
Abstract
Background
Alcohol dehydrogenases (ADHs) in plants are encoded by a multigene family. ADHs participate in growth, development, and adaptation in many plant species, but the evolution and function of the ADH gene family in sugarcane is still unclear.
Results
In the present study, 151 ADH genes from 17 species including 32 ADH genes in Saccharum spontaneum and 6 ADH genes in modern sugarcane cultivar R570 were identified. Phylogenetic analysis demonstrated two groups of ADH genes and suggested that these genes underwent duplication during angiosperm evolution. Whole-genome duplication (WGD)/segmental and dispersed duplications played critical roles in the expansion of ADH family in S. spontaneum and R570, respectively. ScADH3 was cloned and preferentially expressed in response to cold stress. ScADH3 conferred improved cold tolerance in E. coli cells. Ectopic expression showed that ScADH3 can also enhance cold tolerance in transgenic tobacco. The accumulation of reactive oxygen species (ROS) in leaves of transgenic tobacco was significantly lower than in wild-type tobacco. The transcript levels of ROS-related genes in transgenic tobacco increased significantly. ScADH3 seems to affect cold tolerance by regulating the ROS-related genes to maintain the ROS homeostasis.
Conclusions
This study depicted the size and composition of the ADH gene family in 17 species, and investigated their evolution pattern. Comparative genomics analysis among the ADH gene families of S. bicolor, R570 and S. spontaneum revealed their close evolutionary relationship. Functional analysis suggested that ScADH3, which maintained the steady state of ROS by regulating ROS-related genes, was related to cold tolerance. These findings will facilitate research on evolutionary and functional aspects of the ADH genes in sugarcane, especially for the understanding of ScADH3 under cold stress.
Similar content being viewed by others


Background
Plant metabolite production is affected by both plant development and environmental factors. Alcohols are components of plant volatiles and also act as plant stress signaling molecules [1]. Alcohol dehydrogenase (ADH, alcohol: NAD+ oxidoreductase, EC 1.1.1.1) acts as a dimer that relies on NAD(P) co-factors to interconvert ethanol and acetaldehyde and other short linear alcohols/aldehyde pairs [2]. ADHs are involved in seed development [3, 4], fruit development [5], and aerobic metabolism in pollen grain [6]. They also help to protect plants from flooding [7], drought [8], cold [9], and salt [10] stresses.
Plant alcohol dehydrogenase enzyme (ADH-P) was discovered to be active during hypoxia [11]. ADH gene activity is found at all stages of plant growth and under various stress conditions [10]. The ADH gene is involved in different aspects of plant growth and development [12, 13]. Several ADH genes are expressed in plant tissues in a developmentally-regulated manner, especially during fruit ripening. For example, Le-ADH2 is involved in aroma volatilization during fruit ripening [12] . Overexpression of Le-ADH2 improves fruit flavor by increasing alcohol levels (especially Z-3-hexenol) [12]. Tesniere and Verries et al. [13] found that Vv-ADH1 and Vv-ADH3 transcripts in grapes accumulate briefly in young fruits, while Vv-ADH2 transcripts strongly increase in the mature phase named veraison. Expression of the ADH gene is induced by different environmental stresses [14], such as low temperature [9, 15], osmotic [16], drought [8], salt [10], mechanical damage [17], and the exogenous hormone abscisic acid (ABA) [18]. Dolferus et al. [18] found that hypoxia, dehydration, low temperature, and the phytohormone ABA can induce ADH expression in Arabidopsis. Shi et al. [14] demonstrated that alcohol dehydrogenase 1 gene (ADH1) enhances Arabidopsis resistance to abiotic and biotic stresses. Salinity stress induces accumulation of ADH mRNA in soybean (Glycine max) [19], grasspea (Lathyrus sativus L.) [20], and Arabidopsis [21]. The ADH gene is one of the most common cold-induced genes in cereal crops and Arabidopsis [9]. In addition, the ADH levels in octoploid strawberry are highly correlated with cold tolerance [15].
Sugarcane (Saccharum spp.) accounts for 80% of world sugar production [22]. Modern sugarcane cultivars are produced by hybridizations between Saccharum officinarum L. (2n = 80, high sugar content) and Saccharum spontaneum L. (2n = 40–128, disease resistance) [23]. However, adverse environmental factors such as drought, cold, salt, heavy metals, and low temperature can cause great production losses [24]. An effective solution is to identify genetic resources with superior genetic traits and then use molecular biology techniques to breed sugarcane cultivars resistant to stress [25]. However, the ploidy and repetitive genomic characteristics of sugarcane pose a challenge to sugarcane breeding. The development of genome-wide sequencing has aided resistance breeding. Two genomic datasets exist for Saccharum, one for the elite cultivar R570 [26] and the other for S. spontaneum [27].
Few studies have investigated the ADH genes in sugarcane. ADH gene expression upregulated due to waterlogging and waterlogging + nitrogen compounds in both leaf and root tissues [28]. ADH transcripts can also be induced by exogenous hormones (salicylic acid (SA), ABA, and methyl jasmonate (MeJA)) [29, 30]. However, the functions of sugarcane ADH genes are unknown. In the present study, we identified the ADH gene in 17 plant species including eudicots, monocots, basal angiosperms, and mosses. Phylogenetic analysis was used to study the evolutionary history and the origin of ADH genes in plants. We used bioinformatics to analyze the ADH genes in two sugarcane genomes for comparative analysis. These were the haploid genome of the modern sugarcane cultivar R570 [26] and the haploid genome version of the sugarcane ancestor S. spontaneum AP85–441 [27]. The ADH genes in the Sorghum bicolor genome, a common reference for comparative analysis of sugarcane [31], were also analyzed. We cloned and functionally characterized an ADH gene from sugarcane clones. Prokaryotic expression and ectopic expression studies showed that ScADH3 is involved in the defense response to cold stress. These findings increase our understanding of the evolution and functional divergence of the ADH gene family in sugarcane and also the physiological mechanism and regulatory function of ScADH3. The data also provide insight into plant responses to cold.
Results
Identification of ADH genes in plant genomes
The putative ADH or ADH-like protein sequences were submitted to the CDD database, and 151 ADH protein sequences were retrieved from the 17 selected representative plant genomes. These included eudicots (5 species: 43 sequences), monocots (9 species: 95 sequences), basal angiosperms (1 species: 7 sequences), and mosses (2 species: 6 sequences). No ADH was identified in algae (Fig. 1 and Supplementary Table S1). The retained sequences that specifically hit the domain “alcohol_DH_plants (accession: cd08301)” were named ADHs, while the others were named ADH-likes. The copy number of ADH genes varied among the representative lineages of plants, ranging from two in Physcomitrella patens to 32 in S. spontaneum. The genes in S. spontaneum that belong to alleles [27] were designated as the same name followed by the letters “a,” “b,” “c,” and “d,” and duplicated genes were designated as the same name followed by the letters “e” and “f” [27]. Statistics results showed that eight copies of ADH genes are present in eudicots with the exception of Medicago truncatula which possesses nine ADH genes, and Vitis vinifera, which possesses 10 ADH genes. Among the monocots, the ADH gene copy number varied from 6 to 32. The largest number of ADH genes (32) was found in S. spontaneum. In the basal angiosperms, seven copies of ADH genes were identified in Amborella trichopoda. In the mosses, P. patens had two members of ADH genes, and there were four copies in Sphagnum fallax. However, no ADH genes were found in Chondrus crispis, Cyanidioschyzon merolae and Galdieria sulphuraria. One glutathione-dependent formaldehyde dehydrogenase (GSH-FDH) gene, also known as Class III ADH, was found in these three species. The ADH-P gene family is documented to have originated from Class III ADH [2]. Therefore, these Class III ADH genes were reserved for subsequent phylogenetic analysis.

