
Abstract
Background
Bacillus subtilis strain NCD-2 is an excellent biocontrol agent against plant soil-borne diseases and shows broad-spectrum antifungal activities. This study aimed to explore some secondary metabolite biosynthetic gene clusters and related antimicrobial compounds in strain NCD-2. An integrative approach combining genome mining and structural identification technologies using ultra-high-performance liquid chromatography coupled to quadrupole time-of-flight tandem mass spectrometry (UHPLC-MS/MS), was adopted to interpret the chemical origins of metabolites with significant biological activities.
Results
Genome mining revealed nine gene clusters encoding secondary metabolites with predicted functions, including fengycin, surfactin, bacillaene, subtilosin, bacillibactin, bacilysin and three unknown products. Fengycin, surfactin, bacillaene and bacillibactin were successfully detected from the fermentation broth of strain NCD-2 by UHPLC-QTOF-MS/MS. The biosynthetic gene clusters of bacillaene, subtilosin, bacillibactin, and bacilysin showed 100% amino acid sequence identities with those in B. velezensis strain FZB42, whereas the identities of the surfactin and fengycin gene clusters were only 83 and 92%, respectively. Further comparison revealed that strain NCD-2 had lost the fenC and fenD genes in the fengycin biosynthetic operon. The biosynthetic enzyme-related gene srfAB for surfactin was divided into two parts. Bioinformatics analysis suggested that FenE in strain NCD-2 had a similar function to FenE and FenC in strain FZB42, and that FenA in strain NCD-2 had a similar function to FenA and FenD in strain FZB42. Five different kinds of fengycins, with 26 homologs, and surfactin, with 4 homologs, were detected from strain NCD-2. To the best of our knowledge, this is the first report of a non-typical gene cluster related to fengycin synthesis.
Conclusions
Our study revealed a number of gene clusters encoding antimicrobial compounds in the genome of strain NCD-2, including a fengycin synthetic gene cluster that might be unique by using genome mining and UHPLC–QTOF–MS/MS. The production of fengycin, surfactin, bacillaene and bacillibactin might explain the biological activities of strain NCD-2.
Similar content being viewed by others

Background
The Bacillus genus has received considerable attention as a biological resource for the development of microbial pesticides, partly because some or most of its members form stress-resistant spores that do not harm the environment and are useful in pesticide production [1,2,3]. Bacillus subtilis and its closely related species are ubiquitous inhabitants of soil, and are widely recognized as powerful biocontrol agents against plant soil-borne diseases [4]. The mechanisms used by B. subtilis to suppress plant soil-borne diseases include competing with phytopathogens for nutrients and spatial sites, inducing systemic resistance in plants, and inhibiting pathogen growth by producing antimicrobial compounds [5]. The latter is a general characteristic of B. subtilis biocontrol agents and plays an important role in the suppression of plant diseases [6, 7]. B. subtilis produces more than two dozen antimicrobial compounds with amazing structural variety. Based on their biosynthetic pathways, the antimicrobial compounds are divided into small molecular compounds synthesized by the ribosomal pathway, such as bacteriocins, and peptide compounds synthesized by the non-ribosomal pathway, such as lipopeptides and polyketides [8]. Most antimicrobial compounds are secondary metabolites produced by biocontrol of Bacillus spp., and are not necessary for their growth and reproduction but lead to shifts of rhizospheric microbial functional subsystems and affect the availability of nutrients for the plant [9]. Secondary metabolites also function as essential chemical signals for the induction of cellular differentiation in the producing organism and for controlling its metabolism [10, 11].
The genes encoding the secondary metabolites commonly exist in clusters and encode enzyme complexes with multiple functions [12]. The polyketide synthase/non-ribosomal peptide synthetase (PKS/NRPS) gene clusters have been well studied. The PKS pathway polyketides require at least three domains: an acyl transferase, a ketosynthase, and an acyl carrier protein [13]. The NRPS synthetic pathways share a common multicarrier thiotemplate mechanism requiring the cooperation of three basic domains [14]. The adenylation domain selects the cognate amino acid and generates an enzymatically stabilized aminoacyl adenylate. The peptidyl carrier domain is equipped with a 4′-phosphopantetheine prosthetic group, to which the adenylated amino acid substrate is transferred and bonded by a thioester bond. The condensation domain catalyzes the formation of a new peptide bond [13]. The carbon skeleton of the metabolite is synthesized by the core PKS and NRPS enzymes, and is then modified to form the final product with the assistance of various modifying enzymes [15]. The bioactive secondary metabolites produced by the PKS/NRPS pathway in B. subtilis have received extensive studies, such as bacilysin [16], bacilysocin [17], surfactin [18], iturin A [19], fengycin [20], mycosubtilin [21], bacillomycins [8], and difficidin [16].
The conventional method for screening new active products is generally based on biological tests, which is time-consuming and sometimes result in repeatedly screened out the same products [22]. Thus, a more rapid and effective screening strategy to detect secondary metabolites was required [23, 24]. Genome mining is a technology that uses modern bioinformatics to recognize specific functional genes or gene clusters from genome sequences [25]. With the rapid development of DNA sequencing technology and the decrease of sequencing cost, a large number of microbial genome sequences have been determined [26], which makes genome mining an accurate and efficient strategy to find new metabolites [25].
B. subtilis strain NCD-2 is a plant soil-borne disease-suppressive agent producing lipopeptides, fengycin, and surfactin [27]. Fengycin shows strong antifungal activity, and surfactin facilitates the root colonization. Both fengycin and surfactin play important roles in suppression of plant soil-borne diseases by strain NCD-2 [28]. The purpose of this study was to identify potential secondary metabolites in strain NCD-2, reveal the different gene clusters of the secondary metabolites between strain NCD-2 and the reference strain B. velezensis FZB42, and identify the potential secondary metabolites produced by strain NCD-2.
Results
Genomic features of strain NCD-2
A total of 501,671,500 paired-end reads and 5,016,715 clean single reads (412-bp library; paired-ends of 75 bp) were assembled using the software package Velvet [29]. The genome of B. subtilis NCD-2 contains 189 contigs (> 133 bp; N90, 16,187) of 4,644,322 bp, with an average G + C content of 43.5%. The final assembled genome comprises 4444 genes, including 4329 protein-coding genes (418 signal peptide-coding genes), 83 tRNA genes for all 20 amino acids, 30 rRNA genes, and 2 clustered regularly interspaced short palindromic repeats (CRISPR) genes. A total of nine putative gene clusters responsible for antimicrobial metabolite biosynthesis were identified. These gene clusters included PKS and NRPS genes (Fig. 1).

