2 significantly increased leaf width and number of cotyledons, whereas overexpression of miR396a or miR396b resulted in the narrowing of transgenic plants [76]. Therefore, whether miR396 could increase leaf width and number of cotyledons of sweet potato must be investigated because the expression was up-regulated by salt stress. MiR171 cleaves scarecrow-like (SCL) genes, which is a subfamily in the GRAS family [77]. In rice, osa-miR171c is involved in the ABA-dependent pathway and affects stress-related gene expression, which contributes to salt stress tolerance [78]. In roots, both stu-miR171b-3p and rco-MIR171g-p3 were found to target the SCL genes, which may be involved in salt response in sweet potato.
In addition to TFs, many important enzyme and functional protein-encoding genes related to stress responses were also the targets of miRNA, such as AGO, RKS1, FAD7, RD21A, FER2, TUFA and DCL1. Sweet potato nta-miR168d_R-1 was up-regulated, and its target gene is the AGO1 protein, which is involved in miRNA biosynthesis. Stu-miR162a-3 targeting DCL1 was also identified in sweet potato, and miR162 and miR168 are involved in the feedback regulation of miRNA biogenesis and functional pathways by regulating their target mRNAs (DCL1 and AGO1) [79, 80]. In Arabidopsis, AGO1 is involved in miRNA-mediated guided mRNA cleavage process [79]. The increased expression of miR168 inhibits the synthesis of the target gene AGO protein to attenuate miRNA-mediated mRNA cleavage, resulting in increased levels of protein at the translational level, thereby activating and enhancing various physiological functional pathways of the plant [81]. FDA7 was targeted by mes-MIR319e-p5_2ss12GC19GA targets in sweet potato, and antisense expression of Arabidopsis FDA7 gene reduced salt tolerance in tobacco [82]. Significantly down-regulated sweet potato stu-miR396-5p_1ss21TG and osa-MIR818b-p3_2ss5TA18AC targeted to RD21A and TUFA, respectively, thereby playing an important role in plant adaptation to environmental stress [83]. The significantly up-regulated stu-mir172d-5p_1ss2gc of sweet potato targeted FER2, which played a very important role in salt stress resistance in cotton [84]. The function of these sweet potato miRNAs and their corresponding targets participating in environmental stress adaption must be studied further.
Conclusions
In conclusion, through transcriptome, sRNA, and degradome sequencing, hundreds of known and novel miRNAs were identified, and their expression patterns were comprehensively analysed under salt stress treatment. We found that miRNAs specifically targeted many mRNAs, including TFs, signalling proteins, enzymes, etc., in sweet potato, thereby negatively regulating gene expression. This study is the first to demonstrate the miRNA-mediated gene regulation in sweet potato under salt stress, which will provide insights into the understanding of miRNA-mediated salt stress response regulatory networks in sweet potato and construct solid theoretical foundation for molecular breeding of sweet potato by miRNA manipulation.
Methods
Plant materials and salt treatments
The sweet potato cultivar Xu32 used here was same as our previous studies [85, 86], which was bred by Hongmin Li and deposited (deposition number: GPD ganshu 2018–320,002) in the Xuzhou Academy of Agricultural Sciences/Sweet Potato Research Institute, CAAS, Xuzhou, China. Tuberous roots of Xu32 were collected from CAAS and then grown in a greenhouse with a temperature range of 20 °C to 25 °C, a light intensity of 150 μmol/m2.s1 and a photoperiod of 16 h for 1 month. Afterwards, shoots with four to five functional leaves were cut from the tuberous roots and placed in a one-half strength Hoagland solution. After 10 days of culture, plants with fine roots and young leaves were subjected to salinity treatments (150 mM NaCl) for 2 days, whereas the plants in the control were not treated with NaCl. Three replicated samples for the roots and leaves were harvested into liquid nitrogen and stored at − 80 °C for total RNA extraction.