Genome-wide identification of ADHs in 20 species. a Common tree of 20 species used in this study. b The number of identified ADH genes in these 20 species
Using the ExPASy ProtParam tool, the physical and chemical parameters of these ADH proteins were computed (Supplemental Table S1). The number of amino acids in ADH polypeptides ranged from 281 (SsADH-like4) to 881 (FvADH-like3), and the corresponding molecular weight (MW) ranged from 30.18 to 87.59 kDa. The computed theoretical isoelectric points (pI) varied greatly, ranging from 5.07 (AmADH-like3) to 8.82 (SfADH-like3). The results of the grand average of hydropathicity (GRAVY) suggested that 43 of 169 were hydrophilic and the instability index showed that 13 of 151 ADH proteins were unstable. Subcellular locations were predicted by WoLF PSORT program (Supplemental Table S1), and 102 ADH proteins were predicted to be located in the cytoplasm, while 30 ADH proteins were predicted to be localized in the chloroplasts.
Phylogenetic classification of ADH gene family
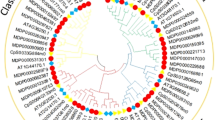
To study the evolution of the ADH gene family, a total of 153 ADHs from 17 plant species and three Class III ADHs from Rhodophyta as outgroups were used to construct a phylogenetic tree using the Maximum Likelihood (ML) method (Fig. 2 and Figure S1). Topology of the ML tree showed that ADH genes clearly cluster into two major groups in plants (Fig. 2), which were designated as Group A and Group B. In the plant clade, Group A possesses the whole land plant lineages, while the mosses were not located in Group B. The result implied that the ADH genes in Group A predated the ADH genes in Group B and had undergone several events of gene duplication during their long-term evolution. These gene duplication events generated multiple copies in plants.

Phylogenetic classification of ADH genes in plants. Phylogenetic tree was constructed using the ML method implemented in RaxML-HPC2. The topology of the ML tree showed that ADH genes in plants can be classified into two major groups, which are designed as Groups A and B. The Rhodophyta was set as the outgroup
The phylogenetic tree results demonstrated that the ADH gene family in plants can be classified into four subgroups, designated as Groups A-1, A-2, B-1, and B-2. Each subgroup can be further subdivided into mosses (Mos-1 and Mos-2), basal angiosperms (Bal-1, Bal-2, Bal-3, and Bal-4), monocots (Mon-1, Mon-2, Mon-3, Mon-4, and Mon-5), and eudicots (Eud-1, Eud-2, Eud-3, Eud-4, and Eud-5) (Figure S1). The Rhodophyta are separated from other embryophytes according to the topology of the ML tree. This may be because the ADH gene family has undergone one gene duplication event (D1) during the evolution of extant terrestrial and seed plants. The generation of four subgroups of the ADH gene family among plants may have been caused by the duplication event. Group A contains all embryophytes, while mosses are absent in Group B. Mosses form two branches in Group A, indicating that at least one gene duplication event occurred in the ancestor of mosses. The basal angiosperm (A. trichopoda) was distributed in each subgroup. The other seed plants also contain two monophyletic groups in Group A or Group B, respectively. These results suggest that gene expansion appeared in these plants during the evolution of the ADH gene family.
Protein motifs and gene structure analysis
A schematic map representing the structure of all 151 ADH proteins from 17 species was constructed from the MEME motif analysis results (Fig. 3). A total of 20 distinct conserved motifs were found. Most ADH members were within the same clade, especially the most closely related members, which usually shared common motif compositions. This indicated potential functional similarities among the ADH proteins. However, some motifs were group-specific: motif 20 was unique to almost all Group A-1; motif 16 was unique to almost all A-2 members; motif 19 was unique to almost all Group B-1 members; motif 17 was unique to almost all Group B-2 members. Most copies of ADHs proteins possessed 15 or 16 motifs. There was no correlation between motif number and protein length. For example, SsADH-like4 had the shortest protein length but its motif number was not the smallest. FvADH-like3 had the longest protein length, and its motif number was not the largest.

Phylogenetic relationships, gene structure and architecture of conserved protein motifs in ADH genes. The phylogenetic tree was constructed based on the full-length sequences of ADH proteins using RaxML-HPC tool. Details of clusters are shown in different colors. The motif compositions of ADH proteins. The motifs, numbers 1–20, are displayed in different colored boxes. Red boxes indicate exons in ADH genes, and black lines indicate introns
The pattern of exon–intron distribution and position of the ADH genes from 17 species were analyzed to study the structural characteristics and evolution of the ADH gene family (Fig. 3). Most ADHs (90 of 151) contained 10 exons, and 30 of 151 ADHs had nine exons. The exon number of ADHs ranged from 1 to 21. In Group A-1, 16 of 31 ADHs contained 10 exons, 12 ADHs contained 8, 9, or 11 exons, and one ADH had 12 exons (Fig. 3). In Groups A-2 and B-1, the number of introns was conserved, 39 of 48 and 13 of 17 ADHs had 10 exons. These genes in Group B-2 contained 6 to 21 introns. These data showed that the intron number of ADHs in Group B-2 was variable.
Chromosomal distribution and duplications to the family expansion
SbADH genes were unevenly distributed on four sorghum chromosomes (Fig. 4). The majority of SbADH genes were located on chromosome 1 (Sb1). The six ShADH genes were randomly distributed on four R570 chromosomes. Chromosome 1 (Sh1) and Chromosome 2 (Sh2) each contained two genes. The distribution of SsADH genes on the 14 chromosomes was uneven. The number of SsADH genes per chromosome varied from one to five (chromosomes 1C (Ss1C)). There was no correlation between chromosome length and the number of ADH genes.

Chromosomal distribution of the ADH gene family in S. bicolor, S. spontaneum and R570. The tandem duplicated genes are represented by red font. The scale bar on the left indicates the chromosome length (Mb)
The types of ADH genes in S. bicolor, S. spontaneum and R570 were identified by the Multiple Collinearity Scan toolkit (MCScanX) software to study the possible gene expansion mechanisms (dispersed, proximal, tandem, and WGD/segmental duplications) in these species (Fig. 5 and Supplemental Table S2). Dispersed and tandem duplications were found in all three species. Proximal duplication was observed in S. bicolor and S. spontaneum, but only S. spontaneum contained the WGD/segmental duplication (Fig. 5). For S. bicolor, tandem duplication played an important role in the gene expansion because more than half of the SbADHs were related to tandem duplication. For R570, dispersed duplication played an important role in the gene expansion. However, for S. spontaneum, more than half of SsADHs were related to WGD/segmental duplication (Fig. 5). Although the rates of the four duplicated types in the three species varied, the dispersed and tandem duplications appear to be the most common mechanisms for ADH family gene expansion.

Expansion mechanisms of the ADH gene family in S. bicolor, R570, and S. spontaneum. The effects of dispersed, proximal, tandem, and WGD/segmental duplications on the expansion of ADH genes in S. bicolor, R570, and S. spontaneum
Synteny analysis of ADH family genes
Synteny analysis can help reveal the phylogenetic relations among ADH family genes. Three comparative syntenic maps between S. bicolor, R570, and S. spontaneum were constructed (Fig. 6 and Supplemental Table S3). A total of 12 SsADH genes showed a syntenic relationship with S. bicolor (Fig. 6a). Five SbADH genes showed a syntenic relationship with R570 (Fig. 6b). To some extent, the observed synteny between ADH genes of R570 and S. bicolor may be regarded as an outcome of the strategy adopted to assemble Saccharum BAC clones. There were 10 orthologous pairs between S. spontaneum and R570 (Fig. 6c). Two SbADH genes (SbADH-like1 and SbADH2) were associated with three or four syntenic gene pairs within S. spontaneum. ShADH genes, ShADH1, ShADH2, ShADH-like1, and ShADH4 were associated with two syntenic gene pairs within S. spontaneum. These results suggest that these genes may have been involved in the evolution of the ADH gene family. Comparing the syntenic blocks, 12 ADH collinear gene pairs (six pairs between S. bicolor and S. spontaneum, three pairs between R570 and S. spontaneum, and three pairs between S. bicolor and R570) were anchored to highly conserved syntenic blocks, which contain more than 100 genes. Only four ADH collinear gene pairs (two pairs between S. bicolor and S. spontaneum, one pair between R570 and S. spontaneum, and one pair between S. bicolor and R570) were located in syntenic blocks that possessed fewer than 30 orthologous gene pairs.