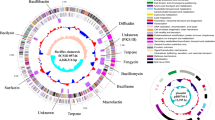
Circular genome of strain NCD-2 with specific features. The circular genome map was created using Circos v0.66 with COG (Cluster of Orthologous Groups of proteins) function annotation. From outside to inside: circle 1, the size of the complete genome; circles 2 and 3, the predicted protein-coding genes on the + and - strands, respectively, where different colours represent different COG function classifications; circle 4, tRNA (green) and rRNA (red); circle 5, G + C content, where peaks outside/inside the circle indicate above or below average GC content, respectively; the inner circle, G + C skew, with G% < C% in purple and with G% > C% in blue. The potato dextrose agar plate inside the representation of the circular genome shows the antifungal activity of strain NCD-2 and its derived strain constructed by atmospheric and room temperature plasma (ARTP) against Botrytis cinerea. The black bars outside the circular genome indicate the secondary metabolite biosynthetic gene clusters
The taxonomic status of strain NCD-2
At present, 272 B. subtilis genome sequences have been deposited in the GenBank database, including 113 whole- and 159 incomplete genome sequences. The genome sizes of the 272 B. subtilis strains range from 2.68 Mb to 5.35 Mb, and the GC contents range from 42.9 to 46.6%. These genome sequences were downloaded from the GenBank database, and their accession numbers were listed in Additional file 1, Table S1. To analyze the evolution of different B. subtilis strains, a phylogenetic tree was constructed based on the complete genome sequences. The 272 strains of B. subtilis were divided into four subspecies, subtilis, inaquosorum, spizizenii, and stercoris due to producing different bioactive secondary metabolites [30]. As shown in Fig. 2, strain NCD-2 (represented by the black bar) clustered with B. subtilis strain UD1022 and was closely related to B. subtilis strains XF-1, BAB-1, HJ5, SX01705, and BSD-2.

Phylogenetic tree of 113 B. subtilis strains based on whole genome alignments. The position of strain NCD-2 in the phylogenetic tree is indicated by a black square mark, and the position of the reference strain B. subtilis NBRC 13719 is indicated by a black circle mark. Single Nucleotide Polymorphisms (SNPs) and short insertions or deletions (indels) within the multiple sequence alignments constructed by the REALPHY pipeline were extracted for subsequent phylogeny reconstruction. The phylogenetic tree was constructed using MEGA 5.0 by the Neighbor-joining method, with a bootstrap of 1000 replications. Bootstrap confidence levels > 50% are indicated at the internodes
Secondary metabolite biosynthetic gene clusters in strain NCD-2
The secondary metabolite biosynthetic gene clusters in the genome of strain NCD-2 were predicted using antiSMASH [31]. In total, nine such clusters were identified (Table 1), including three NRPSs, two terpenes, one hybrid NRPS-TransAT PKS-Other KS, one type III polyketide, one sactipeptide-head to tail gene cluster, and a gene cluster with unknown function. The structural compositions of the gene clusters are shown in Fig. 3. These clusters were composed of core biosynthetic, additional biosynthetic, transport-related, regulatory, and other genes. Among them, clusters 3, 7, 8, and 9 had 100% amino acid sequence homologies with known gene clusters that synthesize bacillaene, bacillibactin, subtilosin, and bacilysin, respectively (Table 1). Gene cluster 1 showed 82% amino acid similarity with a surfactin synthetase gene cluster, and gene cluster 4 showed 93% amino acid similarity with a fengycin biosynthetic gene cluster in B. velezensis strain FZB42. However, gene clusters 2, 5, and 6 did not match any known gene clusters. Clusters 1 and 4 of strain NCD-2 were further compared with their counterparts in the model strain 168 and B. subtilis strains closely related to strain NCD-2 in the phylogenetic tree. The predicted fengycin biosynthetic gene cluster in strain NCD-2 contained three genes, fenEAB, while all the other strains contained five genes, fenCDEAB (Additional file 1, Fig. S1). SrfAB of surfactin was synthesized via typical transcription and translation of srfAB in the 11 strains. However, the same SrfAB was potentially assembled with Gms0366 and Gms0367 and then separately transcribed and translated by gms0366 and gms0367 in strain NCD-2 (Additional file 1, Fig. S2). Therefore, we hypothesized that the structures and functions of fengycin and surfactin from strain NCD-2 might be different from those in other B. subtilis strains.

Schematic diagram of nine secondary metabolite biosynthetic gene clusters in B. subtilis strain NCD-2. antiSMASH was used to predict potential secondary metabolite biosynthetic gene clusters. Different colour blocks represent genes with different functions; the genes marked with dark red, light red, blue, green, and gray are core biosynthetic, additional biosynthetic, transport-related, regulatory, and other genes, respectively
Specificity of the surfactin and fengycin synthetase gene clusters in B. subtilis NCD-2
The surfactin biosynthetic gene cluster gms0365–0368 in strain NCD-2 was analyzed using PRISM, and the core genes were selected for PKS/NRPS analysis. Gms0365 had an identical conserved domain, CATCATCATe, with SrfAA in strain FZB42, in which C, A, T, and Te represent the condensation, adenylation, thiolation, and thioesterase domains, respectively (Fig. 4a). Compared with SrfAB in strain FZB42, Gms0366 in strain NCD-2 lacked the T and E domains, but the amino acid residues for the binding pockets in Gms0366 were exactly the same as those of SrfAB. The residues of the different adenylation domains A6 and A2 from the enzymes Gms0365 and Gms0366, respectively, were exactly the same, and both bound the amino acid leucine. Gms0367 only had T and E domains, and no specific substrate-binding domain. The superposition of the Gms0367 and Gms0366 domains formed a complete SrfAB. The T domain was reversed between Gms0367 and Gms0368. Gms0368 contained CATe domains, in which the thioesterase domain releases linear peptide chains. The domains of Gms0368 were the same as those of SrfAC, but the binding pocket (DAF-LGCV) had one missing residue compared with that of strain FZB42 (DAFXLGCV).

Comparisons of functional domains of core genes involved in synthesizing surfactin and fengycin in NCD-2. The functional domains of the core genes of clusters 1 (a) and 3 (b) in B. subtilis NCD-2. (c) The abbreviations indicate the functions of the corresponding structural domains. (d) The conserved binding pockets for substrates formed by amino acids in different adenylation domains
The fengycin biosynthetic cluster in strain FZB42 contained five genes fenCDEAB (Fig. 4b). However, the same cluster in strain NCD-2 contained only three genes: gms1961, gms1960, and gms1959 (Fig. 4b). Gms1961 corresponded to FenE in strain FZB42 had conserved residues of A8 and A9, which bind amino acids Glu and Val, respectively (Fig. 4b). Gms1960 and Gms1959 had conserved amino acid sequences related to FenA and FenB in strain FZB42, respectively. Interestingly, no homologs of FenC and FenD were identified in the genome of strain NCD-2. Consequently, the amino acid sequences of FenC and FenD of strain FZB42 were compared with the strain NCD-2 proteome using BioEdit, and it was found that their most similar proteins were Gms1961 and Gms1960, respectively (Additional file 1, Tables. S2, S3). This finding led to the hypothesis that Gms1961 and Gms1960 performed the functions of FenC and FenD in strain NCD-2, respectively. Thus, Gms1961 and Gms1960 might both have dual functions in the synthesis of fengycin. Gms1961 in strain NCD-2 had the functions of FenE and FenC in strain FZB42, and Gms1960 had the functions of FenA and FenD. However, it should be pointed out that the FenD domain in strain NCD-2 varied greatly with that of FZB42, and we cannot rule out the possibility that other enzymes in NCD-2 might have similar functions as FenD.