RNA extraction, small RNA library construction and sequencing
Total RNA was extracted from SLC (Xu32 leaf without NaCl treatment as control), SLN (Xu32 leaf with NaCl treatment), SRC (Xu32 root without NaCl treatment as control), and SRN (Xu32 root with NaCl treatment) using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s procedure. Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA USA) were used to analyse the quality and purity of total RNA. Twelve small RNA libraries were constructed following the standard procedures. The raw data have been uploaded into the NCBI database Sequence Read Archive (SRA) and the SRR numbers were from SRR10872161 to SRR10872172. Specifically, the 5′ and 3′ adaptor sequences were successively ligated to the aforementioned RNA fragments with T4 RNA ligase 2 [87]. The resulting RNA was reverse transcribed to cDNA and then subjected to 11 PCR cycles using the adaptor primers. The PCR product fragments with a of size 140–160 bp were isolated on 6% polyacrylamide Tris-borate-EDTA gel. RNA was then sequenced by Illumina HiSeq 2000/2500. In this study, RPKM (reads per kilobase of exon model per million mapped reads) was used to measure the abundance of gene expression. Gene differential expression analysis is the most noteworthy result in small RNA sequencing. |Log2foldchange| ≥ 1 and P value<0.05 was used to identify the differentially expressed genes. The Sweet potato Garden (http://sweetpotato-garden.kazusa.or.jp/) database was used as reference sequence database for transcriptome annotation.
Identification of known and novel miRNAs
The raw data were processed with data cleaning analysis using ACGT101-miR v3.5 (LC Sciences, Huston, TX). In brief, the quality of raw data was measured by Illumina Fast QC to obtain Q30 data. Clean full-length reads were collected after removing all low-quality reads, adapter contaminants, and reads smaller than 18 nt and junk sequences (≥2 N, ≥7A, ≥8C, ≥6G, ≥7 T, ≥10 Dimer, ≥6 Trimer or ≥ 5 Tetramer). In addition, the clean data were filtered using various RNA databases, such as mRNA, RFam (release 9.1) and Repbase (version 15.07) databases, and rRNA, scRNA, snoRNA, snRNA, tRNA, etc. were found and removed as much as possible. The remaining unique sequences were mapped to the precursors in miRBase 21.0. by the fast gapped-read alignment software Bowtie 2 [88]. The unique sequences mapping to specific species mature miRNAs in hairpin arms were identified as known miRNAs. The unannotated sRNAs were expanded to about 250 nt and their structures were predicted using Mfold software (http://unafold.rna.albany.edu/?q=mfold). Novel miRNAs were obtained according to Meyers and Li prediction criteria [39, 89].
Transcriptome and degradome sequencing and analysis
The total RNA was extracted from the four samples (SLC, SLN, SRC and SRN). Then the transcriptome cDNA library construction and sequencing methods are described previously [90]. Clean reads of four transcriptomes were obtained by filtering out adaptor sequences and low-quality reads of the original sequencing data, and then cleaned sequencing data were assembled using Trinity software. Finally, the gene obtained after assembly was compared with the protein sequences in the five public databases (Swiss-Prot, NR, KEGG, KOG and Pfam) (threshold value e ≤ 1e-10). Functional annotation was performed through sequence similarity. RNA of the NaCl-free (CK) and NaCl-treated (Na150) roots and leaves were prepared for degradome sequencing. Total RNA was extracted and captured by beads and connected with a 3′-5′ adaptor. Then, the whole library was constructed by using the mixed reverse transcription of Biotinylated Random Primers and mRNA and amplified by PCR. The constructed library was sequenced by Illumina HiSeq 2000/2500. Raw data obtained by sequencing were used to predict miRNA target genes using the CleaveLand 3.0 program [38] and ACGT301-DGEv1.0 program (LC Sciences, Houston, TX, USA). On the basis of the abundance of the resulting mRNA tags relative to the overall profile of the degradome reads that matched the target [91], all target genes were divided into five categories, namely, Category 0, 1, 2, 3 and 4. The depositing data numbers for the transcriptome and degradome were SRR10854671, SRR10873471 and SRR10873472 in NCBI database.
qRT-PCR detection of expressed miRNAs and target genes
We performed expression verification of six differentially expressed miRNAs and eight target genes by qRT-PCR using TB Green™ Premix Ex Taq™ II (TaKaRa, Japan) and ABI plus sequence detection system (ABI, USA) as previous study [92]. Primer sequences for miRNAs and their targets are listed in Additional file 19: Table S14. Relative expression changes were calculated using the 2−ΔΔCt method [93] with ADP-ribosylation factor (ARF) as an internal control gene. PCR reaction was performed with three biological replicates.
Availability of data and materials
The datasets generated and analysed during the current study are available in the NCBI database SRA repository, SRR10854671, SRR10873471, SRR10873472, and from SRR10872161 to SRR10872172.