Synteny analysis of ADH genes in S. bicolor, R570, and S. spontaneum. a Synteny analysis of ADH genes in S. bicolor and S. spontaneum. b Synteny analysis of ADH genes in S. bicolor and R570. c Synteny analysis of ADH genes in R570 and S. spontaneum. Gray lines in the background indicate the collinear blocks within S. bicolor, R570, and S. spontaneum; red lines highlight the syntenic ADH gene pairs
The Ka/Ks ratios of the ADH gene pairs were calculated to study the evolutionary constraints acting on the ADH gene family (Supplemental Table S3). Only the SsADH-like11/ShADH-like4 gene pair had Ka/Ks > 1. The other orthologous ADH gene pairs had Ka/Ks < 1, suggesting that the ADH gene family might have experienced strong purifying selective pressure during evolution.
Identification and sequence characteristics of ScADH3
AtADH1 (GenBank Acc No. NM_106362.3) is a commonly studied ADH gene. It plays a critical role in response to hypoxic conditions [32], low temperatures, [33] osmotic stress, [34], and pathogen infection [14]. In this study, the full coding sequence of ScADH (GenBank Acc No. MN_879279), which is homologous to AtADH1, was isolated from Saccharum spp. hybrid (ROC22) by reverse transcription-polymerase chain reaction (RT-PCR). Percentage of identity between 6 ShADHs, 32 SsADHs and 1 ScADH showed that this ScADH had high homology with SsADH3a (98.94%), SsADH3b (98.42%), SsADH3c (99.47%), SsADH3d (99.47%), and SsADH3d (98.14%) (Supplemental Table S4). This ScADH was designated as ScADH3. The length of the ORF in ScADH3 was 1137 bp, and it encoded polypeptide of 379 amino acids (Fig. 7). The protein primary structure analysis showed that MW, pI, GRAVY, and instability index (II) of the ScADH3 protein were 40.82 kDa, 6.03, − 0.001, and 29.54. These values suggest that ScADH3 is an acid stable basic hydrophilic protein (Supplemental Table S5). The results of protein secondary structures showed that ScADH3 mainly consisted of a random coil (50.13%), alpha helix (25.07%), and extension chains (24.80%) (Supplemental Table S6). The amino acid sequence of ScADH3 contains a Zn1-binding signature [GHE(X)2G(X)5G(X)2 V], a Zn2 structural motif [GD(X)9,10C(X)2C(X)2C(X)7C], and an NADPH-binding domain [GXG(X)2G] motif (so-called Rossmannfold) (Fig. 7).

Complete cDNA and deduced amino acid sequences of the ScADH3 gene. The full length cDNA was 1452 bp with an ORF encoding 379 amino acids. The purple-shaded portion shows the Zn binding motif, and the green-shaded portion indicates the NADPH binding motif. The underlined sequences show the specific amplification primer pair for ScADH3. * represents the stop codon
Expression of ScADH3 in response to biotic and abiotic stresses
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to detect the expression of ScADH3 under Sporisorium scitamineum, ABA, sodium chloride (NaCl), polyethylene glycol (PEG), and cold (4 °C) stresses (Fig. 8). The expression of ScADH3 was upregulated at 24 h and 48 h, but inhibited at 72 h under S. scitamineum stress. Under ABA and NaCl stresses, ScADH3 was upregulated at all treatment time points. Under PEG stress, the transcript of ScADH3 was induced at 6 h and 12 h. In response to 4 °C stress, the expression of ScADH3 was upregulated at 24 h and 48 h. Of great interest is that the expression of ScADH3 was upregulated at all time points in response to cold stress. Therefore, we were attempted to validate its function in prokaryotic (E. coli) and eukaryotic (N. benthamiana) cells in the following experiments.

qRT-PCR expression analysis of the ScADH3 gene in sugarcane ROC22 plantlets after treatment with Sporisorium scitamineum, 100 μM ABA, 25% polyethylene glycol (PEG) 8000, 250 mM NaCl, and 4 °C. The expression levels of GAPDH were used for normalization. All data points shown are mean ± SE (n = 3). Different lowercase letters indicate a significant difference, as determined by Duncan’s new multiple range test (p < 0.05)
Overexpression of ScADH3 enhances the cold tolerance of E. coli cells
E. coli cells were transformed with the empty vector of pEZYHb or the recombinant vector of pEZYHb-ScADH3 and used to study the tolerance of ScADH3 to cold stress (Fig. 9). In normal conditions, the E. coli cells of pEZYHb or pEZYHb-ScADH3 showed similar and normal growth on solid LB medium. Under cold stress, pEZYHb-ScADH3-transformed bacterial cells with 10− 4-fold dilutions had increased numbers and better survival compared to the untransformed cells, especially after 14 days treatment. These results suggested that ScADH3 may help enhance the tolerance of E. coli cells to cold stress.

Spot assays used for monitoring the growth performance of BL21/pEZYHb and BL21/pEZYHb-ScADH3 transformed E. coli cells on LB plates under cold stress. The growth performance of BL21/pEZYHb and BL21/ pEZYHb-ScADH3 cells on LB plates without supplements or stresses were used as controls. After spotting the sample on LB agar plates, incubation at 4 °C in darkness for 3 days, 7 days, and 14 days was used to study the tolerance of BL21/pEZYHb and BL21/ pEZYHb- ScADH3 cells under cold stress
Overexpression of ScADH3 improves the cold tolerance of transgenic N. benthamiana
Transgenic N. benthamiana overexpressing ScADH3 was used for additional analysis of ScADH3 function in cold tolerance. The phenotypes of wild type (WT) and transgenic N. benthamiana were similar (Fig. 10a). However, in the cold treatment, the WT had a more severe water-soaking phenotype than the transgenic plants. Excessive ROS will cause oxidative stress, which will negatively affect cell integrity [35]. Histochemical staining showed that the WT leaves showed deeper staining by DAB than those of the transgenic plants (Fig. 10b). This indicates that the transgenic plants had lower levels of ROS in response to cold stress. To study the molecular mechanisms underlying the enhanced cold tolerance by overexpressing ScADH3, qRT-PCR was used to detect the mRNA abundance of the ROS-related genes NtSOD, NtPOD, and NtCAT in the WT and transgenic lines. Except for NtSOD, transcript levels of NtPOD and NtCAT were higher in the transgenic lines than in the WT (Fig. 10c–e), indicating that overexpression of ScADH3 led to an up-regulation of the ROS-related genes. These results demonstrated that overexpression of ScADH3 improved cold tolerance of the transgenic tobacco plants.

Overexpression of ScADH3 enhances the cold tolerance of transgenic tobacco. a Phenotypes of 2-month-old plants of transgenic line (OE) and the wild type (WT) before and after freezing treatment (− 2 °C for 3 h). b Histochemical staining with DAB (upper panels) for detection of in situ accumulation of H2O2 in the transgenic lines (OE) and the wild type (WT) before and after cold treatment. c-e are qRT-PCR expression analysis of NtPOD (c), NtSOD (d), and NtCAT (e) in the (OE) and the wild type (WT). Ubiquitin was used as the reference gene. All data points shown are mean ± SE (n = 3). Different lowercase letters indicate a significant difference, as determined by Duncan’s new multiple range test (p < 0.05)
Discussion
Alcohol dehydrogenases (ADHs, alcohol: NAD+ oxidoreductase, EC 1.1.1.1) can utilize NADH as a reducing agent to catalyze the reduction of acetaldehyde to ethanol [36, 37]. ADH genes are involved in a wide range of metabolic processes including hypoxia (CH3CHO detoxification) [2], C-balance (PDH bypass) [38, 39], and scent [40, 41]. This study identified and analyzed the evolution and function of ADHs.