PCR amplification using the primer set targeting the fenE and dacC genes produced the expected 1032 bp fragment in strain NCD-2 but not in strain FZB42 due to the extremely large size of the target (16,555 bp) (Fig. 5a-b). Sequencing of the 1032 bp fragment and alignment with the gene locus gms1959–1962 confirmed the lack of fenC and fenD homologs in this cluster (Fig. 5c-d). Compared to wild-type NCD-2, the in-frame deletion mutant of gms1961 completely lost fengycin production (Fig. 6a-c).

PCR and sequence of the fragment between fenE and dacC. a Schematic diagram used to design primers according to conserved bases from NCD-2 and B. velezensis FZB42; b PCR of the fenE-dacC fragment using the genomic templates NCD-2 and FZB42, with 16S rDNA as an internal reference control; c Schematic diagram of the constructed sequencing vector by ligating the fenE-dacC fragment to the pEASY-Blunt Zero vector, d BLAST of the fenE-dacC fragments from NCD-2 and pEASY-Blunt Zero fenE-dacC, in which the two sequences of fenE-dacC were complete same

The role of gms1961 in synthesizing fengycin. a FPLC of the lipopeptides of strain NCD-2 and Δgms1961, b quantitative production of fengycin in strain NCD-2 and Δgms1961, where the error bars represent the standard deviation and asterisks depict significant differences as measured by the t-test (**p < 0.01), c Extract Ions Using Dialog (XIC) and UHPLC-QTOF-MS of fengycin from NCD-2 and Δgms1961. The lipopeptide fengycin exhibited a difference at 25–50 min between strains NCD-2 and Δgms1961, and only the precursor related to m/z 725.4 was same (the light purple line), but the fragments were absolutely different from those of fengycin from strain NCD-2
We further compared the fengycin synthetase gene cluster of NCD-2 with other 11 corresponding clusters from B. subtilis strains closely related to strain NCD-2 (Additional file 1, Fig. S1). All the strains contained the fengycin biosynthetic gene cluster fenCDEAB (also ppsABCDE), except strain NCD-2, which contained fenEAB, suggesting that the fengycin biosynthetic gene cluster of strain NCD-2 is unique.
MS/MS of fengycin and surfactin in NCD-2
Fengycin was separated from the lipopeptide extract of strain NCD-2 using fast protein liquid chromatography (FPLC) (Additional file 1, Fig. S3). QTOF–MS/MS analysis revealed five fractions in the fengycin cluster (Fig. 7a–e), which had mass-to-charge ratio (m/z) values of 732.4, 746.4, 725.4, 739.4, and 767.4 (secondary MS), representing fengycin A, fengycin B, fengycin A2, fengycin B2, and fengycin C, respectively. The typical MS/MS spectra showed the distributions of key fragmentation ions (α and β), representing the linear N-terminal and the cyclic C-terminal segments, respectively, of diverse fengycin species (Fig. 7a–e and Additional file 1, Fig. S4a-b). The MS/MS spectrum of the fengycin ion at m/z 732.4 yielded two intense product ions at m/z 966.5 and 1080.5, representing fengycin A (Fig. 5a), while the MS/MS spectrum of the fengycin ion at m/z 746.4 (Fig. 7b) yielded key product ions at m/z 994.5 and 1108.6, representing fengycin B (Fig. 7b). The MS/MS spectrum of the fengycin ion at m/z 725.4 yielded two intense product ions at m/z 952.4 and 1066.5, representing fengycin A2 (Fig. 7c), while the MS/MS spectrum of the fengycin ion at m/z 739.4 (Fig. 7d) yielded key product ions at m/z 980.5 and 1094.5 representing fengycin B2 (Fig. 7d). The MS/MS spectrum of the fengycin ion at m/z 767.4 yielded two intense product ions at m/z 994.5/1008.5 and 1108.6/1122.6 representing fengycin C (Fig. 7e). Five classes of fengycins were identified based on the key product ions of β-hydroxy fatty acid (β-OH FA) with chain lengths varying from C12 to C20 (Table 2, Figs. S5–S9). The MS/MS spectrum of the surfactin ion at m/z 1008.7 yielded one intense product ion at m/z 685.5 (Fig. 7f and Additional file 1, Fig. S4c). Based on this key product ion, one class of compounds was identified: surfactins (m/z values of 994.6, 1008.7, 1022.7 and 1036.7) with fatty acid chains varying from C11 to C15 (Fig. S10).

MS/MS spectra of protonated cyclic fengycin and surfactin ions. a m/z 732.4, b m/z 746.4, c m/z 725.4, d m/z 739.4, e m/z 767.4, and f m/z 1008.7
Detection of other antimicrobial active compounds in NCD-2
Besides the fengycin and surfactin, other four antimicrobial compounds bacillaene, bacilysin, bacillibactin and subtilosin were also extracted from the fermentation broth of strain NCD-2 by using different extracting methods, respectively. However, only bacillaene and bacillibactin were detectable from the extracts by UHPLC-QTOF-MS (Fig. 8a, b).

MS/MS spectra of protonated cyclic bacillaene and bacillibactin ions. a m/z 581.4, b m/z 883.3
Discussion
B. subtilis has the potential to produce two dozen antimicrobial substances, and 5–8% of the B. subtilis genome contributes to the production of antimicrobial substances [32]. Some of these substances inhibit the growth of pathogens and the germination of spores. The lipopeptide mixture of B. subtilis C232 inhibits the formation of Verticillium dahliae microsclerotia [33], and the volatile compounds secreted by B. subtilis JA inhibit the conidial formation and mycelial growth of Glomus etunicatum [34].
However, certain antimicrobial compounds are synthesized only in response to external stimulation or under special conditions, this made it difficult to harvest all antimicrobial compounds produced by a given Bacillus strain using traditional cultivation and extraction methods [22]. Genome mining allows the prediction of metabolites based on genome sequences, including both identified antimicrobial compounds and novel antibiotics that have not been previously described. For example, the new NRPS antibiotic coelichelin was identified by genomic analysis from Streptomyces coelicolor [35]. Pseudomycoicidin in Bacillus pseudomycoides DSM 12442 was discovered by genome mining and through heterologous expression of its BGC in Escherichia coli [36].