Abbreviations
- AGO:
-
Argonaute
- AUX/IAA:
-
Auxin/ indole-3-acetic acid
- CT:
-
Cycle threshold
- GO:
-
Gene ontology
- MFE:
-
Minimum free energy
- MFEI:
-
Minimum free energy index
- miRNAs:
-
microRNAs
- ncRNA:
-
Non-coding RNA
- pre-miRNA:
-
precursor miRNA
- qRT-PCR:
-
quantitative real-time PCR
- RISC:
-
RNA-induced silencing complex
- rRNA:
-
ribosomal RNA
- snoRNA:
-
small nucleolar RNA
- snRNA:
-
small nuclear RNA
- TF:
-
Transcription factor
- tRNA:
-
transfer RNA
References
Munns R, Tester M. Mechanisms of salinity tolerance. Annu Rev Plant Biol. 2008;59:651–81.
Butcher K, Wick AF, Desutter T, Chatterjee A, Harmon J. Soil salinity: a threat to global food security. Agron J. 2016;108(6):2189.
Zhao Q, Zhang H, Wang T, Chen SX, Dai SJ. Proteomics-based investigation of salt-responsive mechanisms in plant roots. J Proteome. 2013;82:230–53.
Flowers TJ, Muscolo A. Introduction to the special issue: halophytes in a changing world. AoB Plants. 2015;7:plv020.
Reid RW, Luo Y, Yan S, Miller TE, Song BH. Transcriptome dataset of halophyte beach morning glory, a close wild relative of sweet potato. Front Plant Sci. 2016;7:1267.
Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–87.
Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17(4):196–203.
Zhang BH, Wang QL. MicroRNA-based biotechnology for plant improvement. J Cell Physiol. 2015;230:1–15.
Kurihara Y, Watanabe Y. Arabidopsis microRNA biogenesis through dicer-like 1 protein functions. Proc Natl Acad Sci U S A. 2004;101:12753–8.
Xu L, Wang Y, Zhai L, Xu Y, Wang L, Zhu X, et al. Genome-wide identification and characterization of cadmium- responsive microRNAs and their target genes in radish (Raphanus sativus L.) roots. J Exp Bot. 2013;64:4271–87.
Sunkar R, Chinnusamy V, Zhu J, Zhu JK. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007;12:301–9.
Sun R, Wang Q, Ma J, He Q, Zhang B. Differentiated expression of microRNAs may regulate genotype-dependent traits in cotton. Gene. 2014;547(2):233–8.
Debernardi JM, Mecchia MA, Vercruyssen L, Smaczniak C, Kaufmann K, Inze D, et al. Post-transcriptional control of GRF transcription factors by microRNA miR396 and GIF co-activator affects leaf size and longevity. Plant J. 2014;79(3):419–26.
Sunkar R, Kapoor A, Zhu JK. Posttranscriptional induction of two cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006;18:2051–65.
Sahito ZA, Wang L, Sun Z, Yan Q, Zhang X, Jiang Q, et al. The miR172c-NNC1 module modulates root plastic development in response to salt in soybean. BMC Plant Biol. 2017;17(1):229.
Luan M, Xu M, Lu Y. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene. 2015;55:178–85.
Macovei A, Tuteja N. MicroRNAs targeting dead-box helicases are involved in salinity stress response in rice (Oryza sativa L.). BMC Plant Biol. 2012;12(1):1–12.
Gao P, Zhu Y, Bai X, Yang L, Lv D, Pan X, et al. Osa-MIR393: a salinity- and alkaline stress-related microRNA gene. Mol Biol Rep. 2011;38(1):237–42.
Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001–19.
Zhou M, Li D, Li Z, Hu Q, Yang C, Zhu L, et al. Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol. 2013;161(3):1375–91.
Yuan S, Li Z, Li D, Yuan N, Hu Q, Luo H. Constitutive expression of rice microRNA528 alters plant development and enhances tolerance to salinity stress and nitrogen starvation in creeping bentgrass. Plant Physiol. 2015;169(1):576–93.
Mondal TK, Panda AK, Rawal HC, Sharma TR. Discovery of microRNA-target modules of African rice (Oryza glaberrima) under salinity stress. Sci Rep. 2018;8(1):570.
Ning LH, Du WK, Song HN, Shao HB, Qia WC, Amr Sheteiwy MS, et al. Identification of responsive miRNAs involved in combination stresses of phosphate starvation and salt stress in soybean root. Environ Exp Bot. 2019;167:103823.
