
Abstract
Background
Puccinia graminis f. sp. tritici (Pgt) race TTKSK and its lineage pose a threat to barley production world-wide justifying the extensive efforts to identify, clone, and characterize the rpg4-mediated resistance locus (RMRL), the only effective resistance to virulent Pgt races in the TTKSK lineage. The RMRL contains two nucleotide-binding domain and leucine-rich repeat (NLR) resistance genes, Rpg5 and HvRga1, which are required for resistance. The two NLRs have head-to-head genome architecture with one NLR, Rpg5, containing an integrated C-terminal protein kinase domain, characteristic of an “integrated sensory domain” resistance mechanism. Fast neutron mutagenesis of line Q21861 was utilized in a forward genetics approach to identify genetic components that function in the RMRL or Rpg1 resistance mechanisms, as Q21861 contains both genes. A mutant was identified that compromises both RMRL and Rpg1-mediated resistances and had stunted seedling roots, designated required for P. graminis resistance 9 (rpr9).
Results
The rpr9 mutant generated in the Q21861 background was crossed with the Swiss landrace Hv584, which carries RMRL but contains polymorphism across the genome compared to Q21861. To map Rpr9, a Hv584 x rpr9 F6:7 recombinant inbred line (RIL) population was developed. The RIL population was phenotyped with Pgt race QCCJB. The Hv584 x rpr9 RIL population was genotyped with the 9 k Illumina Infinium iSelect marker panel, producing 2701 polymorphic markers. A robust genetic map consisting of 563 noncosegregating markers was generated and used to map Rpr9 to an ~ 3.4 cM region on barley chromosome 3H. The NimbleGen barley exome capture array was utilized to capture rpr9 and wild type Q21861 exons, followed by Illumina sequencing. Comparative analysis, resulting in the identification of a 1.05 Mbp deletion at the chromosome 3H rpr9 locus. The identified deletion contains ten high confidence annotated genes with the best rpr9 candidates encoding a SKP1-like 9 protein and a F-box family protein.
Conclusion
Genetic mapping and exome capture rapidly identified candidate gene/s that function in RMRL and Rpg1 mediated resistance pathway/s. One or more of the identified candidate rpr9 genes are essential in the only two known effective stem rust resistance mechanisms, present in domesticated barley.
Similar content being viewed by others

Background
The obligate biotrophic fungal pathogen, Puccinia graminis, causes the disease stem rust on a broad range of primary hosts including more than 365 species of cereals and grasses. While three forma specialis of Puccinia graminis cause disease on barley [1], wheat stem rust caused by P. graminis f. sp. tritici is considered one of the most serious disease of wheat and barley, because of historic epidemics that devastated crops, especially in the Upper Midwestern US in the early to mid-twentieth century. In the mid-twentieth century wheat breeders began pyramiding several resistance (R)-genes into varieties, providing durable resistance, which ended the occurrence of major epidemics on wheat. Stem rust was also managed by genetic resistance in barley, but only a single resistance gene, Rpg1, was deployed in the Midwestern US, protecting barley cultivars since 1942 [2].
The remarkably durable Rpg1 gene was identified in 2002 by a positional cloning effort and was predicted to encode a cytoplasmic localized, constitutively expressed protein with two serine/threonine protein kinase domains, designated pK1 and pK2 [3]. The pK1 domain appeared to be a pseudokinase, lacking phosphorylation activity, whereas the pK2 domain was characterized as an active kinase. However, both domains were shown to function in stem rust resistance [4]. Interestingly, it was also shown that the RPG1 protein was systemically phosphorylated in vivo, five minutes post inoculation with avirulent stem rust urediniospores [5], but was not induced by virulent isolates. The RPG1 phosphorylation was required to elicit resistance response/s. Also, ubiquitin ligase mediated RPG1 protein degradation occurred between 20 and 24 h post inoculation (HPI) with avirulent isolates and was required to elicit the resistance response/s [6].
Although Rpg1 maintained a remarkable level of durable resistance, for a single stem rust resistance gene deployed over vast cereal crop acreage, a new pathotype of Pgt designated race QCC (later given the five letter nomenclature QCCJB) virulent on barley containing Rpg1 was identified in North Dakota in 1989 [7]. Pgt race QCCJB increased in prevalence and became one of the most common virulence types in North America causing disease epidemics [8]. The threat to barley production by this new stem rust race virulent on Rpg1 prompted the evaluation of over 18,000 barley accessions from the USDA National Small Grains collection with the best source of Pgt race QCCJB resistance discovered in the unimproved barley line Q21861 from Queensland, Australia via CMMYT [9]. Genetic studies revealed that a single recessive gene, designated rpg4, conferred the resistance. The rpg4 gene was also shown to be temperature sensitive providing resistance at lower (17–22 °C) temperatures [10], but was completely ineffective at temperatures above 27 °C. The rpg4 gene was genetically mapped to the long arm of barley chromosome 5H [11].