Evolution of ADH gene family in plants
We identified 151 ADH genes, including 43 in five dicots, 95 in nine monocots, 7 in A. trichopoda, and 6 in two bryophytes. No ADH-P gene was identified in algae (C.s crispus, C. merolae, and G. sulphuraria). However, one Class III ADH gene was obtained from all these three species. The ADH-P gene family originated from Class III ADH [2]. It is thought that the ADH-P gene may only be present in terrestrial plants. The copy number of the ADH gene in plants varies widely but mostly ranges from 6 to 10. The largest number (32) was found in S. spontaneum. P. patens had the smallest number (2). The results indicated that the ADH gene family has undergone several rounds of gene duplication events during its evolution. The phylogenetic tree suggests that the origin of the ADH gene can be traced back to a common ancestor of lower plants and higher plants before their divergence. The formation of actual multigene families in plants was probably driven by duplication events.
As the phylogenetic tree of the examined plants indicated, the ADH gene families were phylogenetically clustered into four groups (Groups A-1, A-2, B-1, and B-2). The duplication event (D1) resulted in the separation of Rhodophyta and other embryophytes (Figure S1). The Group A-1 and A-2 only contained all the representative plants whereas the Group B-1 and B-2 only contained the seed plants. The results implied that B-1 and B-2 may have divided before the divergence of seed plants, and the mosses might have lost ADHs belonging to these groups during their evolution. WGDs are considered to be important evolutionary events [42]. Some WGDs, associated with the origin of the recent common ancestor of extant angiosperms (ε), pan-core eudicots (γ), and monocots (ρ), have been revealed by integrated synteny, age estimates of gene duplication, and phylogenomic analysis [43, 44]. WGD, occurring at 150–270 Mya, is considered to be the most recent evolutionary event and produced terrestrial plants, which are the common ancestors of current angiosperms [43,44,45]. A. trichopoda is strongly supported as the single living representative species of the sister lineage to all other extant flowering plants. It provides a unique reference for inferring the genome content and structure of the most recent common ancestor of living angiosperms [46]. In B-1 and B-2, the divergence appears to have occurred before the emergence of A. trichopoda, indicating that all these ADH genes experienced duplication. There is a clear separation between the monocot and eudicot lineages in the four groups. These results suggest that additional duplications occurred subsequently to the angiosperm radiation. It also suggests that before monocots and eudicots split (165 Mya), the ADH genes may have undergone divergence or functional specialization.
Comparative genomics of ADH gene family in S. bicolor, S. spontaneum and R570
Within the three focal genomic references (S. bicolor, S. spontaneum and R570), the genomic features of the predicted ADHs specifically with regard to their motif, gene structure, chromosomal arrangement, duplicated types, and synteny were investigated.
Substantial differentiations may have occurred during the evolution of gene families. The divergence of their protein sequences is usually used to study their phylogenetic topology and reveal their evolutionary process. Similarly, gene structure is commonly used to investigate and infer evolutionary processes [47, 48]. In this study, the gene structures were analyzed to study the structural characteristics and evolution of the ADH genes (Fig. 3). Among S. bicolor, S. spontaneum and R570, the intron number of ADH genes ranged from 0 to 16, and 26 of the 47 ADH genes had nine introns. In the common ancestor, the standard number of introns in plant ADH genes is nine, and these are located at similar positions in ADH genes within the entire plant kingdom [2]. In melon, Jin et al. (2016) found that the intron numbers of ADH genes ranged from 2 to17 per gene [5]. In Gossypium, the ADHB, C and D loci carried genes with nine introns [49]. We speculate that a basic gene model consisting of nine introns was the structure of ancestral ADH genes. Based on phylogenetic analysis, we infer that the age, in duplicated descending order, for the four groups is A-1, A-2, B-1, and B-2 (Figure S1). Compared to other groups, the exon/intron organization in B-2 genes is more diverse, which may because ADHs in Group B-2 mainly originated from recent single-gene duplications rather than ancient WGD events. The number of introns of ADH genes in B-2 varied, suggesting that the insertion and/or loss of introns, the gain of an intron or fusion of exons all might have occurred during evolution.
Chromosome distribution analysis demonstrated that ADHs (SbADHs, SsADHs, and ShADHs) in the three reference genomes were unevenly distributed on chromosomes 1, 2, 4, and 5, respectively. We suggest that the chromosome distributions in these three plant species are conserved. Synteny analysis showed that there are at least four collinear blocks between each pair in the three species. Previous studies showed that inversions and rearrangements could be predicted among chromosomes Ss2 (A, B, C, and D) of AP85–441 and Sb5 of sorghum [27]. However, SsADH3a, SsADH3b, SsADH4a, SsADH4f, and SbADH2 were not located in the rearranged chromosomal regions. Rody et al. [22] found that the genes that are located in the non-arranged chromosomal regions between AP85–441 and sorghum may represent conserved sources of disease resistance in sugarcane and sorghum. The disease resistance function of ADH requires further study.
Dispersed, tandem, proximal, and WGD/segmental duplications were identified among the ADH genes within the three focal genomic references. These four gene duplication events guided the evolution of gene families encoding proteins at the gene and chromosomal levels [50]. A tandem duplication is the result of a single unequal crossing event and/or multiple iterations during DNA repair [51]. Many plant gene families involved in plant disease resistance and glucosinolate biosynthesis have copy number variations caused by tandem duplication [52]. In our study, tandem duplication appeared to be the major force in the gene expansion of SbADHs. Dispersed duplication as a small-scale gene duplication event, also played an important role in gene family expansion [53]. About 67% of the ShADHs in R570 were expanded by dispersed duplication. WGD is a large-scale duplication event. Almost all angiosperms have experienced at least one WGD [43]. In S. spontaneum, more than half of SsADHs were related to WGD/segmental duplication. The results showed that tandem duplication, dispersed duplication, and WGD/segmental duplication were the main driving forces for the expansion of the ADH gene family in S. bicolor, S. spontaneum and R570, respectively.
The function of ScADH3 under various stresses
ScADH3, with an ORF length of 1137 bp, was cloned by RT-PCR amplification. This gene encodes a 379 amino acid polypeptide. Sequence analysis showed that ScADH3 contained a Zn1-binding signature, the Zn2 structural motif and the NADPH-binding domain motif (so-called Rossmannfold). The results suggest that ScADH3 appears to be a zinc-dependent ADH belonging to the plant ADH protein family and the medium-chain dehydrogenase/reductase (MDR) superfamily [54, 55].
qRT-PCR analysis suggested that ScADH3 responds to a variety of stresses. Pathuri et al. [56] found that Blumeria graminis f.sp. hordei can induce an increase of ADH enzyme activity. Under S. scitamineum attack, transcripts of ScADH3 in this study were induced at 24 h and 48 h. Infection by pathogenic bacteria can possibly induce the expression of ADH genes and increase ADH enzyme activity. ABA helps to regulate adaptive responses to stresses [57]. ADH1 was used as a stress responsive marker gene, especially in the ABA-responsive signaling pathway in Arabidopsis [58]. We also found that ScADH3 was induced by ABA, and its expression pattern was similar to that of the ADH gene in Arabidopsis [14]. ScADH3 is also up-regulated under abiotic stresses (PEG, NaCl, and 4 °C). These results are consistent with studies on other species showing that the expression of AtADH1 is up-regulated by salt, dehydration [18, 36, 59], and cold stress [8, 9, 60]. Based on these results, we believe that ScADH3 may be involved in the abiotic and biotic stress responses of sugarcane, especially under cold stress, since the expression of this gene was continually upregulated by 4 °C stress.