Gao XN, Cong YT, Yue JR, Xing ZY, Wang Y, Chai XJ. Small RNA, transcriptome, and degradome sequencing to identify salinity stress responsive miRNAs and target genes in Dunaliella salina. J Appl Phycol. 2019;31(2):1175–83.
Bovell-Benjamin A. Sweet potato: a review of its past, present, and future role in human nutrition. Adv Food Nutr Res. 2007;52:1–59.
Wang F, Tong W, Zhu H, Kon W, Peng R, Liu Q, et al. A novel Cys 2 /his 2 zinc finger protein gene from sweet potato, IbZFP1, is involved in salt and drought tolerance in transgenic Arabidopsis. Planta. 2016;243:783–97.
Yang Z, Sun J, Chen Y, Zhu P, Xu T. Genome-wide identification, structural and gene expression analysis of the bzip transcription factor family in sweet potato wild relative Ipomoea trifida. BMC Genet. 2019;20(1):41.
Kim SH, Ahn YO, Ahn MJ, Jeong JC, Lee HS, Kwak SS. Cloning and characterization of an orange gene that increases carotenoid accumulation and salt stress tolerance in transgenic sweet potato cultures. Plant Physiol Biochem. 2013;70:445–54.
Wang LJ, He SZ, Zhai H, Liu DG, Wang YN, Liu QC. Molecular cloning and functional characterization of a salt tolerance-associated gene IbNFU1 from sweet potato. J Integr Agric. 2013;12:27–35.
Liu DG, He SZ, Zhai H, Wang LJ, Zhao Y, Wang B, et al. Overexpression of IbP5CR enhances salt tolerance in transgenic sweet potato. Plant Cell Tissue Org. 2014;117:1–16.
Liu DG, Wang LJ, Liu CL, Song XJ, He SZ, Zhai H, et al. An Ipomoea batatas iron-sulfur cluster scaffold protein gene, IbNFU1, is involved in salt tolerance. PLoS One. 2014;9:e93935.
Liu DG, Wang LJ, Zhai H, Song XJ, He SZ, Liu QC. A novel a/b-hydrolase gene IbMas enhances salt tolerance in transgenic sweet potato. PLoS One. 2014;9:e115128.
Liu DG, He SZ, Song XJ, Zhai H, Liu N, Zhang DD, et al. IbSIMT1, a novel salt-induced methyltransferase gene from Ipomoea batatas, is involved in salt tolerance. Plant Cell Tissue Org. 2015;120:701–15.
Wang FB, Zhai H, An YY, Si ZZ, He SZ, Liu QC. Overexpression of IbMIPS1 gene enhances salt tolerance in transgenic sweet potato. J Integr Agric. 2015;15(2):271–81.
Zhai H, Wang FB, Si ZZ, Huo JX, Xing L, An YY, et al. A myo-inositol-1-phosphate synthase gene, IbMIPS1, enhances salt and drought tolerance and stem nematode resistance in transgenic sweet potato. Plant Biotechnol J. 2016;14(2):592–602.
Hoen PAC, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RH, de Menezes RX, et al. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36(21):e141.
Ambady S, Wu Z, Dominko T. Identification of novel microRNAs in Xenopus laevis metaphase II arrested eggs. Genesis. 2012;50(3):286–99.
Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2008;25(1):130–1.
Li B, Duan H, Li J, Deng XW, Yin W, Xia X. Global identification of miRNAs and targets in Populus euphratica under salt stress. Plant Mol Biol. 2013;81(6):525–39.
Lopez-Gomollon S, Mohorianu I, Szittya G, Moulton V, Dalmay T. Diverse correlation patterns between microRNAs and their targets during tomato fruit development indicates different modes of microRNA actions. Planta. 2012;236(6):1875–87.
Kawashima CG, Yoshimoto N, Maruyama-Nakashita A, Tsuchiya YN, Saito K, Takahashi H, et al. Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. Plant J. 2009;57(2):313–21.
He L, Tang R, Shi X, Wang W, Cao Q, Liu X, et al. Uncovering anthocyanin biosynthesis related microRNAs and their target genes by small RNA and degradome sequencing in tuberous roots of sweet potato. BMC Plant Biol. 2019;19(1):232.