A strain of stem rust, Pgt race TTKSK (also called as Ug99), emerged in Uganda, Africa in 1998 [12] that was considered highly virulent because it carried a unique combination of virulence genes enabling it to infect more than 80% of the wheat grown worldwide [13, 14] and more than 97% of barley cultivars, including those having Rpg1 [15]. The threat posed by Pgt race TTKSK, mainly to wheat production, raised concerns for world food security [16]. Fortunately, for barley production, which is especially vulnerable to races virulent on commercial wheat varieties due to inoculum build up, the rpg4-mediated resistance locus (RMRL) in barley line Q21861 also confers effective resistance against Pgt race TTKSK [17] and its lineage.
Line Q21861 also contains resistance to isolates of rye stem rust, including Pgs isolate 92-MN-90, which was designated the Rpg5 gene. The single dominant resistant gene was shown to be tightly linked to rpg4 [18] and it was initially reported that rpg4 and Rpg5 could be the same gene despite their different inheritance, recessive vs dominant, respectively. The high-resolution recombinant lines used for positional cloning of Rpg5 [19, 20] and rpg4 behaved similarly to Pgt race QCCJB at the seedling stage and Pgt race TTKSK at both the seedling and adult plant stages suggesting that the same gene/s function in RMRL-mediated Pgt race QCCJB and TTKSK resistance.
Utilizing positional cloning, Rpg5 was delimited to an ~ 70 kb region of the genome harboring three tightly linked genes including two nucleotide-binding domain and leucine-rich repeat (NLR) genes and an actin depolymerization factor. Allele analysis and post transcriptional gene silencing determined that the Rpg5 rye stem rust resistance gene, one of the NLRs, contained an additional C-terminal integrated serine / threonine protein kinase (STPK) domain [21]. When Rpg5 was originally identified, it was determined to be distinct from rpg4 because high-resolution mapping based on 5232 gametes and RFLP markers showed rpg4 segregating 0.11 cM away from Rpg5 with an actin depolymerization factor, HvAdf2, reported as the best rpg4 candidate gene [19, 22]. However, genomic sequence comparison at RMRL identified several SNP markers that when used to saturate the region showed that HvAdf2 was not rpg4. The SNP marker saturation also determined that the Rpg5 gene is required for rpg4-mediated wheat stem rust resistance [21] and is the functionally polymorphic R-gene at the RMRL [20].
The cloning and characterization of RMRL determined that the two resistances, rpg4-mediated wheat stem rust and Rpg5-mediated rye stem rust resistance mechanisms depend on Rpg5 as the R-gene component [23] since its polymorphic nature in resistant and susceptible barley lines governs immune response. Another NLR gene Rga1 in the RMRL locus is also required for QCCJB resistance yet didn’t contain resistance defining sequence polymorphism. Interestingly, the recent identification of the required for rpg4-mediated resistance 1 (Rrr1), gene proximal to RMRL region was reported to be required for RMRL mediated resistance. Rrr1 is also required for Rpg1-mediated resistance in the presence of the RMRL introgression [24]. These data suggest that the RMRL and Rpg1 resistance mechanisms interact or have common signaling components. We hypothesize that the only two effective stem rust resistance genes characterized in barley to date, Rpg1 and RMRL, confer broad spectrum resistance mediating early defense response mechanisms with spatial, temporal and functional hallmarks of non-host or slow rusting resistance mechanisms [25, 26]. Both resistances appear to be non-hypersensitive response (HR) resistance mechanism (Solanki et al., unpublished) indicative of a pathogen associated molecular pattern (PAMP) triggered immunity (PTI)-like resistance response. However, Rpg1-mediated resistance was previously suggested to be a (HR) dependent resistance mechanism more indicative of an effector triggered immunity (ETI) response [7], but our recent histology data suggests otherwise (Solanki et al., unpublished).
The plant innate immunity system has been separated into distinct layers with PTI acting as the first line of defense which has been classified as an active form of non-host resistance because of the recognition of conserved microbial molecules providing resistance to broad taxa of potential pathogens. This early defense mechanism is triggered at the cell surface by recognition of extracellular PAMP effectors, that induce responses including H2O2 accumulation, pathogen related (PR) gene expression, callose deposition at the point of ingress, and sometimes a low amplitude PCD response that is typically limited to a small number of cells surrounding the infection site [27]. These general PTI responses are activated by conserved transmembrane cell surface receptor complexes known to contain pattern recognition receptors (PRRs). The PRRs typically contain an extracellular receptor domain, a transmembrane domain and an intracellular kinase-signaling domain and are known as the receptor-like kinases (RLKs). Because, the broad Rpg1-mediated resistance responses are so rapid, within minutes of avirulent spores reaching the plant surface [6] it was hypothesized that there must be a cell surface receptor or receptor complex that recognizes the pathogen and relays the message to the cytoplasm activating Rpg1 phosphorylation and protein degradation. One of the main targets of the forward genetics approach employed in this study was to identify this putative cell surface receptor or members of the complex.
Host specific pathogens counter evolved virulence effectors to evade PTI responses by masking their PAMPs from detection, as observed for the chitin binding effector proteins [28, 29], or by blocking the signaling pathways as is seen with effectors that inhibit FLS2 PRR-mediated signaling following flg22 perception [30]. Fungal pathogens appear to secrete effectors into the host cell cytoplasm, or into the apoplast as is observed with the fungal chitin binding effectors, however, characterization of these mechanisms have yet to be elucidated at the functional level. Plants counter-evolved cytoplasmic localized immunity receptors, typically with NLR protein domain architecture, that recognize the presence of these effectors and elicit the hallmark higher amplitude PCD immunity response in plants, referred to as the hypersensitive response (HR) [31]. Once an effector is recognized by a cognate NLR immunity receptor, then it becomes an avirulence protein. For biotrophic pathogens that require living host cells to feed, these effectors that elicit programmed cell death (PCD) responses no longer facilitate disease, but hinder the pathogen by eliciting the plant immunity responses [32].