Several lipopeptide antibiotics, including fengycin and surfactin have been identified in B. subtilis NCD-2 by using traditional cultivation and extraction methods [28]. Fengycin showed strong antifungal abilities against V. dahliae and B. cinerea. In this study, seven additional secondary metabolite gene clusters were found by genome mining, and some of them were identified using MS/MS. In total, B. subtilis NCD-2 had the potential to produce at least 9 kinds of secondary metabolites including surfactin, bacillaene, fengycin, bacillibactin, subtilosin, bacilysin, two terpenes, and one unknown product. Surfactin exhibits antibacterial, antiviral, antitumor and hemolytic action [37]. Bacillaene inhibits bacterial growth by inhibiting prokaryotic protein synthesis [38]. Fengycin shows specific antifungal activity against filamentous fungi [39]. Bacillibactin functions as a siderophore to compete for irons with environmental microbes especially under the iron deficiency conditions. B. subtilis expresses genes involved in the synthesis for bacillibactin to pirate other microbial iron [40].. Subtilosin possesses antibacterial activity against a diverse range of bacteria [41]. Bacilysin exhibits antimicrobial activities against both bacteria and Candida albicans [42]. However, only fengycin, surfactin, bacillaene and bacillibactin were successfully detected from the extract of strain NCD-2 by UHPLC-MS/MS (Figs. 7, 8). Bacilysin and subtilosin remained undetectable. A likely reason for their undetectability is the low expression level of their biosynthetic gene clusters under the experimental conditions.
B. velezensis FZB42 is a model strain of plant beneficial rhizobacteria. Thirteen gene clusters involved in the non-ribosomal and ribosomal synthesis of secondary metabolites with putative antimicrobial action have been identified within the genome of strain FZB42, including fengycin. The mechanism of fengycin synthesis has been well studied in B. velezensis strain FZB42 [43]. B. subtilis 168 has the entire gene cluster for synthesizing fengycin, but it can not produce fengycin because of a deficiency of a native sfp gene [44]. The BGC repository MIBiG (Minimum Information about a Biosynthetic Gene cluster) includes only has one fengycin biosynthetic gene cluster from B. velezensis FZB42 [45, 46]. Therefore, the fengycin biosynthetic gene cluster of strain NCD-2 was compared with that of B. velezensis FZB42. Fengycin comprises a peptide ring circled by 10 amino acids with a fatty acid chain tail. The fengycin biosynthetic gene clusters in most producing strains consist of the genes fenCDEAB (38 kb), which encode 5 enzymes, among which FenC recognizes and carries glutamate and ornithine; FenD recognizes and carries tyrosine and threonine, FenE recognizes and carries glutamate and valine; FenA recognizes and carries proline, glutamine, and tyrosine; and FenB recognizes and carries isoleucine. FenCDEAB recognizes 10 amino acids and carries them to the β-OH FA chain to form fengycin [47,48,49]. However, NCD-2 only had fenEAB, but no fenC and fenD, compared with the typical cluster structure of fenCDEAB in strain FZB42 strain and other 10 Bacillus strains (Fig. 4b and Additional file 1, Fig. S1). To exclude the errors introduced by genome sequencing or assembly, the fragment between fenE and dacC was cloned and sequenced, and it was confirmed that fenC and fenD were lost in strain NCD-2 (Fig. 5a-d). To identify the enzymes FenC and FenD in the NCD-2 genome, their amino acid sequences from FZB42 were selected to screen for homologs by scanning the local NCD-2 proteome using BioEdit. The protein Gms1961 in strain NCD-2 had the greatest similarity to FenC at an amino acid sequence level (Additional file 1, Table S2). The Gms1961 protein contained 2550 amino acids, with a molecular weight of 287.50 kDa. The substrate bound by the adenylation domain of the Gms1961 protein was predicted (Additional file 1, Table S4). The adenylation A9 domain bound valine and N5-hydroxyornithine, with the latter being a transitional form of ornithine combined with the adenylation domain [50]. The UHPLC-QTOF-MS/MS results of the fengycins revealed the amino acid ornithine at position 2 in all of the examined structures (Fig. 7a–e), indicating the presence of a protein that transports ornithine in the NCD-2 strain. We thus hypothesized that Gms1961 functions as FenC and FenE. The same analysis was performed using the Gms1960 protein, which had the greatest similarity with FenD (Additional file 1, Table S3). However, the FenD domains in Gms1960 and FZB42 varied greatly. Therefore, it was hypothesized that Gms1960 or other enzymes function similarly to FenD.
Although lacking two important genes, fenC and fenD, strain NCD-2 was capable of producing 26 homologs of 5 types of fengycins (A, B, A2, B2, and C) (Additional file 1, Fig. S4). The amino acids at positions 6 and 10 in the cyclic peptide ring of fengycin determine its structural type. When the amino acid at position 6 was valine with isoleucine or valine at position 10, fengycin B or fengycin B2, respectively, was produced (Fig. 7a, b and Additional file 1, Fig. S4); however, when the amino acid at position 6 was alanine with isoleucine or valine at position 10, fengycin A or fengycin A2, respectively, was produced (Fig. 7c, d and Additional file 1, Fig. S4). When the amino acid at position 6 was isoleucine or leucine with valine at position 10, fengycin C was produced (Fig. 7e and Additional file 1, Fig. S4). Each fengycin type had different homologs according to the number of carbon atoms in its β-OH FA chain, and the molecular weights of each homologs differed by 14 (−CH2) [51]. Compared to the short-chain varieties, long-chain fatty acids increase the hydrophobic activities of lipopeptides, making them more likely to have membrane-bound antimicrobial effects [52]. A B. circulans strain produces four fengycin homologs, but only fengycins with C16 and C17 carbon atoms in their β-OH FA chains have antibacterial activity [53]. Among the 26 fengycin homologs produced by strain NCD-2, 14 fengycins had more than 16 carbon atoms in their β-OH FA chains, which might be the most important composition for antimicrobial function. The B. siamensis SCSIO 05746 strain produces a large number of fengycin homologs, including 19 homologs of fengycin B [54]. Using MS/MS analysis, the five fengycins produced by the NCD-2 strain were divided into 26 homologs (Fig. 7a–e and Additional file 1, Fig. S5-S9). Therefore, NCD-2 is currently the strain with the largest number of known fengycin homologs [55].
During microbial synthesis of secondary metabolites, such as lipopeptide, the relatively high energy-consuming process of protein synthesis takes priority [56]. Excessive energy consumption is not conducive to the normal growth of microbes, and, generally, microbes produce antibiotics in large amounts only under stress, such as encountering pathogens [57]. We hypothesized that the essential biosynthetic genes fenEAB involved in fengycin synthesis were retained, while another two important biosynthetic genes fenCD were lost in the long-term evolution of strain NCD-2. Five fengycins were still produced. Gms1961 might serve the dual roles of FenC and FenE, indicating that NCD-2’s fengycin biosynthetic process is unique to the strain and is more energy-efficient than the process used in the other strains.
Conclusions
Genome mining and UHPLC–QTOF–MS/MS analysis revealed 9 gene clusters encoding antimicrobial compounds in the genome of Bacillus subtilis NCD-2. Among them, the fengycin biosynthetic gene cluster containing fenEAB genes is unique to strain NCD-2 compared with the other tested B. subtilis strains. Strain NCD-2 might employ a unique mechanism for synthesizing fengycin, which may shed new light on the synthesis and evolution of antimicrobial lipopeptides through the NRPS pathway.