The ADH gene is one of the most common cold-induced genes in cereals and Arabidopsis [9]. Considering the qRT-PCR expression analysis results, we performed cold stress experiments in transformed E. coli and N. benthamiana overexpressing the ScADH3 gene. Prokaryotic expression analysis showed that ScADH3 enhanced the tolerance of E. coli cells to cold (4 °C) stress. This suggested that even when ScADH3 is expressed in prokaryotes, it can play a positive role in the response to low temperature stress. In a eukaryotic expression system, after exposure to freezing, transgenic tobacco plants had less foliar damage than wild-type tobacco. The level of ROS is an indicator of the magnitude of stress severity and stress tolerance. A lower level of ROS following stress exposure is an indicator of improved tolerance [61]. Therefore, detecting the level of ROS in plants can increase our understanding of plant tolerance. Histochemical staining clearly showed that, after cold treatment, the transgenic ScADH3 overexpressing tobacco accumulated less H2O2 than the WT. Lower ROS levels may explain why transgenic plants have increased ability to withstand damage from cold stress. The balance between ROS generation and scavenging is the major determinant of ROS homeostasis during stress [62, 63]. Hence the transcription levels of three hydrogen peroxide genes (NtPOD, NtSOD, and NtCAT) were detected to study the molecular mechanisms of transgenic and wild-type plants under cold stress. Under cold stress, the transcript expression of two hydrogen peroxide genes (NtPOD and NtCAT) in the transgenic lines was significantly higher than that in the WT. This suggests that the transgenic plants may be better able to eliminate ROS. This also explains why the accumulation of ROS in transgenic lines is low. In summary, these results indicate that ScADH3 may control ROS accumulation by regulating antioxidant-scavenging to achieve enhanced cold resistance.
Conclusions
We identified 151 ADH proteins in 17 plant genomes (five eudicots, nine monocots, one basal angiosperm, and two mosses). Phylogenetic analysis showed that ADH genes subfamilies of ADH genes underwent distinct gene duplication patterns during angiosperm evolution. Comparative genomics analysis demonstrated the close evolutionary relationship of ADH gene families in S. bicolor, R570 and S. spontaneum. Function analysis implied that ScADH3 is involved in cold tolerance, which involved modulation of the homeostasis of reactive oxygen species (ROS) by regulating ROS-related genes. These findings reveal evolutionary and functional aspects of the ADH genes in sugarcane and identify genes that may be useful for genetic manipulation.
Methods
Plant materials and treatments
The sugarcane cultivar ROC22 (Saccharum spp. hybrid) was used for gene cloning and gene expression analysis. This was provided by the Key Laboratory of Sugarcane Biology and Genetic Breeding, Ministry of Agriculture (Fuzhou, China).
To study the expression of the sugarcane ScADH3 gene in response to ABA, abiotic, and biotic stress, uniform four-month-old tissue cultured plantlets of sugarcane were grown in water for 1 week at 28 °C with a 16:8 h (L:D) photoperiod. For biotic stress, ROC22 was inoculated with 0.5 μL of a 0.01% (v/v) Tween-20 suspension containing 5 × 106 smut spores·mL− 1; the control was inoculated with 0.01% (v/v) Tween-20 in sterile distilled water [64,65,66]. Five buds were randomly selected at 0 h, 24 h, 48 h, and 72 h after inoculation. For abiotic stresses, under 25% PEG 8000 and 250 mM NaCl treatments, the samples were collected at 0 h, 6 h, 12 h, and 24 h. For the plantlets under 4 °C treatment, sampling times of 0 h, 6 h, 24 h, and 48 h were established. For exogenous hormone stress, the whole plantlets treated by 100 μM ABA were harvested at 0 h, 3 h, 6 h, and 12 h. Three biological replicates were prepared for each treatment. All of the harvested plant tissues were frozen in liquid nitrogen and stored at − 80 °C until total RNA extraction.
Identification of ADH family genes and conserved residue analysis in plant genomes
Twenty sequenced plant genomes were collected and screened for ADH genes to obtain a representation of the major plant lineage (Supplementary Table S7). Among them, 15 plant genomes were obtained from Phytozome (https://phytozome.jgi.doe.gov/) [67]. The genome data of S. spontaneum were downloaded from the following link: http://www.life.illinois.edu/ming/downloads/Spontaneum_genome/ [27]. The monoploid reference R570 genome came from the Sugarcane Genome Hub (http://sugarcane-genome.cirad.fr/) [26]. Three genomes of Rhodophyta were collected from Ensembl (http://plants.ensembl.org/index.html) [68]. The HMM profile of ADH_N (PF08240) was downloaded from the Pfam protein family database (Pfam; http://pfam.sanger.ac.uk/) [69]. Using HMMER v3 [70] with the Hidden Markov Model (HMM) corresponding to the raw ADH_N, ADH protein candidates from the 20 plant genomes were obtained. The first transcript isoform was selected if two or more transcripts were annotated for the same gene from alternative splicing. Putative ADH protein sequences were submitted to CDD (https://www.ncbi.nlm.nih.gov/cdd) [71] to confirm the domain. Finally, sequences with complete domains were preserved. ExPASy (http://web.expasy.org/protparam/) was used to predict the basic properties (MW, pI, GRAVY, and instability index) of these ADH genes encoded proteins. Subcellular localizations were predicted by WoLF PSORT (https://wolfpsort.hgc.jp/).
Multiple sequence alignment and phylogenetic analysis
MUSCLE v3.7 [72] was used to conduct a protein multiple sequence alignment (MSA) analysis with default parameters. The MSA was used to generate a ML phylogenetic tree with the RaxML-HPC tool [73]. The bootstrap value was set to 1000 repetitions. The resulting treefile was visualized with FigTree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and EvolView (https://www.evolgenius.info/evolview/#login) [74].
Chromosomal distribution and gene duplication
MapGene2Chrom (MG2C) software (http://mg2c.iask.in/mg2c_v2.1/) was applied to map the chromosomal positions of the ADH genes in S. bicolor, R570 and S. spontaneum. Synteny block and gene duplication events were determined and analyzed by the MCScanX with the default parameters [75]. The Ka/Ks was calculated by TBtools [76].
Determination of protein motif distribution and gene structure
The online MEME program (http://meme-suite.org/tools/meme) (Bailey et al., 2006) was used to analyze protein motifs with the parameters of maximum motif number with 20, minimum motif width with 6, maximum motif width with 50, and distribution of motif occurrences with zero or one per sequence. The protein motifs and gene structures were drawn by iTOL (https://itol.embl.de/).
Gene isolation and protein structure analysis
The total RNA of all samples was extracted using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA). According to manufacturer specifications, first-strand cDNA synthesis was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Fermentas, Shanghai, China). For qRT-PCR analysis, the Prime-Script™ RT Reagent Kit (Perfect For Real Time) (TaKaRa, Dalian China) was used to perform first-strand cDNA synthesis.
The sequence of a putative ScADH3 was obtained from our previous RNA sequencing data in sugarcane [77]. The primers for ScADH3 were designed by the NCBI primer designing tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Supplementary Table S8). Based on the specifications for Ex Taq (TaKaRa, Dalian, China), RT-PCR reaction was constructed. The first-strand cDNA of ROC22 was chosen as the amplification template. The amplification reaction was 94 °C for 4 min; 94 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min 20 s, 35 cycles; and 72 °C for 10 min. Gel-purification of the production of RT-PCR and cloning into pMD19-T vector (TaKaRa, Dalian, China) was then conducted. The recombinant plasmid pMD19-T-ScADH3 was transformed into E. coli DH5α competent cells and sequenced (Sangon, Shanghai, China). Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used to calculate the percent identity matrix between 6 ShADHs, 32 SsADHs and ScADH3. The primary and secondary structure analysis of ScADH3 was predicted by ExPASy (http://web.expasy.org/protparam/) and prabi (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_gor4.html), respectively.
Expression pattern under S. scitamineum, ABA, NaCl, PEG, and 4 °C stresses
The relative expression levels of ScADH3 under different exogenous stresses were detected using qRT-PCR. Beacon Designer 8.12 software was employed to design the qRT-PCR primers of ScADH3. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was selected as the reference gene [78, 79]. The qRT-PCR reaction system (SYBR Green Master Mix: 10 μL, 10 μM forward and reverse primers: 0.8 μL, 20 × diluted cDNA template: 1.0 μL, and sterile distilled water: 7.4 μL) was constructed with reference to the manual of SYBR Green Master Mix (TaKaRa). The reaction procedure was as follows: 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The 2-ΔΔCt method was used to normalize the relative expression level of qRT-PCR data [80]. Each of the samples had three biological replicates. Three technical replicates were performed. All primers used in qRT-PCR are listed in Supplementary Table S8. Data Processing System v9.50 software (China) was used to conduct the statistical analysis. Data were expressed as the mean ± standard error (SE), Significance (p < 0.05) was calculated using one-way ANOVA, followed by Duncan’s new multiple range test.