Fast neutron (FN) irradiation mutagenesis results in genomic DNA deletions from 1 bp (base pair) to several Megabase [33,34,35]. A FN induced mutant in the Q21861 background was identified with compromised RMRL specific Pgt race QCCJB resistance [36] and Rpg1 specific Pgt race HKHJC resistance, designated as the required for P. graminis resistance 9 (rpr9) mutant. The rpr9 mutant still retains a functional RMRL locus and Rpg1 gene, thus, the mutant phenotype was due to a deletion within, or containing, an important gene required for RMRL and Rpg1 function. This mutant also has a stunted root phenotype, short roots at early growth stages. Mapping of the mutation/s responsible for the rpr9 and stunted root phenotypes in a Hv584 x rpr9 RIL mapping population determined that the two mutant phenotypes co segregated to the same genetic interval. Thus, our identification of the rpr9 mutant gene/s also identified candidate genes effecting the physiology of root development as well.
Here we report on the identification of ten candidate rpr9 genes via genetic mapping and exome capture using a “mapping by sequencing” approach. To rapidly identify the deletion within or containing the Rpr9 gene, the barley NimbleGen (Roche) exome capture array was utilized on the rpr9 mutant and wildtype Q21861 and comparative sequence analysis identified a 1.052 Mbp deletion in the rpr9 delimited region. Based on the barley POPSEQ positions of the genes in the deletion block and the intact genes flanking the deletion, a genetic to physical comparison identified ten high confidence genes classified into a family of four peroxidases, two NAD(P)H-quinone oxidoreductases, a SKP1-like 9 protein, a F-box family protein, a L-tyrosine decarboxylase and a RNAase protein. We propose that our top candidates encode the SKP1-like 9 and F-box proteins which were shown to work together as component of a SCF (SKP/Cullin/F-box and ring finger protein Rbx1) E3 ubiquitin ligase complex which have been shown to play roles in early plant immunity and root development pathways. RPG1 resistance relies on protein degradation after pathogen recognition, linking to the findings reported here, considering that our candidate mutant genes are components of the SCF E3 ubiquitin ligase complex and disrupt Rpg1-mediated resistance. The genetic analyses and tools developed here will facilitate the identification and functional validation of the rpr9 gene and the gene/s responsible for the stunted root phenotype.
Results
Identification of mutants that compromise RMRL and Rpg1 mediated resistance responses
The phenotyping of the Q21861 fast neutron (FN) irradiated population at the M2 generation with Pgt race QCCJB identified ten putative mutant individuals with compromised RMRL-mediated resistance that were designated as containing the required for P. graminis resistance 8–17 (rpr8-rpr17) genes. The original rpr9 M2 mutant exhibited an average susceptible infection type (IT) of 3–2 (Fig. 1a) with Pgt race QCCJB. A rpr9 M3 individual was backcrossed to wt Q21861 for four generations, which required phenotyping at each BCF2 generation with Pgt race QCCJB. The resulting BC4F2 rpr9 mutant with the background mutations cleaned up exhibited ITs ranging from 2 to 3 to 3- with a mode of 3–2 compared to the highly resistant wild type line Q21861, which showed ITs ranging from 0; to 0;12 with a mode of 0; (Fig. 1a; Table 1). The line Q21861 is resistant to Pgt race QCCJB due to the RMRL and is also resistant to Pgt race HKHJC due to the Rpg1 specific resistance [3]. Interestingly, rpr9 also exhibited susceptible ITs to Pgt race HKHJC ranging from 2, 1 to 3- with a mode of 3-, 2, 1 compared to the resistant wild type line Q21861 which shows ITs ranging from 0;1 to 0;12 with a mode of;1 (Table 1). Thus, the rpr9 mutant generated in the wheat stem rust resistant barley line Q21861 background, which carries both the Rpg1 and RMRL resistance genes [3, 21, 22, 37], is compromised for resistance (Table 1) to both Pgt race QCCJB, specifically avirulent on RMRL [17] and HKHJC that is specifically avirulent on Rpg1 [3, 19]. The Hv584/rpr9 F2:6 RIL population was phenotyping with Pgt race QCCJB (Additional file 1: Table S1) and used for rpr9 and the stunted root mutant QTL mapping rpr9 qualitative gene mapping.