Methods
Microorganisms and culture conditions
B. subtilis NCD-2 was routinely grown at 37 °C on Luria Bertani medium. For lipopeptide, bacillaene, bacilysin, bacillibactin and subtilosin production, strain NCD-2 was grown in Landy broth [58], PA medium [59], MSA medium [60], and TSB medium [61] at 30 °C and 180 rpm. The phytopathogen Botrytis cinerea BC-10 was used for antifungal activity test following the method described by Guo et al. [28] with some modifications. Briefly, a 6-mm diameter disc of B. cinerea was placed in the center of a 9-cm potato dextrose agar (PDA) plate, and the plate was inoculated 2 cm from the center with B. subtilis NCD-2 using a sterilized toothpick. Finally, the diameter of the inhibition zone was measured after a 3-d incubation at 25 °C.
Genome sequencing of strain NCD-2
The Illumina Solexa platform (BGI, Shenzhen, China) was used for whole-genome sequencing following the method described by Karim [62] with some modifications. The quality of reads was checked using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) [63], and paired-end reads were trimmed using Sickle (https://github.com/najoshi/sickle), and were assembled using the software package Velvet [29]. QUAST 5.02 was used to assess the quality of contigs and scaffolds [64]. The assembled scaffolds were annotated using Prokka (version v.1.13) [65]. Annotation of the genome of strain NCD-2 was performed using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline (http://www.ncbi.nim.nih.gov/genome/annotation_prok/) utilizing GeneMark, Glimmer, and tRNAscan-SE tools [66], and functional annotation was carried out using the Rapid Annotations by subsystems Technology (RAST) server with the seed database [67]. Finally, the genome of strain NCD-2 was deposited in the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/) under the GenBank accession number CP023755.
Evolutionary analysis, signal peptide and CRISPR repeat detection
Whole-genome sequences of B. subtilis and closely related species were downloaded from the NCBI database, and the REALPHY website (http://realphy.unibas.ch) [68] was used to perform genome-wide comparisons with the default parameters. A phylogenetic analysis was conducted using MEGA5 [69] with the Maximum Composite Likelihood parameter model [70]. A phylogenetic tree was constructed using the Neighbor-joining algorithm method with bootstrap values based on 1000 replications. The signal peptide was predicted using the SignalP-5.0 website (www.cbs.dtu.dk/services/SignalP-5.0/) [71]. CRISPR repeats were detected using CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index) [72].
Prediction and specificity analysis of secondary metabolite biosynthetic gene clusters
Secondary metabolite biosynthetic gene clusters for strain NCD-2 were detected using antiSMASH (http://antismash.secondarymetabolites.org) [31, 73] and PRISM (http://grid.adapsyn.com/prism/) [74] with the default parameters. The functional domain predictions for PKS/NRPS in the predicted gene clusters were analyzed using the PKS/NRPS Analysis Website (http://nrps.igs.umaryland.edu/) [75]. Typical PKS and NRPS sequences were selected for genomic and proteomic scanning after using BioEdit software to create a local BLAST based on strain NCD-2’s genome and proteome, respectively.
Detection of FenC and FenD loss in the genome of strain NCD-2
FenC and FenD are two important enzymes for synthesizing fengycin. A pair of degenerate primers targeting fenE (5′- CCRTCCATKAYGATATGATG − 3′) and dacC (5′- TGACAGAATGRYGGGMGGAAC − 3′) were designed based on the conserved bases of fenE and dacC in strain NCD-2 and B. velezensis strain FZB42. The 16S rDNA (27-F/1492R) primers were used as a positive control [76]. The amplification procedure included a denaturation step at 95 °C for 2 min, followed by 32 cycles of 20 s strand separation at 95 °C, 20 s annealing at 55 °C, and 90 s elongation at 72 °C, and an elongation step of 5 min at 72 °C. The target fragment from NCD-2 was purified by a gel extraction kit (Sangon, Shanghai, China), ligated to blunt-ended vector (Transgen, Beijing, China) and sequenced by BGI company (Shenzhen, China). The fragment sequence was deposited at NCBI with the accession number of MT984302.
Separation of lipopeptides by FPLC
Lipopeptides were extracted using the method described by Guo et al. [28]. Briefly, strain NCD-2 or derived strain Δgms1961 were cultured in 100 mL Landy broth [58] at 30 °C for 72 h with shaking at 180 rpm. The cell-free supernatant was obtained by centrifugation at 8000×g for 30 min at 4 °C. The supernatant was adjusted to pH 2.0 with 6 mol/L HCl and stored for 12 h at 4 °C. After centrifugation at 10,000×g, for 20 min, the resulting pellet was extracted with 10 mL methanol under continuous magnetic stirring for 2 h. The obtained extracts were sterilized by passing through 0.45-μm filters (Millex-GV, Millipore, Billerica, MA, USA) to obtain crude lipopeptides. The crude lipopeptides were separated and purified using an AKTA Purifier (GE Healthcare, Uppsala, Sweden) with a SOURCE 5RPC ST 4.6/150 column as described previously [77]. The lipopeptides were eluted with solvent A [2% acetonitrile containing 0.065% trifluoroacetic acid (TFA) (V/V)] and solvent B [80% acetonitrile containing 0.05% TFA (V/V)] using a linear gradient of 0–100% acetonitrile over 57 min at a flow rate of 1 mL/min. The detection wavelength was 215 nm. All the main peaks were automatically collected by FPLC. Finally, each peak was concentrated using a rotary evaporator and was analyzed using UHPLC-QTOF–MS/MS.
UHPLC–QTOF–MS/MS
UHPLC–QTOF–MS/MS analysis was conducted on a hybrid quadrupole time-of-flight tandem mass spectrometer (AB SCIEX TripleTOF 5600 Q-TOF/MS, Foster City, CA, USA) with an HPLC (Shimadzu, Kyoto, Japan) equipped with LC-30 AD binary pumps, a SIL-30 AC autosampler, and a CTO-30 AC column oven. A C18 reversed phase LC column (Shim-pack GIST 2-μm particles, 2.1 mm × 100 mm) was used for separation. The mobile phases A and B were water and acetonitrile, respectively, with 0.1% formic acid in both phases and with the optimized linear gradient elution procedure as follows: 0.0–0.5 min, 30% B; 0.5–50 min, 60% B; 50–52 min, 95% B; 52–55 min, 95% B; 55–55.1 min, 30% B; and 55.1–60 min, 30% B. The injection volume was 20 μL with a flow rate of 0.30 mL/min. The column oven was set at 40 °C. MS analysis was performed using a 5600 TripleTOF system equipped with a DuoSpray™ Ion Source, and the data were processed using Analyst TF 1.7 software (Applied Biosystems Sciex, Toronto, ON, Canada). PeakView™ software 2.0 (Applied Biosystems Sciex, Toronto, ON, Canada) was used to investigate and interpret the mass spectral data, with special tools for processing accurate mass data and structural elucidation. The DuoSpray™ ion source was used in positive ion mode. The instrumental parameters were set as follows: ion spray voltage floating, 5000 V; nebulizing gas, 50 psi; heater gas, 50 psi; curtain gas, 35 psi; temperature, 350 °C; declustering potential, 100 V; collision energy, TOF MS experiments: 10.0 V. TOF-MS/MS experiments: rolling collision energy, with collision energy spread 5 V. The data was acquired using Information Dependent Acquisition for a single run analysis with m/z range of 200–2000 in TOF MS and 50–1600 in MS/MS.