Cold stress tolerance assay using transformed E. coli BL21 (DE3) cells
Prokaryotic expression in E. coli BL21 (DE3) cells is usually employed to study the function of genes under abiotic stresses [64, 81,82,83,84]. In this study, the prokaryotic expressive vector of pEZYHb-ScADH3 was constructed by LR ClonaseTM II Enzyme Mix (Invitrogen) and transformed to E. coli BL21 (DE3) competent cells. E. coli BL21 with the empty vector pEZYHb was used as the control. When E. coli BL21 cells containing different vectors were grown to OD600 of 0.6, 1.0 mmol L− 1 isopropyl β-D-thiogalactoside (IPTG) was added to induce protein production. After continuous growth at 37 °C for 12 h, we adjusted the concentration of the culture to OD600 of 0.6 and used LB medium to dilute the samples by 10− 3- and 10− 4-fold. Finally, 10 μL from each of the 10− 3- and 10− 4-fold dilutions of the sample was spotted on LB agar plates. For the cold tolerance assay, samples were spotted on LB agar plates, and they were placed at 4 °C in darkness. After 3 days, 7 days, and 14 day at 4 °C, the plates were cultured at 37 °C overnight and then photographed.
Cold tolerance assay of N. benthamiana overexpressing ScADH3 gene
The overexpression vector pEarleyGate 203-ScADH3, which contained the 35S promoter, was constructed by gateway cloning technique and then transformed into Agrobacterium strain GV1301 competent cells. For transformation into N. benthamiana, the leaf-disc method was employed [85]. Transgenic lines were selected by glufosinate ammonium and further confirmed by RT-PCR amplification.
Seeds of three transgenic tobacco lines (OE) at the T2 generation were germinated and selected on 1/2 Murashige and Skoog (MS) medium with glufosinate ammonium and the WT were germinated on 1/2 MS medium. One month later, the seedlings were transferred to soil pots and kept in a growth chamber (16:8 (L:D) photoperiod, and 25 °C). The cold treatment method was similar to that used by Geng and Liu [61] with slight modifications. We used two-month-old tobacco plants (OE and WT) that were subjected to − 2 °C for 3 h. The leaves were collected after cold treatment for phenotypic observation, physiological measurement and gene expression analysis. To characterize the function of ScADH3, the phenotypic variations were observed by comparing the wild-type and the transgenic plants. We judged the tolerance of tobacco to cold stress based on the water-soaking degree of leaves [61]. Histochemical staining with 3, 3′-diaminobenzidine (DAB) was used to detect the accumulation of H2O2 in tobacco leaves [86, 87]. Briefly, freshly prepared solutions of 1 mg ml− 1 DAB (in 50 mM potassium phosphate, pH 3.8) were used to incubate the leaves. After incubation for 12 h in darkness at room temperature, the chlorophyll was removed with 75% ethanol in a boiling water bath, and the leaves were then photographed. Transcript analyses of the three tobacco hydrogen peroxide-related genes (NtPOD, NtSOD, and NtCAT) were conducted on the treated N. benthamiana leaves according to Geng and Liu [61], and the NtUbiquitin was treated as a reference gene [61]. The 2-ΔΔCt method was used to normalize the relative expression level of qRT-PCR data [80]. RT-PCR was used to detect whether ScADH3 had been overexpressed in N. benthamiana, with the RNA of treated leaves and ScADH3 specific primers. The NtEF1-α was treated as control. The RT-PCR procedure was 94 °C for 4 min; 94 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min 30 s, 35 cycles; and 72 °C for 10 min. Each of the samples had three biological replicates. Three technical replicates were performed. All primers used in qRT-PCR are listed in Supplementary Table S8. All experiments had three biological replicates. All samples, after treatment, were frozen in liquid nitrogen and stored at − 80 °C.
Availability of data and materials
The data supporting the conclusions of this article are within the paper.
Abbreviations
- ADH:
-
Alcohol dehydrogenases
- WGD:
-
Whole-genome duplication
- ROS:
-
Reactive oxygen species
- ABA:
-
Abscisic acid
- PEG:
-
Polyethylene glycol
- NaCl:
-
Sodium chloride
- SA:
-
Salicylic acid
- MeJA:
-
Methyl jasmonate
- MW:
-
Molecular weight
- pI:
-
isoelectric points
- GRAVY:
-
Grand average of hydropathicity
- Mos:
-
Mosses
- Bal:
-
Basal angiosperms
- Mon:
-
Monocots
- Eud:
-
Eudicots
- MDR:
-
Medium-chain dehydrogenase/reductase
- HMM:
-
Hidden Markov Model
- MSA:
-
Multiple sequence alignment
- MG2C:
-
MapGene2Chrom
- MCScanX:
-
Multiple Collinearity Scan toolkit
- IPTG:
-
Isopropyl β-D-thiogalactoside
- OE:
-
Transgenic tobacco lines
- WT:
-
Wild type
- MS:
-
Murashige and Skoog
- DAB:
-
3, 3′-diaminobenzidine
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- qRT-PCR:
-
quantitative reverse transcription polymerase chain reaction
- SE:
-
Standard error
References
Singh RK, Srivastava S, Chidley HG, Nath P, Sane VA. Overexpression of mango alcohol dehydrogenase (MiADH1) mimics hypoxia in transgenic tomato and alters fruit flavor components. Agri Gene. 2018;7:23–33.
Strommer J. The plant ADH gene family. Plant J. 2011;66(1):128.
Hanson AD, Jacobsen JV, Zwar JA. Regulated expression of three alcohol dehydrogenase genes in barley aleurone layers. Plant Physiol. 1984;75(3):573–81.
MacNicol PK, Jacobsen JV. Regulation of alcohol dehydrogenase gene expression in barley aleurone by gibberellin and abscisic acid. Physiol Plant. 2001;111(4):533–9.
Jin Y, Zhang C, Liu W, Tang Y, Qi H, Chen H, Cao S. The alcohol dehydrogenase gene family in melon (Cucumis melo L.): bioinformatic analysis and expression patterns. Front Plant Sci. 2016;7:670.
Bucher M, Brander KA, Sbicego S, Mandel T, Kuhlemeier C. Aerobic fermentation in tobacco pollen. Plant Mol Biol. 1995;28(4):739–50.
Bailey-Serres J, Voesenek LA. Flooding stress: acclimations and genetic diversity. Annu Rev Plant Biol. 2008;59:313–39.
Diab A, Kantety R, Ozturk Gokce N, Benscher D, Nachit M, Sorrells M. Drought-inducible genes and differentially expressed sequence tags associated with components of drought tolerance in durum wheat. Sci Res Essays. 2008;3:9–26.
Lindlöf A, Bräutigam M, Chawade A, Olsson B, Olsson O. Identification of cold-induced genes in cereal crops and Arabidopsis through comparative analysis of multiple EST sets. In: Hochreiter S, Wagner R, editors. Bioinformatics Research and Development. Berlin, Heidelberg: Springer Berlin Heidelberg; 2007. p. 48–65.
Yi SY, Ku SS, Sim H-J, Kim S-K, Park JH, Lyu JI, So EJ, Choi SY, Kim J, Ahn MS, et al. An alcohol dehydrogenase gene from Synechocystis sp. confers salt tolerance in transgenic tobacco. Front Plant Sci. 2017;8:1965.
Hageman RH, Flesher D. The effect of an anaerobic environment on the activity of alcohol dehydrogenase and other enzymes of corn seedlings. Arch Biochem Biophys. 1960;87(2):203–9.
Speirs J, Lee E, Holt K, Yong-Duk K, Steele Scott N, Loveys B, Schuch W. Genetic manipulation of alcohol dehydrogenase levels in ripening tomato fruit affects the balance of some flavor aldehydes and alcohols. Plant Physiol. 1998;117(3):1047–58.
Tesniere C, Verries C. Molecular cloning and expression of cDNAs encoding alcohol dehydrogenases from Vitis vinifera L. during berry development. Plant Sci. 2000;157(1):77–88.