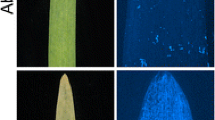
Stem rust seedling disease and root length phenotyping on wild type Q21861 and Q21861 rpr9 mutant seedlings. a Seedling disease assay on the resistant barley line Q21861-wildtype and loss of RMRL resistance mutant Q21861-rpr9. b A representative picture of three germinated seeds and their root length determination by the Ragdoll test for wildtype Q21861 and the Q21861 rpr9 mutant. The short root length phenotype was found to be associated with the rpr9 mutant compared to wild type Q21861
Identification of stunted root growth phenotype associated with rpr9
Germination of rpr9 mutant seed on petri plates lead to the observation that they exhibited a stunted root growth compared to wildtype Q21861 seed (Fig. 1b). After four rounds of backcrossing the mutant to wildtype Q21861 and selection for susceptibility to Pgt race QCCJB the stunted root phenotype was still present suggesting that it could be determined by the same mutation that compromised RMRL- and Rpg1-mediated resistance. The calculated mean root length of fourteen rpr9 mutants was 16.5 mm, whereas wt Q21861 had a mean root length of 25.64 mm. The difference in root length was found to be statistically significant (two tailed P < 0.05) between rpr9 and wt Q21861 using an unpaired t-test (Table 2). Further, root length was recorded for three seeds of each of the 81 Hv584/rpr9 RIL individuals, the parental lines rpr9 and Hv584 and wildtype Q21861. The average root length data from germinated seeds were used for QTL mapping (Fig. 2; Additional file 1: Table S1).

QTL and qualitative mapping of the Rpr9 region showing the deletion identified within the region. a The Y-axis represents LOD values and X-axis the chromosome 3H map with markers designated below. Three replications and average of Pgt race QCCJB infection types with stunted root length QTL mapping are shown. The colors representing individual reps and averages are shown in the upper right. Blue indicate flanking and red indicate markers associated with the QTL. b The seven recombinants delimiting rpr9 with Hv584/rpr9 designations are listed on the left with the Q21861-rpr9 and Hv584 parental lines and Q21861 wild type. The horizontal bars represent the genotypes of each (orange = Q21861 genotype and green = Hv584 genotype for markers listed above. The general reaction to Pgt race QCCJB is shown on the right (R = resistant and S = susceptible). Black Xs represent recombination regions. The horizontal bars above shows the cosegregating blocks of markers with the most distal and proximal rpr9 cosegregating markers based on genome sequence positions denoted with a red ovals and other markers shown as vertical black lines. Blue ovals denote flanking markers with the smallest physical interval. White ovals denote the next segregation blocks proximally and distally. All markers are shown in their approximate physical positions. The asterisk denotes the SCRI_RS_27,417 marker that is presents within the rpr9 deletion. c The deletion covering ten high confidence annotated genes, with functions listed below and the top rpr9 candidates indicated in bold lettering. The horizontal white bar represents the genome sequence of cv Morex with red arrows representing the relative positions and directionality of annotated rpr9 candidate genes. Black arrows the flanking genes that were present in both wildtype Q21861 and the rpr9 mutant delimiting the deletion. The black lines below denote the minimum (0.773 Mbp) and maximum (1.052 Mbp) deletion size. The 100 kilobase scale is shown below
Linkage mapping and QTL analysis for rpr9 and stunted root phenotype
To determine if the rpr9 phenotype was contributed by a single recessive mutation, the Swiss landrace line Hv584 that contains a functional RMRL [38] was crossed with the rpr9 mutant. Fifty-four Hv584/rpr9 F2 individuals independent from the F2 individuals advanced for the RIL population development were phenotyped with Pgt race QCCJB showing a 12 resistant: 42 susceptible ratio which fit the expected 1:3 ratio (χ2 = 0.637) for a single recessive rpr9 mutation. The recessive phenotype was observed upon fast neutron mutagenesis and deletion of the Rpr9 gene. The mapping of the rpr9 stem rust susceptibility and stunted root growth phenotype was performed utilizing the barley iSelect 9 k Infinium chip genotyping data of the Hv584/rpr9 F6 RIL population made up of 81 individuals. A robust genetic linkage map was generated with 2701 polymorphic markers distributed across the seven-barley chromosome representing ~ 30% polymorphism for the 9000 iSelect markers on the 9 k Infinium chip (Additional file 2: Table S2).
Following removal of co-segregating markers, the final linkage map used for QTL analysis consisted of 563 non-redundant loci spread across all seven barley chromosomes (Additional file 3: Figure S1). QTL analysis identified an ~ 3.4 cM region harboring the Rpr9 gene as well as the mutation resulting in the stunted root phenotype. We identified a significant QTL (LOD = 24,α0.05 = 6) on barley chromosome 3H for seedling Pgt race QCCJB resistance (Additional file 4: Figure S2) delimited by the co-segregating block represented by non-redundant flanking markers BOPA1_12_30616 (12_30616) and SCRI_RS_159006 (Fig. 2a) covering POPSEQ positions from 59.63 cM to 75.92 cM representing a physical region of ~ 59.78 Mbp from positions 525,056,438 -584,839,846 bp on chromosome 3H. This QTL harbors two SNP markers SCRI_RS_178907 (LOD = 21.99) and BOPA1_861–268 (LOD = 18) representing two different co-segregating blocks of 49 and 4 markers respectively (Additional file 2: Table S2) underlying the rpr9 region. For the fine mapping using rpr9 phenotyping as a qualitative marker seven critical recombinants (Hv584/rpr9–09, Hv584/rpr9–60, Hv584/rpr9–35, Hv584/rpr9–25, Hv584/rpr9–46, Hv584/rpr9–73 and Hv584/rpr9–70) were identified, further delimit it to the co-segregating block of 49 markers represented by SCRI_RS_178907/ BOPA1_12_31262 on the QTL map (Fig. 2a and b). The most proximal marker within the rpr9 cosegragating markers based on the genome assembly was SCRI_RS_178907 (POPSEQ position ~ 59 cM, physical start position on chr. 3H at 525,394,569 bp) and most distal markers was BOPA2_12_31262 (POPSEQ position ~ 68 cM, end position on barley physical map is chr. 3H at 567,974,409 bp) representing a physical region of ~ 57.43 Mbp on chr. 3H (Fig. 2b). This co-segregating block was flanked by the markers BOPA2_12_30616 (525,056,438 bp) 0.7 cM proximal of rpr9 and BOPA1_861_268 (582,486,654 bp) 0.7 cM distal of rpr9 delimiting the region to an ~ 57.44 Mbp region (Fig. 2b).