Detection of bacillaene, bacilysin, bacillibactin and subtilosin
For bacillaene, strain NCD-2 was cultured in 100 mL Landy broth at 30 °C for 72 h with shaking at 180 rpm, and bacillaene was extracted with methanol using the method described by Reddick et al. [78]. For bacilysin, strain NCD-2 was cultured in 100 mL PA medium at 30 °C for 72 h with shaking at 180 rpm, and bacilysin was extracted with ice-cold ethanol as described by Wu et al. [59]. For bacillibactin, strain NCD-2 was cultured in 100 mL MSA medium at 30 °C for 72 h, and bacillibactin was extracted with ethanol as described by Li et al. [60]. For subtilosin, strain NCD-2 was cultured in 100 mL TSB medium at 30 °C for 72 h, and subtilosin was extracted with precipitation with 65% ammonium sulphate as described by Charles et al. [61]. The extracts were detected by UHPLC-QTOF-MS/MS as described above.
Availability of data and materials
The datasets used and analyzed in the current study are available from the corresponding author on reasonable request. The genome of strain NCD-2 was deposited at NCBI (https://www.ncbi.nlm.nih.gov) under the GenBank accession number CP023755 (https://www.ncbi.nlm.nih.gov/nuccore/CP023755.1?report=fasta) and the fragment fenE-dacC sequence of NCD-2 was deposited at NCBI with the accession number MT984302. All B. subtilis strain with the assembly level of complete genome or chromosome could be obtained at NCBI by their RefSeq assembly accession numbers which were listed in Table S1. The genome of B. velezensis FZB42 was downloaded at NCBI under the GenBank accession number CP000560.2 (https://www.ncbi.nlm.nih.gov/nuccore/CP000560.2).
Abbreviations
- UHPLC-QTOF-MS/MS:
-
Ultra-high-performance liquid chromatography coupled to quadrupole-time-of-flight tandem mass spectrometry
- A domain:
-
Adenylation domain
- C domain:
-
Condensation domain
- T domain:
-
Thiolation domain
- Te:
-
Thioesterase domain
- E domain:
-
Epimerization domain
- N90:
-
The minimum contig length to cover 90% of the genome
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- PDA:
-
Potato dextrose agar
- BGC:
-
Biosynthetic gene cluster
- FPLC:
-
Fast protein liquid chromatography
- m/z :
-
mass-to-charge ratio
- TFA:
-
Trifluoroacetic acid
- β-OHFA:
-
β-hydroxy-fatty acid
References
Wang P, Guo Q, Ma Y, Li S, Lu X, Zhang X, Ma P. DegQ regulates the production of fengycins and biofilm formation of the biocontrol agent Bacillus subtilis NCD-2. Microbiol Res. 2015;178:42–50.
Fan H, Ru J, Zhang Y, Wang Q, Li Y. Fengycin produced by Bacillus subtilis 9407 plays a major role in the biocontrol of apple ring rot disease. Microbiol Res. 2017;199:89–97.
Wu Y, Wang Y, Zou H, Wang B, Sun Q, Fu A, Wang Y, Wang Y, Xu X, Li W. Bacillus amyloliquefaciens probiotic SC06 induces autophagy to protect against pathogens in macrophages. Front Microbiol. 2017;8:469.
Sonenshein AL. Control of sporulation initiation in Bacillus subtilis. Curr Opin Microbiol. 2000;3(6):561–6.
Moszer I, Jones L, Moreira S, Fabry C, Danchin A. SubtiList: the reference database for the Bacillus subtilis genome. Nucleic Acids Res. 2002;30(1):62–5.
Torres MJ, Brandan CP, Sabate DC, Petroselli G, Errabalsells R, Audisio MC. Biological activity of the lipopeptide-producing Bacillus amyloliquefaciens PGPBacCA1 on common bean Phaseolus vulgaris L. pathogens. Biol Control. 2017;105:93–9.
Agustín L-B, Raunel T-V, Gerardo C, Kohei K, Katsuhiro K, Enrique G, Leobardo S-C. Effects of bacillomycin D homologues produced by Bacillus amyloliquefaciens 83 on growth and viability of Colletotrichum gloeosporioides at different physiological stages. Biol Control. 2018;127:145–54.
Stein T. Bacillus subtilis antibiotics: structures, syntheses and specific functions. Mol Microbiol. 2005;56(4):845–57.
Kröber M, Wibberg D, Grosch R, Eikmeyer F, Verwaaijen B, Chowdhury SP, Hartmann A, Pühler A, Schlüter A. Effect of the strain Bacillus amyloliquefaciens FZB42 on the microbial community in the rhizosphere of lettuce under field conditions analyzed by whole metagenome sequencing. Front Microbiol. 2014;5:252.
Beppu T. Secondary metabolites as chemical signals for cellular differentiation. Gene. 1992;115(1–2):159–65.
Chaudhary AK, Dhakal D, Sohng JK. An insight into the “-omics” based engineering of streptomycetes for secondary metabolite overproduction. Biomed Res Int. 2013;2013:968518.
Ichikawa N, Sasagawa M, Yamamoto M, Komaki H, Yoshida Y, Yamazaki S, Fujita N. DoBISCUIT: a database of secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2012;41(D1):D408–14.
Chen XH, Koumoutsi A, Scholz R, Borriss R. More than anticipated - production of antibiotics and other secondary metabolites by Bacillus amyloliquefaciens FZB42. J Mol Microbiol Biotechnol. 2009;16(1–2):14–24.
Stein T, Vater J, Kruft V, Otto A, Wittmannliebold B, Franke P, Panico M, Mcdowell RA, Morris HR. The multiple carrier model of nonribosomal peptide biosynthesis at modular multienzymatic templates. J Biol Chem. 1996;271(26):15428–35.
Du L, Lou L. PKS and NRPS release mechanisms. Nat Prod Rep. 2010;27(2):255–78.
Arguellesarias A, Ongena M, Halimi B, Lara Y, Brans A, Joris B, Fickers P. Bacillus amyloliquefaciens GA1 as a source of potent antibiotics and other secondary metabolites for biocontrol of plant pathogens. Microb Cell Factories. 2009;8(1):63.
Tamehiro N, Okamotohosoya Y, Okamoto S, Ubukata M, Hamada M, Naganawa H, Ochi K. Bacilysocin, a novel phospholipid antibiotic produced by Bacillus subtilis 168. Antimicrob Agents Chemother. 2002;46(2):315–20.
Carrillo C, Teruel JA, Aranda FJ, Ortiz A. Molecular mechanism of membrane permeabilization by the peptide antibiotic surfactin. Biochim Biophys Acta. 2003;1611(1–2):91–7.