Shi H, Liu W, Yao Y, Wei Y, Chan Z. Alcohol dehydrogenase 1 (ADH1) confers both abiotic and biotic stress resistance in Arabidopsis. Plant Sci. 2017;262:24–31.
Davik J, Koehler G, From B, Torp T, Rohloff J, Eidem P, Wilson RC, Sonsteby A, Randall SK, Alsheikh M. Dehydrin, alcohol dehydrogenase, and central metabolite levels are associated with cold tolerance in diploid strawberry (Fragaria spp.). Planta. 2013;237(1):265–77.
Kato-Noguchi H. Osmotic stress increases alcohol dehydrogenase activity in maize seedlings. Biol Plant. 2000;43(4):621–4.
Kato-Noguchi H. Wounding stress induces alcohol dehydrogenase in maize and lettuce seedlings. Plant Growth Regul. 2001;35(3):285–8.
Dolferus R, Jacobs M, Peacock WJ, Dennis ES. Differential interactions of promoter elements in stress responses of the Arabidopsis Adh gene. Plant Physiol. 1994;105(4):1075–87.
Sobhanian H, Razavizadeh R, Nanjo Y, Ehsanpour AA, Jazii FR, Motamed N, Komatsu S. Proteome analysis of soybean leaves, hypocotyls and roots under salt stress. Proteome Sci. 2010;8(1):19.
Chattopadhyay A, Subba P, Pandey A, Bhushan D, Kumar R, Datta A, Chakraborty S, Chakraborty N. Analysis of the grasspea proteome and identification of stress-responsive proteins upon exposure to high salinity, low temperature, and abscisic acid treatment. Phytochemistry. 2011;72(10):1293–307.
Manak MS, Paul AL, Sehnke PC, Ferl RJ. Remote sensing of gene expression in Planta: transgenic plants as monitors of exogenous stress perception in extraterrestrial environments. Life Support Biosph Sci. 2002;8(2):83–91.
Rody HVS, Bombardelli RGH, Creste S, Camargo LEA, Van Sluys M-A, Monteiro-Vitorello CB. Genome survey of resistance gene analogs in sugarcane: genomic features and differential expression of the innate immune system from a smut-resistant genotype. BMC Genomics. 2019;20(1):809.
D'Hont A, Ison D, Alix K, Roux C. Determination of basic chromosome numbers in the genus Saccharum by physical mapping of ribosomal RNA genes. Genome. 2011;41:221–5.
Patade VY, Bhargava S, Suprasanna P. Salt and drought tolerance of sugarcane under iso-osmotic salt and water stress: growth, osmolytes accumulation, and antioxidant defense. J Plant Interact. 2011;6(4):275–82.
Ming R, Moore P, Wu K-K, D'Hont A, Tew T, Mirkov T, Da Silva J, Jifon J, Rai M, Schnell R, et al. Sugarcane improvement through breeding and biotechnology. In: Janick J, editor. Plant Breeding Reviews vol. 27, pp. 15-118. Hoboken: Wiley; 2005. https://doi.org/10.1002/9780470650349.ch2.
Garsmeur O, Droc G, Antonise R, Grimwood J, Potier B, Aitken K, Jenkins J, Martin G, Charron C, Hervouet C, et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat Commun. 2018;9(1):2638.
Zhang J, Zhang X, Tang H, Zhang Q, Hua X, Ma X, Zhu F, Jones T, Zhu X, Bowers J, et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat Genet. 2018;50(11):1565–73.
Jain R, Singh SP, Singh A, Singh S, Chandra A, Solomon S. Response of foliar application of nitrogen compounds on sugarcane grown under waterlogging stress. Sugar Tech. 2016;18(4):433–6.
Huang N, Ling H, Zhang X, Mao H, Su Y, Su W, Liu F, Xu L, Chen R, Que Y. A small GTP-binding gene scran from sugarcane is involved in responses to various hormone stresses and Sporisirium scitamineum challenge. Sugar Tech. 2018;20:669–80.
Su W, Huang N, Ling H, Liu F, Zeng R, Su Y, Wu Q, Gao S, Que Y. Cloning and expression of ScADH from sugarcane. Chin J Appl Environ Biol. 2017;23(3):474–81.
Van Bel M, Diels T, Vancaester E, Kreft L, Botzki A, Van de Peer Y, Coppens F, Vandepoele K. PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2018;46(D1):D1190–6.
Yang CY. Hydrogen peroxide controls transcriptional responses of ERF73/HRE1 and ADH1 via modulation of ethylene signaling during hypoxic stress. Planta. 2014;239(4):877–85.
Jarillo JA, Leyva A, Salinas J, Martinez-Zapater JM. Low temperature induces the accumulation of alcohol dehydrogenase mRNA in Arabidopsis thaliana, a chilling-tolerant plant. Plant Physiol. 1993;101(3):833–7.
Xiong L, Lee H, Ishitani M, Zhu JK. Regulation of osmotic stress-responsive gene expression by the LOS6/ABA1 locus in Arabidopsis. J Biol Chem. 2002;277(10):8588–96.
Choudhury FK, Rivero RM, Blumwald E, Mittler R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017;90(5):856–67.
Chang C, Meyerowitz EM. Molecular cloning and DNA sequence of the Arabidopsis thaliana alcohol dehydrogenase gene. P Natl Acad Sci USA. 1986;83(5):1408–12.
Chung HJ, Ferl RJ. Arabidopsis alcohol dehydrogenase expression in both shoots and roots is conditioned by root growth environment. Plant Physiol. 1999;121(2):429–36.
Wei Y, Lin M, Oliver DJ, Schnable PS. The roles of aldehyde dehydrogenases (ALDHs) in the PDH bypass of Arabidopsis. BMC Biochem. 2009;10:7.
Zabalza A, van Dongen JT, Froehlich A, Oliver SN, Faix B, Gupta KJ, Schmalzlin E, Igal M, Orcaray L, Royuela M, et al. Regulation of respiration and fermentation to control the plant internal oxygen concentration. Plant Physiol. 2009;149(2):1087–98.
Garabagi F, Strommer J. Distinct genes produce the alcohol dehydrogenases of pollen and maternal tissues in Petunia hybrida. Biochem Genet. 2004;42(5–6):199–208.
Garabagi F, Duns G, Strommer J. Selective recruitment of Adh genes for distinct enzymatic functions in Petunia hybrida. Plant Mol Biol. 2005;58(2):283–94.
Leitch AR, Leitch IJ. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320(5875):481.
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473(7345):97–100.
Jiao Y, Li J, Tang H, Paterson AH. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell. 2014;26(7):2792–802.
Edger PP, Pires JC. Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosom Res. 2009;17(5):699–717.
Amborella Genome P. The amborella genome and the evolution of flowering plants. Science. 2013;342(6165):1467.
Zhao M, He L, Gu Y, Wang Y, Chen Q, He C. Genome-wide analyses of a plant-specific LIM-domain gene family implicate its evolutionary role in plant diversification. Genome Biol Evol. 2014;6(4):1000–12.
Feng G, Burleigh JG, Braun EL, Mei W, Barbazuk WB. Evolution of the 3R-MYB gene family in plants. Genome Biol Evol. 2017;9(4):1013–29.
Small RL, Wendel JF. Copy number lability and evolutionary dynamics of the Adh gene family in diploid and tetraploid cotton (Gossypium). Genetics. 2000;155(4):1913–26.
Maher C, Stein L, Ware D. Evolution of Arabidopsis microRNA families through duplication events. Genome Res. 2006;16(4):510–9.
Hofberger JA, Nsibo DL, Govers F, Bouwmeester K, Schranz ME. A complex interplay of tandem- and whole-genome duplication drives expansion of the L-type lectin receptor kinase gene family in the brassicaceae. Genome Biol Evol. 2015;7(3):720–34.
Parniske M, Wulff BB, Bonnema G, Thomas CM, Jones DA, Jones JD. Homologues of the Cf-9 disease resistance gene (Hcr9s) are present at multiple loci on the short arm of tomato chromosome 1. Mol Plant-Microbe Interact. 1999;12(2):93–102.