Interestingly, the only significant seedling short root QTL (LOD = 6, α0.05 = 5.74) was also flanked by the markers BOPA1_12_30616 (LOD = 4.4) and BOPA1_861–268 (LOD = 4.5) on chromosome 3H (Fig. 2a, Additional file 5: Figure S3). Thus, from here forth we will refer to the rpr9 locus as governing, RMRL and Rpg1-mediated resistance as well as the stunted root phenotype.
Identification of candidate deleted genes using exome capture
As multiple wt and mutant genotypes are barcoded and sequenced in parallel on a single Illumina Nextseq flow cell, of the ~ 700 million reads generated, a total of 115,741,280 sequencing reads were identified as wildtype Q21861 and 104,846,733 reads were from the rpr9 mutant, representing a very balanced exome capture sequencing library. After QC and trimming the sequences averaged 151 bases per read. Alignment of the reads to the Morex draft genome sequence resulted in 88.79% of the wt Q21861 and 82.60% of the rpr9 reads aligning to the reference barley genome.
The pipeline that calculates captured unique sequence coverage across all exome capture targets (Additional file 6: Table S3) identified a large block of deleted genes within the rpr9 region delimited by the genetic map (Fig. 3).

A graphical representation of ratio of exome capture read in rpr9 mutant and Q21861 wildtype. The X axis represents the physical sequence position (x Mbp) on barley chromosome 3H in the rpr9 region, whereas Y axis represents the ratio of total exome capture coverage reads of 487 high confidence annotated genes between rpr9 mutant and Q21861-wt. Blue circles represents each gene in the region. Red circles represent the deleted genes in the rpr9 region with a very low coverage ratio (< 0.05)
The deletion spanned a physical region of 1.052 Mbp based on the positions of two genes, HORVU3Hr1G074910 (end position on the barley physical map ch. 3H - 560,448,070) and HORVU3Hr1G075060 (start position on the barley physical map ch. 3H - 561,500,252) that flank the deletion yet are still present in the rpr9 mutant (Fig. 2c) based on the coverage ratio (cutoff ratio 0.05) of rpr9 and Q21861 genes present in the QTL region (Additional file 6: Table S3). The region present between the most proximal and distal deleted genes, HORVU3Hr1G074920 (start position on the barley physical map ch. 3H – 560,659,267) and HORVU3Hr1G075050 (end position on the barley physical map ch. 3H – 561,438,786), respectively, on the barley physical map [39], show a minimum deletion of ~ 0.773 Mbp (Fig. 2c). Within this ~ 0.773 Mb deletion, eleven HC genes are annotated in barley reference genome. This block of eleven genes includes a predicted leucine-rich repeat receptor-like protein kinase (gene model HORVU3Hr1G074930) yet sequencing reads aligned to this gene model. The IPK barley high confidence gene list released in 2016 is utilized for exome data analysis and this receptor like kinase gene is annotated twice on chromosome 3H as HORVU3Hr1G074930 (560,737,798-560,739,850) and HORVU3Hr1G067050 (509,350,259-509,352,312). Upon thorough inspection we concluded that gene model HORVU3Hr1G074930 was identical to HORVU3Hr1G067050 and appears to be a mis-annotated gene in the rpr9 deletion region on chromosome 3H. Thus, the deletion contained ten HC deleted genes (Table 3) including a SKP1-like 9 protein encoding, a NAD(P)H-quinone oxidoreductase subunit H, a NAD(P)H-quinone oxidoreductase subunit 1, a RNAase encoding, one L-tryrosine decarboxylase, an F-box family protein encoding gene and four peroxidase superfamily genes. Interestingly in our 9 k iSelect co-segregating marker panel for Hv584/rpr9, we identified a locus SCRI_RS_27,417 starting at the ch. 3H physical position 560,935,764 bp on the rpr9 containing co-segregation block (de novo genetic position 57.55 cM) (Additional file 2: Table S2). This marker is situated in the identified rpr9 deletion region according to the barley genome sequence. We found the physical position of marker SCRI_RS_27,417 in the 3′ untranslated region (UTR) of annotated mRNA HORVU3Hr1G074960.2 (560934358–560,935,865, chr. 3H) encoding for a peroxidase superfamily protein, present in the mapped rpr9 locus. In our deletion analysis this gene had 4835 exome capture reads in wild type Q21861 across the HORVU3Hr1G074960 and 269 exome capture reads in rpr9 mutants, thus giving a coverage ratio of 0.05 between rpr9 and Q21861. However, in Q21861, coverage was across the entire coding determining sequence (CDS), while in rpr9, the 269 exome capture reads specifically aligned to the 3′ terminus of the gene with a very specific terminus at position 988 in the CDS. This read alignment suggested that the gene was at one end of the deletion suggesting that the gene ordering in the region may not be correct based on the genome sequence (Additional file 7: Figure S4).