Yu GY, Sinclair JB, Hartman GL, Bertagnolli BL. Production of iturin a by Bacillus amyloliquefaciens suppressing Rhizoctonia solani. Soil Biol Biochem. 2002;34(7):955–63.
Jacques P, Hbid C, Destain J, Razafindralambo H, Paquot M, De Pauw E, Thonart P. Optimization of biosurfactant lipopeptide production from Bacillus subtilis S499 by plackett-burman design. Appl Biochem Biotechnol. 1999;77–79:223–33.
Moyne A, Cleveland TE, Tuzun S. Molecular characterization and analysis of the operon encoding the antifungal lipopeptide bacillomycin D. FEMS Microbiol Lett. 2004;234(1):43–9.
Tulp M, Bohlin L. Rediscovery of known natural compounds: nuisance or goldmine? Bioorg Med Chem. 2005;13(17):5274–82.
Oman TJ, van der Donk WA. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat Chem Biol. 2010;6(1):9–18.
Lane AL, Moore BS. A sea of biosynthesis: marine natural products meet the molecular age. Nat Prod Rep. 2011;28(2):411–28.
Rutledge PJ, Challis GL. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol. 2015;13(8):509–23.
van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30(9):418–26.
Guo Q, Li S, Lu X, Li B, Ma P. PhoR/PhoP two component regulatory system affects biocontrol capability of Bacillus subtilis NCD-2. Genet Mol Biol. 2010;33(2):333–40.
Guo Q, Dong W, Li S, Lu X, Wang P, Zhang X, Wang Y, Ma P. Fengycin produced by Bacillus subtilis NCD-2 plays a major role in biocontrol of cotton seedling damping-off disease. Microbiol Res. 2014;169(7–8):533–40.
Zerbino D, Birney E. Velvet : algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008;18(5):821–9.
Dunlap CA, Bowman MJ, Zeigler DR. Promotion of Bacillus subtilis subsp. inaquosorum, Bacillus subtilis subsp. spizizenii and Bacillus subtilis subsp. stercoris to species status. Anton Leeuw Int J Gen Mol Microbiol. 2020;113(1):1–12.
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41(W1):W204–12.
Sansinenea E, Ortiz A. Secondary metabolites of soil Bacillus spp. Biotechnol Lett. 2011;33(8):1523–38.
Yu D, Fang Y, Tang C, Klosterman SJ, Tian C, Wang Y. Genomewide transcriptome profiles reveal how Bacillus subtilis lipopeptides inhibit microsclerotia formation in Verticillium dahliae. Mol Plant-Microbe Interact. 2019;32(5):622–34.
Xiao X, Chen H, Chen H, Wang J, Ren C, Wu L. Impact of Bacillus subtilis JA, a biocontrol strain of fungal plant pathogens, on arbuscular mycorrhiza formation in Zea mays. World J Microbiol Biotechnol. 2008;24(7):1133–7.
Challis GL, Ravel J. Coelichelin, a new peptide siderophore encoded by the Streptomyces coelicolor genome: structure prediction from the sequence of its non-ribosomal peptide synthetase. FEMS Microbiol Lett. 2000;187(2):111–4.
Basichipalu S, Dischinger J, Josten M, Szekat C, Zweynert A, Sahl H, Bierbaum G. Pseudomycoicidin, a class II lantibiotic from Bacillus pseudomycoides. Appl Environ Microbiol. 2015;81(10):3419–29.
Seydlová G, Svobodová J. Review of surfactin chemical properties and the potential biomedical applications. Central Eur J Med. 2008;3(2):123–33.
Patel PS, Huang S, Fisher S, Pirnik D, Aklonis C, Dean L, Meyers E, Fernandes P, Mayerl F. Bacillaene, a novel inhibitor of procaryotic protein synthesis produced by Bacillus subtilis. J Antibiot. 2006;48(9):997.
Ramarathnam R, Bo S, Chen Y, Fernando WG, Xuewen G, de Kievit T. Molecular and biochemical detection of fengycin- and bacillomycin D-producing Bacillus spp., antagonistic to fungal pathogens of canola and wheat. Can J Microbiol. 2007;53(7):901–11.
Miethke M, Klotz O, Linne U, May JJ, Beckering CL, Marahiel MA. Ferri-bacillibactin uptake and hydrolysis in Bacillus subtilis. Mol Microbiol. 2006;61(6):1413–27.
Thennarasu S, Lee DK, Poon A, Kawulka KE, Vederas JC, Ramamoorthy A. Membrane permeabilization, orientation, and antimicrobial mechanism of subtilosin a. Chem Phys Lipids. 2005;137(1–2):38–51.
Kenig M, Abraham EP. Antimicrobial activities and antagonists of bacilysin and anticapsin. Microbiology. 1976;94(1):37–45.
Fan B, Wang C, Song X, Ding X, Wu L, Wu H, Gao X, Borriss R. Bacillus velezensis FZB42 in 2018: the gram-positive model strain for plant growth promotion and biocontrol. Front Microbiol. 2018;9:2491.
Jin P, Wang H, Liu W, Miao W. Characterization of lpaH2 gene corresponding to lipopeptide synthesis in Bacillus amyloliquefaciens HAB-2. BMC Microbiol. 2017;17(1):227.
Chen X, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, Morgenstern B, Voss B, Hess W, Reva O, et al. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol. 2007;25(9):1007–14.
Koumoutsi A, Chen X, Henne A, Liesegang H, Hitzeroth G, Franke P, Vater J, Borriss R. Structural and functional characterization of gene clusters directing nonribosomal synthesis of bioactive cyclic lipopeptides in Bacillus amyloliquefaciens strain FZB42. J Bacteriol. 2004;186(4):1084–96.
Chen C, Chang L, Chang Y, Liu S, Tschen JS. Transposon mutagenesis and cloning of the genes encoding the enzymes of fengycin biosynthesis in Bacillus subtilis. Mol Gen Genomics. 1995;248(2):121–5.
Lin G, Chen C, Tschen JS, Tsay S, Chang Y, Liu S. Molecular cloning and characterization of fengycin synthetase gene fenB from Bacillus subtilis. J Bacteriol. 1998;180(5):1338–41.
Lin TP, Chen CL, Chang LK, Tschen JS, Liu ST. Functional and transcriptional analyses of a fengycin synthetase gene, fenC, from Bacillus subtilis. J Bacteriol. 1999;181(16):5060–7.
Lautru S, Deeth RJ, Bailey LM, Challis GL. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat Chem Biol. 2005;1(5):265–9.
Bie XM, Lü FX, Lu ZX, Huang XQ, Shen J. Isolation and identification of lipopeptides produced by Bacillus subtilis fmbJ. Sheng wu gong cheng xue bao. 2006;22(4):644–9.
Tripathi L, Irorere VU, Marchant R, Banat IM. Marine derived biosurfactants: a vast potential future resource. Biotechnol Lett. 2018;40(11–12):1441–57.
Sivapathasekaran C, Mukherjee S, Samanta R, Sen R. High-performance liquid chromatography purification of biosurfactant isoforms produced by a marine bacterium. Anal Bioanal Chem. 2009;395(3):845–54.