Taylor J, Raes J. Small-scale gene duplications. In: Ryan Gregory T, editor. The evolution of the genome pp. 289–32. Amsterdam: Elsevier Academic Press; 2005. https://doi.org/10.1016/B978-012301463-4/50007-3.
Persson B, Krook M, Jornvall H. Characteristics of short-chain alcohol dehydrogenases and related enzymes. Eur J Biochem. 1991;200(2):537–43.
McKie JH, Jaouhari R, Douglas KT, Goffner D, Feuillet C, Grima-Pettenati J, Boudet AM, Baltas M, Gorrichon L. A molecular model for cinnamyl alcohol dehydrogenase, a plant aromatic alcohol dehydrogenase involved in lignification. Biochim Biophys Acta. 1993;1202(1):61–9.
Pathuri IP, Reitberger IE, Hückelhoven R, Proels RK. Alcohol dehydrogenase 1 of barley modulates susceptibility to the parasitic fungus Blumeria graminis f.sp. hordei. Plant Signal Behav. 2011;62(10):3449–57.
Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR. Abscisic acid: emergence of a core signaling network. Annu Rev Plant Biol. 2010;61:651–79.
Miura K, Lee J, Jin JB, Yoo CY, Miura T, Hasegawa PM. Sumoylation of ABI5 by the Arabidopsis SUMO E3 ligase SIZ1 negatively regulates abscisic acid signaling. P Natl Acad Sci USA. 2009;106(13):5418.
Papdi C, Abraham E, Joseph MP, Popescu C, Koncz C, Szabados L. Functional identification of Arabidopsis stress regulatory genes using the controlled cDNA overexpression system. Plant Physiol. 2008;147(2):528–42.
Christie PJ, Hahn M, Walbot V. Low-temperature accumulation of alcohol dehydrogenase-1 mRNA and protein activity in maize and rice seedlings. Plant Physiol. 1991;95(3):699–706.
Geng J, Liu JH. The transcription factor CsbHLH18 of sweet orange functions in modulation of cold tolerance and homeostasis of reactive oxygen species by regulating the antioxidant gene. J Exp Bot. 2018;69(10):2677–92.
Miller G, Suzuki N, Ciftci-Yilmaz S, Mittler R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010;33(4):453–67.
Huang XS, Wang W, Zhang Q, Liu JH. A basic helix-loop-helix transcription factor, PtrbHLH, of Poncirus trifoliata confers cold tolerance and modulates peroxidase-mediated scavenging of hydrogen peroxide. Plant Physiol. 2013;162(2):1178–94.
Liu F, Sun TT, Wang L, Su WH, Gao SW, Su YC, Xu LP, Que YX. Plant jasmonate ZIM domain genes: shedding light on structure and expression patterns of JAZ gene family in sugarcane. BMC Genomics. 2017;18(1):771.
Que Y, Xu L, Wu Q, Liu Y, Ling H, Liu Y, Zhang Y, Guo J, Su Y, Chen J, et al. Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genomics. 2014;15:996.
Huang N, Ling H, Su Y, Liu F, Xu L, Su W, Wu Q, Guo J, Gao S, Que Y. Transcriptional analysis identifies major pathways as response components to Sporisorium scitamineum stress in sugarcane. Gene. 2018;678:207–18.
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(Database issue):D1178–86.
Kersey PJ, Allen JE, Allot A, Barba M, Boddu S, Bolt BJ, Carvalho-Silva D, Christensen M, Davis P, Grabmueller C, et al. Ensembl genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018;46(D1):D802–d808.
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018;47(D1):D427–32.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37.
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45(D1):D200–d203.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Subramanian B, Gao S, Lercher MJ, Hu S, Chen WH. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019;47(W1):W270–w275.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R, TBtools - an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020. https://doi.org/10.1016/j.molp.2020.06.009.
Que Y, Su Y, Guo J, Wu Q, Xu L. A global view of transcriptome dynamics during Sporisorium scitamineum challenge in sugarcane by RNA-Seq. PLoS One. 2014;9(8):e106476.
Iskandar H, Simpson R, Casu R, Bonnett G, Maclean D, Manners J. Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Mol Biol Report. 2004;22:325–37.
Ling H, Wu QB, Guo JL, Xu LP, Que YX. Comprehensive selection of reference genes for gene expression normalization in sugarcane by real time quantitative RT-PCR. PLoS One. 2014;9(5):e97469.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–8.
Guo JL, Xu LP, Fang JP, Su YC, Fu HY, Que YX, Xu JS. A novel dirigent protein gene with highly stem-specific expression from sugarcane, response to drought, salt and oxidative stresses. Plant Cell Rep. 2012;31(10):1801–12.
Su YC, Xu LP, Xue BT, Wu QB, Guo JL, Wu LG, Que YX. Molecular cloning and characterization of two pathogenesis-related β-1,3-glucanase genes ScGluA1 and ScGluD1 from sugarcane infected by Sporisorium scitamineum. Plant Cell Rep. 2013;32(10):1503–19.
Liu F, Huang N, Wang L, Ling H, Sun TT, Ahmad W, Muhammad K, Guo JL, Xu LP, Gao SW, et al. A novel L-ascorbate peroxidase 6 gene, ScAPX6, plays an important role in the regulation of response to biotic and abiotic stresses in sugarcane. Front Plant Sci. 2018;8:2262.
Hua-Ying M, Wen-Ju W, Wei-Hua S, Ya-Chun S, Feng L, Cong-Na L, Ling W, Xu Z, Li-Ping X, You-Xiong Q. Genome-wide identification, phylogeny, and expression analysis of Sec14-like PITP gene family in sugarcane. Plant Cell Rep. 2019;38(5):637–55.
Muller AJ, Mendel RR, Schiemann J, Simoens C, Inze D. High meiotic stability of a foreign gene introduced into tobacco by Agrobacterium-mediated transformation. Mol Gen Genet. 1987;207(1):171–5.
Wang J, Sun PP, Chen CL, Wang Y, Fu XZ, Liu JH. An arginine decarboxylase gene PtADC from Poncirus trifoliata confers abiotic stress tolerance and promotes primary root growth in Arabidopsis. J Exp Bot. 2011;62(8):2899–914.
Su W, Huang L, Ling H, Mao H, Huang N, Su Y, Ren Y, Wang D, Xu L, Muhammad K, et al. Sugarcane calcineurin B-like (CBL) genes play important but versatile roles in regulation of responses to biotic and abiotic stresses. Sci Rep. 2020;10(1):167.
Acknowledgments
We are grateful to the reviewers for their helpful comments on the original manuscript. We would like to thank editors for their efficient works.
Funding
This work was funded by National Key R&D Program of China (2019YFD1000500 and 2018YFD1000503), National Natural Science Foundation of China (31871688, 31671752, 31101196 and 31340060), Natural Science Foundation of Fujian Province, China (2015 J06006), and the Sugar Crop Research System of China (CARS-17). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: WHS and YXQ. Performed the experiments: WHS, YJR, DJW and HCT. Analyzed the data: WHS, JFF and CZ. Wrote the paper: WHS. Revised the final version of the paper: LPX, YCS, KM and YXQ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. This is to confirm that no specific permits was needed for the described experiments, and this study did not involve any endangered or protected species.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Phylogenetic relationship of ADH gene family in major groups of plants.
Additional file 2: Table S1.
The detailed information of ADH genes included in this study. Table S2. The gene type of ADH genes in S. bicolor, R570, and S. spontaneum. Table S3. One-to-one orthologous relationships between S. bicolor, R570, and S. spontaneum. Table S4. Percentage of identity between 6 ShADHs, 32 SsADHs, and 1 ScADH was calculated using Clustal Omega. Table S5. Primary structure analysis of ScADH3. Table S6. Secondary structure analysis of ScADH3. Table S7. Sources of ADH genes from sequenced plant included in this study. Table S8. Primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Su, W., Ren, Y., Wang, D. et al. The alcohol dehydrogenase gene family in sugarcane and its involvement in cold stress regulation. BMC Genomics 21, 521 (2020). https://doi.org/10.1186/s12864-020-06929-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-06929-9