Discussion
In wheat, nearly 60 stem rust resistance genes have been identified and hundreds of different races have been typed using single R-gene differentials. However, in barley only five stem rust resistance genes have been identified and of these only two, Rpg1 and the rpg4/Rpg5 complex or RMRL, have been shown to be effective. However, recent association mapping using landraces and wild barley populations [40] have identified some novel stem rust resistances. Although, Rpg1 and RMRL confer effective resistances they still do not resemble the strong race specific resistances identified in wheat. The Rpg1 and RMRL genes/loci confer broad-spectrum resistances, which are more similar to incomplete or slow rusting gene action and when combined currently provide resistance to all known races of wheat stem rust. We speculate that barley is a recent non-host to wheat stem rust and has not undergone a prolonged evolutionary molecular arms race with the adapted pathogen. This relatively short co-evolutionary history between barley and wheat stem rust has resulted in limited resistance sources in barley compared to the large number of race specific R-genes and multiple pathogen races correlating to interactions with specific resistances in wheat. The Rpg1- and RMRL-mediated resistance mechanisms may be forms of non-host resistance that do not fall into the usual class of typical race specific NLR rust R-genes. These race specific NLR resistances are typically post-haustorial resistance mechanisms that elicit strong ETI mechanisms associated with strong HR responses as characterized in the classic flax-flax rust model system [41] and the majority of stem rust resistance genes characterized in wheat.
In barley, the RMRL provides resistance against a broad spectrum of stem rust races including the highly virulent race TTKSK. To identify conserved genes that function in RMRL or Rpg1 resistance, FN irradiation of barley line Q21861 (RMRL+ and Rpg1+) was used to induce the deletion mutant rpr9 that is susceptible to Pgt race QCCJB which is specifically avirulent on RMRL and is also susceptible to Pgt race HKHJC which is specifically avirulent on Rpg1 containing barley lines. The rpr9 mutant was utilized for genetic mapping via a Hv584 x rpr9 cross followed by exome capture and mapping-by-sequencing to rapidly identify candidate Rpr9 genes that underlie the mutant phenotypes. A combine approach of QTL and genetic mapping was used to identify the rpr9 region of the barley genome. QTL mapping using the 9 k iSelect marker panel delimited rpr9 to a ~ 59.78 Mbp region on chromosome 3H between the SNP markers BOPA2_12_30616 and SCRI_RS_159006 located at physical map positions from 525,056,438 -584,839,846 bp respectively. For fine mapping of the rpr9 QTL we identified seven critical recombinants enabled to us to further resolve the position of rpr9 on a co-segregating block of 49 loci covering an ~ 57 Mbp physical region. We also determined that the rpr9 mutant exhibited a stunted root phenotype, as after cleaning the genetic background of rpr9 with four rounds of backcrossing, both the stem rust susceptibility and stunted root phenotype were retained suggesting that the mutation-giving rise to both mutant phenotypes may not be distinct.
Root length is controlled by interaction between many cellular factors [42], thus identification of the only significant QTL for the stunted root phenotype in the Hv584/rpr9 RIL population at the rpr9 deletion region flanked by the two segregation blocks represented by SNP markers BOPA2_12_30616 and BOPA1_861–268 located at physical map positions from 525,056,438 -582,486,654 bp respectively, indicates that the same mutation event is pleiotropic and compromise both, the RMRL and Rpg1 mediated resistance responses as well as the seedling root length growth. The distribution of root length in the RIL population was found to have a normal distribution rather than following a bimodal distribution (Additional file 8: Figure S5) suggesting that although the only significant root length QTL mapped to the rpr9 deletion-locus causing shorter root length, the natural polymorphism present between Q21861 and Hv584 contributes to the segregating and continuous distribution of the root lengths within the RIL population. However, none of the natural polymorphism between Q21861 and Hv584 had a significant effect that was detected in the population, suggesting that our artificially induced polymorphism, using fast neutron mutagenesis, provided a greater phenotypic effect than any of the natural variation contributing to this complexly inherited trait.