Zhang J, Huang M, Liang J, Pan Y, Cheng L, Wu J, et al. Genome-wide mining for microRNAs and their targets in Betula luminifera using high-throughput sequencing and degradome analyses. Tree Genet Genomes. 2016;12(5):99.
Zhao T, Li G, Mi S, Li S, Hannon GJ, Wang XJ, et al. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 2007;21(10):1190–203.
Brodersen P, Voinnet O. The diversity of RNA silencing pathways in plants. Trends Genet. 2006;22(5):268–80.
Wu L, Zhou HY, Zhang QQ, Zhang J, Ni F, Liu C, et al. DNA methylation mediated by a microRNA pathway. Mol Cell. 2010;38:465–75.
Mi SJ, Cai T, Hu YG, Hodges E, Ni F, Wu L, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell. 2008;133(1):116–27.
Montgomery TA, Howell MD, Cuperus JT, Li D, Hansen JE, Alexander AL, et al. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell. 2008;133(1):0–141.
Takeda A, Iwasaki S, Watanabe T, Utsumi M, Watanabe Y. The mechanism selecting the guide strand from small RNA duplexes is different among argonaute proteins. Plant Cell Physiol. 2008;49(4):493–500.
Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA. Conservation and divergence of plant microRNA genes. Plant J. 2006;46(2):243–59.
Zhang B, Pan X, Cox S, Cobb G, Anderson T. Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci. 2006;63(2):246–54.
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot. 2009;103(1):29–38.
Liu HH, Tian X, Li YJ, Wu CA, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2018;14(5):836–43.
Zhuang Y, Zhou XH, Liu J. Conserved miRNAs and their response to salt stress in wild eggplant Solanum linnaeanum roots. Int J Mol Sci. 2014;15(1):839–49.
Xie FL, Wang QL, Sun RR, Zhang BH. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J Exp Bot. 2015;66(3):789–804.
Dodd AN, Kudla J, Sanders D. The language of calcium signaling. Annu Rev Plant Biol. 2010;61(1):593–620.
Tao XC, Lu YT. Loss of ATCRK1 gene function in Arabidopsis thaliana decreases tolerance to salt. J Plant Biol. 2013;56(5):306–14.
Choi HI, Park HJ, Park JH, Kim S, Im MY, Seo HH, et al. Arabidopsis calcium-dependent protein kinase atcpk32 interacts with abf4, a transcriptional regulator of abscisic acid-responsive gene expression, and modulates its activity. Plant Physiol. 2005;139(4):1750–61.
Woodrow P, Pontecorvo G, Ciarmiello LF, Annunziata MG, Fuggi A, Carillo P. Transcription factors and genes in abiotic stress. Crop Stress and its Management: Perspectives and Strategies. Dordrecht: Springer Netherlands; 2011. p. 317–57.
Nicolas M, Cubas P. TCP factors: new kids on the signaling block. Curr Opin Plant Biol. 2016;33:33–41.
Takasaki H, Maruyama K, Kidokoro S, Ito Y, Fujita Y, Shinozaki K, et al. The abiotic stress-responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol Gen Genomics. 2010;284(3):173–83.
Hussain RM, Ali M, Feng X, Li X. The essence of NAC gene family to the cultivation of drought-resistant soybean (Glycine max l. merr.) cultivars. BMC Plant Biol. 2017;17:1.
Ning K, Chen S, Huang H, Jiang J, Yuan H, Li H. Molecular characterization and expression analysis of the SPL gene family with bpspl9 transgenic lines found to confer tolerance to abiotic stress in Betula platyphylla suk. Plant Cell, Tissue Organ Culture (PCTOC). 2017;130:469–81.
Hou H, Jia H, Yan Q, Wang X. Overexpression of a SBP-box gene (VpSBP16) from chinese wild vitis species in Arabidopsis improves salinity and drought stress tolerance. Int J Mol Sci. 2018;19(4):940.
Aukerman MJ, Sakai H. Regulation of flowering time and floral organ identity by a microRNA and its apetala2-like target genes. Plant Cell. 2003;15(11):2730–41.
Gutterson N, Reuber TL. Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr Opin Plant Biol. 2004;7(4):465–71.
Shukla RK. Expression of CAP2, an APETALA2-family transcription factor from chickpea, enhances growth and tolerance to dehydration and salt stress in transgenic tobacco. Plant Physiol. 2006;142(1):113–23.