Pan H, Tian X, Shao M, Xie Y, Huang H, Hu J, Ju J. Genome mining and metabolic profiling illuminate the chemistry driving diverse biological activities of Bacillus siamensis SCSIO 05746. Appl Microbiol Biotechnol. 2019;103(10):4153–65.
Ongena M, Jacques P. Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol. 2008;16(3):115–25.
Bhat A, Chakraborty R, Adlakha K, Agam G, Chakraborty K, Sengupta S. Ncl1-mediated metabolic rewiring critical during metabolic stress. Life Sci Alliance. 2019;2(4):e201900360. https://doi.org/10.26508/lsa.201900360.
Bae J, Park J, Hahn M, Kim M, Roe J. Redox-dependent changes in RsrA, an anti-sigma factor in Streptomyces coelicolor: zinc release and disulfide bond formation. J Mol Biol. 2004;335(2):425–35.
Landy M, Warren GH. Bacillomycin; an antibiotic from Bacillus subtilis active against pathogenic fungi. Proc Soc Exp Biol Med Soc Exp Biol Med (New York). 1948;67(4):539–41.
Wu L, Wu H, Chen L, Xie S, Zang H, Borriss R, Gao X. Bacilysin from Bacillus amyloliquefaciens FZB42 has specific bactericidal activity against harmful algal bloom species. Appl Environ Microbiol. 2014;80(24):7512–20.
Li Y, Jiang W, Gao R, Cai Y, Guan Z, Liao X. Fe (III)-based immobilized metal-affinity chromatography (IMAC) method for the separation of the catechol siderophore from CD36. 3 Biotech. 2018;8(9):392.
Shelburne C, An F, Dholpe V, Ramamoorthy A, Lopatin D, Lantz M. The spectrum of antimicrobial activity of the bacteriocin subtilosin a. J Antimicrob Chemother. 2007;59(2):297–300.
Karim A, Poirot O, Khatoon A, Aurongzeb M. Draft genome sequence of a novel Bacillus glycinifermentans strain having antifungal and antibacterial properties. J Glob Antimicrob Resist. 2019;19:308–10.
Andrews S. FastQC A quality control tool for high throughput sequence data; 2010.
Alexey G, Vladislav S, Nikolay V, Glenn T: QUAST: quality assessment tool for genome assemblies. In: 2013; 2013: 1072–1075.
Torsten S. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9.
Disz T, Akhter S, Cuevas DA, Olson R, Overbeek R, Vonstein V, Stevens R, Edwards R. Accessing the SEED genome databases via web services API: tools for programmers. BMC Bioinformatics. 2010;11(1):319.
Aziz RK, Bartels D, Best AA, Dejongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass EM, Kubal M. The rast server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Haubold B, Klotzl F, Pfaffelhuber P. Andi: fast and accurate estimation of evolutionary distances between closely related genomes. Bioinformatics. 2015;31(8):1169–75.
Tamura K, Peterson DS, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Xu X, Reid N. On the robustness of maximum composite likelihood estimate. J Stat Plan Inference. 2011;141(9):3047–54.
Armenteros JJA, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, Nielsen H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–3.
Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, Pourcel C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46(W1):W246–51.
Medema MH, Blin K, Cimermancic P, De Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39(2):W339–46.
Skinnider MA, Dejong CA, Rees PN, Johnston CW, Li H, Webster ALH, Wyatt MA, Magarvey NA. Genomes to natural products PRediction informatics for secondary Metabolomes (PRISM). Nucleic Acids Res. 2015;43(20):9645–62.
Bachmann BO, Ravel J. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217.
Byers H, Stackebrandt E, Hayward C, Blackall LL. Molecular investigation of a microbial mat associated with the great Artesian Basin. FEMS Microbiol Ecol. 1998;25(4):391–403.
Li B, Lu X, Guo Q, Qian C, Li S, Ma P. Isolation and identification of lipopeptides and volatile compounds produced by Bacillus subtilis strain BAB-1, vol. 43; 2010.
Reddick J, Antolak S, Raner G. PksS from Bacillus subtilis is a cytochrome P450 involved in bacillaene metabolism. Biochem Biophys Res Commun. 2007;358(1):363–7.
Acknowledgements
We would like to thank professor Liqun Zhang from China Agricultural University whose comments and suggestions greatly improved the quality of this article.
Funding
The genome sequencing and data analyses in this study were financially supported by the earmarked fund for National Key R & D Projects (2017YFD0200400) and the China Agriculture Research System (CARS-18-15), the experimental material was supported by the Natural Science Foundation of Hebei Province (C2019301101), the National Natural Science Foundation of China (31572051 and 31601680), the PhD Fund of Hebei Academy of Agriculture and Forestry (C19R01003), and the Special Fund for Agro-scientific Research in the Public Interest, China (201503109). The funding body didn’t participate in the design of the study, collection, analysis and interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
ZHS, QGG, and PM designed the experiments. ZHS, XYC, and XML performed all the experiments. ZHS and XYC analyzed the data. ZHS, QGG, and PM wrote the manuscript. ZHS, XYC, QGG, XML, SZL, XYL, ZYZ, PPW, LHD, WSZ and PM reviewed the final manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1 : Fig. S1
. Fengycin biosynthetic gene clusters of different strains that have a close relation with NCD-2 or the model strains. Fig. S2. Surfactin biosynthetic gene clusters of different strains that have a close relation with NCD-2 or the model strains. Fig. S3. Elution of lipopeptides separated from the crude methanolic extract using an AKTA Purifier. Fig. S4. Primary structures of fengycins and surfactins. Fig. S5. Fengycin A of a β-OH FA with a chain length varying from C14 to C19 identified based on key product ions. Fig. S6. Fengycin B of a β-OH FA with a chain length varying from C12 to C19 identified based on key product ions. Fig. S7. Fengycin A2 of a β-OH FA with a chain length varying from C15-C18 identified based on key product ions. Fig. S8. Fengycin B2 of a β-OH FA with a chain length varying from C14-C18 identified based on key product ions. Fig. S9. Fengycin C of a β-OH FA with a chain length varying from C18-C20 identified based on key product ions. Fig. S10. Surfactin of a fatty acid with a chain length varying from C11-C15 identified based on key product ions. Fig. S11 Original, full-length gel images. Table S1. All B. subtilis strains with the assembly level of chromosome and their RefSeq assembly accessions. Table S2. Homologues of FenC of FZB42 detected by scanning the local NCD-2 proteome in BioEdit. Table S3. Homologues of FenD of FZB42 detected by scanning the local NCD-2 proteome in BioEdit. Table S4. Adenylation domain binding amino acids predicted by PRISM.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Su, Z., Chen, X., Liu, X. et al. Genome mining and UHPLC–QTOF–MS/MS to identify the potential antimicrobial compounds and determine the specificity of biosynthetic gene clusters in Bacillus subtilis NCD-2. BMC Genomics 21, 767 (2020). https://doi.org/10.1186/s12864-020-07160-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-07160-2