Using the recently developed barley exome capture array followed by Illumina sequencing of the rpr9 mutant compared with the wt Q21861, a deletion with a maximum length of ~ 1.05 Mbp was identified on chromosome 3H between the physical position of 560,448,070–561,500,252 bp underlying the rpr9 delimited region. The deleted region contains ten high confidence genes along with a mis-annotated LRK. The ten high confidence genes were classified as a block of four peroxidases, two NAD(P)H-quinone oxidoreductases and one each of SKP1- like 9, a F-box family protein, a L-tyrosine decarboxylase, and a RNase family protein (Fig. 2c, Table 3). Interestingly in the 9 k iSelect co-segregating marker panel for Hv584/rpr9, we identified the SNP marker SCRI_RS_27,417 that was present in the identified rpr9 deleted region, thus warranting further examination of the annotated deletion. Our analysis identified the position of the SNP marker in the 3′ UTR of annotated gene HORVU3Hr1G074960 (560934358–560,935,865, chr. 3H, CDS- 1074 bp). Since exome capture reads covered the entire CDS in wildtype Q21861 and abruptly started at position 988 in the last exon extending into the 3’UTR it appears that this gene is actually located at the terminal end of the deletion. This suggests that although the gene content of the region is correct the ordering of the genes at the region are incorrect. We have observed such discrepancies in the Morex draft genome assembly when fine mapping and sequencing of the rcs5 spot blotch resistance locus [43] and RMRL (Solanki et al., Unpublished).
The genetic mapping and exome capture data shows that the stunted root phenotype and rpr9 disease susceptibility phenotype are either governed by two different genes present in the deletion block or a single gene within the deletion is responsible for both mutant phenotypes. If a single gene controls both phenotypes, then it represents an interesting example of a pleiotropic effect. However, there are many examples of pleiotropic genes effecting both abiotic and biotic resistances as well as developmental processes [44, 45]. It has been hypothesized that since the rpr9 mutant compromises both the very early Rpg1-mediated stem rust resistance mechanisms [5] as well as the early responses induced by RMRL there may be a cell surface receptor responsible for the early perception of the pathogen resulting in early pathogen perception at the leaf surface and rapid intracellular responses within minutes of the pathogen contacting the leaf surface. Therefore, upon our initial characterization of the deletion region the receptor-like kinase gene present in the region was of great interest and our initial top candidate gene. However, in the recently released IPK barley high confidence gene list utilized for exome data analysis, this receptor like kinase gene is annotated twice on chromosome 3H as HORVU3Hr1G074930.1 (560,737,798-560,739,850) and HORVU3Hr1G067050.1 (509,350,259-509,352,312) and both gene annotations are identical. We concluded that it is a mis-annotation at the 560,737,798-560,739,850 (HORVU3Hr1G074930.1) position present inside the rpr9 region, further indicating the need to refine the barley whole genome assembly and annotation. Thus, we focused on the other candidate genes within the deleted region. Interestingly, two of the candidate genes are predicted to encode a SKP1 (S-phase kinase- associated protein) and F-box proteins, known to be part of functional components of the multiprotein E3 ubiquitin ligase complex known as SCF (SKP/Cullin/F-box and ring finger protein Rbx1). SCF type E3 ubiquitin ligases are well characterized [46] and in SCF complexes the Cullin and Rbx1 proteins constitutes a scaffold core which is connected to the F-box proteins through SKP1 [47]. In the SCF complex F box proteins determine the specificity for ubiquitination thus are considered substrate recognition components [48]. In Arabidopsis 21 SKP genes (ASK) were predicted [49], however in yeast and human only one functional SKP1 gene is present [50]. We found that in barley sixteen high confidence SKP genes were predicted upon a genome search (Additional file 9: Table S4).
Many studies suggested the role of SCF complex in regulation of plant immunity. Recent evidence of E3 ubiquitin ligase MUSE1 and MUSE2 controlling the dual NLR defense responses mediated by SNC1-SIKIC2 NLR pair indicate the importance of ubiquitination mediated regulation to regulate the fine switch between partner NLRs [51]. In barley Rpg5 and Rga1 NLR pair may have similar regulation mediated by SCF complex for Pgt resistance. In tobacco it has been reported that NbSGT1 (Nicotiana benthamiana suppressor of G2 allele of SKP1) a highly conserved co-chaperone component of the SCF complex, interacts directly with NbSKP1 (a component of the SCF ubiquitin ligase complex) and NbRar1 [52, 53]. Rar1 is required for TNL (Toll/interlukin 1 receptor-Nucleotide binding-Leucine rich repeats) N gene mediated resistance in tobacco and also has been shown to function downstream of powdery mildew recognition and upstream of plant hypersensitive cell death responses upon H2O2 accumulation via several CNLs (CC-NB-LRR) including Mla mediated resistance in barley [54]. TNL and CNL proteins are involved in pathogen recognition and defense mechanisms yet have diverse signaling pathways. TNLs are rare in monocots and absent in cereals whereas CNLs are found in dicots. Thus, Rar1 represents a signaling factor functioning in common pathways evoked by TNL and CNL resistance genes. In tobacco upon SKP1 or SGT1 silencing, N gene mediated resistance has been shown to be compromised for TMV resistance. Triticum aestivum SKP1 (TSK1) was found to be expressed in young root and spikes and a low-level expression was detected in leaves. TSK1 overexpression in Arabidopsis was shown to increase abscisic acid (ABA) responsive phenotypes such as stomatal closure, root growth and seed germination and enhanced drought tolerance [55], thus possibly functioning as a positive regulator of ABA signaling .