Li Y, Zhang H, Zhang Q, Liu Q, Zhai H, Zhao N, et al. An AP2/ERF gene, IbRAP2-12, from sweet potato is involved in salt and drought tolerance in transgenic Arabidopsis. Plant Sci. 2019;281:19–30.
Martín-Trillo M, Cubas P. TCP genes: a family snapshot ten years later. Trends Plant Sci. 2010;15(1):0–39.
Huo Y, Xiong W, Su K, Li Y, Yang Y, Fu C, et al. Genome-wide analysis of the TCP gene family in switchgrass (Panicum virgatum L.). Int J Genomics. 2019;2019:8514928.
Zhou M, Luo H. Role of microRNA319 in creeping bentgrass salinity and drought stress response. Plant Signal Behav. 2014;9(4):e28700.
Qin Z, Chen J, Jin L, Duns GJ, Ouyang P. Differential expression of miRNAs under salt stress in Spartina alterniflora leaf tissues. J Nanosci Nanotechnol. 2015;15(2):1554–61.
Lian C, Li Q, Yao K, Zhang Y, Meng S, Yin W, et al. Populus trichocarpa PtNF-YA9, a multifunctional transcription factor, regulates seed germination, abiotic stress, plant growth and development in Arabidopsis. Front Plant Sci. 2018;9:954.
Jia X, Ding N, Fan W, Yan J, Gu Y, Tang X, et al. Functional plasticity of mir165/166 in plant development revealed by small tandem target mimic. Plant Sci. 2015;233:11–21.
Yan J, Zhao C, Zhou J, Yang Y, Wang P, Zhu X, et al. The mir165/166 mediated regulatory module plays critical roles in aba homeostasis and response in Arabidopsis thaliana. PLoS Genet. 2016;12(11):e1006416.
Wang L, Gu X, Xu D, Wang W, Wang H, Zeng M, et al. Mir396-targeted AtGRF transcription factors are required for coordination of cell division and differentiation during leaf development in Arabidopsis. J Exp Bot. 2011;62(2):761–73.
Ma Z, Hu X, Cai W, Huang W, Zhou X, Luo Q, et al. Arabidopsis mir171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 2014;10(8):e1004519.
Yang W, Fan T, Hu X, Cheng T, Zhang M. Overexpressing Osa-mir171c decreases salt stress tolerance in rice. J Plant Biol. 2017;60(5):485–92.
Vaucheret H, Vazquez F, Crété P, Bartel DP. The action of argonaute1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004;18(10):1187–97.
Xie Z, Kasschau KD, Carrington JC. Negative feedback regulation of dicer-like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr Biol. 2003;13(9):784–9.
Vaucheret H. Ago1 homeostasis involves differential production of 21-nt and 22-nt mir168 species by mir168a and mir168b. PLoS One. 2009;4(7):e6442.
Im YJ, Han O, Chung GC, Cho BH. Antisense expression of an Arabidopsis ω-3 fatty acid desaturase gene reduces salt/drought tolerance in transgenic tobacco plants. Mol Cell. 2002;13(2):264–71.
Singh BN, Mishra RN, Agarwal PK, Mamta G, Suresh N, Sopory SK, et al. A pea chloroplast translation elongation factor that is regulated by abiotic factors. Biochem Biophys Res Commun. 2004;320(2):523–30.
Xu Y, Magwanga RO, Cai X, Zhou Z, Wang X, Wang Y, et al. Deep transcriptome analysis reveals reactive oxygen species (ros) network evolution, response to abiotic stress, and regulation of fiber development in cotton. Int J Mol Sci. 2019;20:8.
Yu Y, Xu T, Li X, Tang J, Ma D, Li Z, et al. NaCl-induced changes of ion homeostasis and nitrogen metabolism in two sweet potato (Ipomoea batatas L.) cultivars exhibit different salt tolerance at adventitious root stage. Environ Exp Bot. 2016;129:23–36.
Yu Y, Wang A, Li X, Kou M, Wang W, Chen X, et al. Melatonin-stimulated triacylglycerol breakdown and energy turnover under salinity stress contributes to the maintenance of plasma membrane H+–ATPase activity and K+/Na+ homeostasis in sweet potato. Front Plant Sci. 2018;9:256.
Khajuria C, Williams CE, Bouhssini ME, Whitworth RJ, Richards S, Stuart JJ, et al. Deep sequencing and genome-wide analysis reveals the expansion of MicroRNA genes in the gall midge Mayetiola destructor. BMC Genomics. 2013;14(1):187.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9.
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, et al. Criteria for annotation of plant microRNAs. Plant Cell. 2009;20(12):3186–90.
Xie J, Deng L, Zhou Y, Yao S, Zeng K. Analysis of changes in volatile constituents and expression of genes involved in terpenoid metabolism in oleocellosis peel. Food Chem. 2017;243:269–76.
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008;18(10):758–62.
Lu Y, Sun J, Yang Z, Zhao C, Zhu M, Ma D, et al. Genome-wide identification and expression analysis of glycine-rich RNA-binding protein family in sweet potato wild relative Ipomoea trifida. Gene. 2019;686:177–86.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;25:402–8.
Acknowledgements
We thank Lianchuan Biotechnology (Hangzhou, China) for supplying small RNA, transcriptome and degradome sequencing service.
Funding
This work was supported jointly by the projects of the National Key R&D Program of China (2018YFD1000705, 2018YFD1000700), the National Natural Science Foundation of China (Grant No. 31701481), the China Agriculture Research System (CARS-10-B03), the Natural Science Foundation of Jiangsu Higher Education Institutions of China (19KJA510010) and Jiangsu Overseas Visiting Scholar Program for University Prominent Young & Middle-aged Teachers and Presidents. These funding bodies supported this research from inception to completion; ie, the design, data collection, analysis and interpretation of the data, and the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
TX and ZL conceived and designed this experiment. ZY, JS and TD carried out the experiments. ZY and PZ analyzed the data and wrote the manuscript. LL, HK and MZ helped to analyze the data. TX, HK and QC helped to revise the manuscript. QC offered the plant material. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Summary of sequence data generated for sweet potato transcriptome and quality filtering.
Additional file 2: Table S2.
Assembly statistics of reads.
Additional file 3: Table S3.
Distribution of the assembled genes and transcript length.
Additional file 4: Table S4.
Blast analysis of non-redundant unigenes against public databases.
Additional file 5: Table S5.
Summary of sRNA sequencing data in sweet potato.
Additional file 6: Table S6.
Identification of known miRNAs by high-throughput sequence.
Additional file 7: Figure S1.
Distribution of conserved miRNAs in species.
Additional file 8: Figure S2.
Quantitative distribution and expression level analysis of miRNA family in four libraries (SRC, SRN, SLC and SLN). (A) Analysis of expression levels between miRNA families. (B) Distribution of conserved miRNAs in the miRNA family of SRC, SRN, SLC and SLN libraries.
Additional file 9: Table S7.
Identification of novel miRNAs by high-throughput sequence.
Additional file 10: Figure S3.
Analyses of the length distribution and nucleotide base bias of novel miRNAs. (A) Size distribution of novel miRNA. (B) Percentage of the first nucleotide bias in novel miRNAs. (C) Novel miRNA nucleotide bias at first position.
Additional file 11: Figure S4.
Predicted secondary structures of potential novel miRNAs from sweet potato. Sequences indicated in green correspond to predicted miRNA.
Additional file 12: Table S8.
Summary of the comparison of known and novel miRNAs between the control and salt libraries in sweet potato.
Additional file 13: Table S9.
Known and novel miRNA targets identified by degradome sequencing.
Additional file 14: Table S10.
CleaveL and pipeline category of degradome.
Additional file 15: Table S11.
Target prediction results of the differentially expressed miRNAs between sweet potato DSN and DSC degradome libraries. (XLS 30 kb)
Additional file 16: Table S12.
Targets of salt-responsive differentially expressed miRNAs.
Additional file 17: Table S13.
Information of the significantly enriched GO terms for differentially expressed miRNA target genes.
Additional file 18: Figure S5.
Functional analysis of target genes of differentially expressed miRNAs. (A) Significant enrichment of target genes in cellular component classification. (B) Significant enrichment of target genes in molecular function classification. (C) Significant enrichment of target genes in biological process classification. (D) KEGG analysis of differentially expressed miRNA target genes in response to salt stress.
Additional file 19: Table S14.
Primer sequences of miRNA and their targets used for qRT-PCR.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, Z., Zhu, P., Kang, H. et al. High-throughput deep sequencing reveals the important role that microRNAs play in the salt response in sweet potato (Ipomoea batatas L.). BMC Genomics 21, 164 (2020). https://doi.org/10.1186/s12864-020-6567-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6567-3