In barley five high confidence (HORVU3Hr1G075050.2, HORVU1Hr1G077600.2, HORVU4Hr1G052070.1, HORVU0Hr1G030450.1, HORVU1Hr1G068570.1) and one low confidence (HORVU3Hr1G079580.6) F-box genes were mined from the IPK database. In Arabidopsis 694 F-BOX genes were predicted [48] making it one of the largest protein families. F-box proteins show high functional diversity and are involve in many functions including pathogen perception and circadian rhythm. Arabidopsis DOR (DrOught tolerance Repressor) is a SFL (S-locus F box like) family protein and is expressed in the stomatal guard cells and shown to control the ABA biosynthetic pathway and negatively controls ABA induced stomatal closure under drought stress [56]. In another study the Arabidopsis F-box-Nictaba was shown to be a pathogen inducible gene and overexpression in plants showed resulted in reduced infection by Pseudomonas syringae pv. tomato strain DC3000 [57]. It was also shown that the F-box protein ACIF1 (Avr9/Cf-9–Induced F-Box 1) silencing suppressed the hypersensitive response triggered by a diversity of pathogen elicitors such as Avr9, AvrPto and the P50 helicase of TMV [58]. Thus, the E3 ubiquitin ligase SCF complex components identified within the deletion in the rpr9 mutant region are certainly strong rpr9 candidate genes possibly functioning downstream in the cellular pathways governed by the CNL Rpg5 and dual kinase protein Rpg1. It has been determined that Rpg1-mediated resistance is dependent upon RPG1 ubiquitination and subsequent E3-mediated degradation [6], thus the deleted SCF complex proteins may be responsible for the E3 ubiquitination required for Rpg1 degradation post interaction with avirulent Pgt isolates. The Rpg1 protein degradation 24 h post infection is required for the resistant reaction to occur, thus, if these E3 ubiquitin ligase SCF complex components are Rpr9, then loss of Rpg1 degradation could be expected in the rpr9 mutant post infection with Pgt race HKHJ if this hypothesis is correct (Fig. 4).

A hypothetical model of candidate rpr9 gene disrupting the RMRL, Rpg1 and root length signaling pathway. Our topmost candidate rpr9 genes are SCF and F-box protein coding, associated with ubiquitin mediated protein degradation pathway. Rapid phosphorylation of Rpg1 dual kinase protein followed by protein degradation is required for Pgt race HKHJ resistance. Similarly, Ubiquitination plays an important role in root development and hormone signaling. Thus, we hypothesize rpr9 mutation disrupts the Rpg1, RMRL and hormone signaling pathway by inhibiting the possible intermediate ubiquitination step resulting in compromised phenotype
Although the SCF complex proteins are considered our best candidates there are other deleted genes in the region that are associated with biotic stress responses. Two other candidates are the NQO (NAD(P)H-quinone oxidoreductase) subunit H and subunit 1 that were previously show to be involved in electron transport in photosystem II and quinone detoxification [59, 60]. Also, the Arabidopsis L-tyrosine decarboxylase (TyrDC) was shown to be induced by wounding, drought stress and fungal effector perception [61,62,63] and is known as the first enzyme in the benzylisoquinoline alkaloids pathway. These metabolites act as antimicrobial compounds and cell wall reinforcement agents to provide immune function [64]. Peroxidases are well characterized in plant defense responses and have been shown to be involved in removal of hydrogen peroxide at the cell wall and initiate the wounding defense responses [27, 65,66,67]. Thus, future functional studies are warranted to validate the rpr9 gene among the list of the ten candidate genes identified. Although Rpg1 and RMRL confer some differential specificity [15, 68], the rpr9 mutant suggests the two mechanisms contain common resistance component/s probably downstream in a converging resistance pathway.
Conclusions
Virulent races of Pgt emerging in Africa, Asia, Europe including recent reports of Pgt in the United Kingdom after nearly 60 years, on barberry plants adjacent to barley fields, underlines the reoccurrence of this dangerous foe. In the present research we have successfully utilized the combination of fast neutron mutagenesis, genetic mapping and exome capture sequencing to rapidly identify conserved genes involve in RMRL- and Rpg1-mediated resistance responses, the only two effective Pgt resistances in barley which provide protection against a broad spectrum of Pgt races. We mapped and identified candidate rpr9 genes, which is not only required for both RMRL- and Rpg1-mediated Pgt resistance but also confer a stunted root phenotype. Further validation of the candidate genes will provide insight into the functional mechanisms controlling both phenotypes possibly by one or distinct genes underlying rpr9 mutation. The identification of genes involved in the resistance signaling mechanisms will fill knowledge gaps in downstream signaling mechanism in NLR pathways.
Methods
Mutant screens
The barley line Q21861 (PI584766) was utilized for the forward genetics screen because it carries the RMRL as well as the Rpg1 stem rust resistance gene. After three generations of single seed descent and seed increase the genetic quality Q21861 seed (~ 3 kg) was irradiated with fast neutrons (3.5 or 4.0 Gy using protocol 563) at the FAO/IAEA Seibersdof SNIF facility near Vienna, Austria. Q21861 M1 seeds were planted and allowed to self and ~ 6000 spikes containing M2 seed were harvested from individual plants. In the greenhouse, 20–30 M2 seed from individual spikes were planted in cones filled with a peat moss:perlite (3,1 v/v) potting mix (#1 Sunshine Mix, Fisons, Vancouver, Canada). Plants were inoculated with pathotype Pgt-QCCJB when the first leaves were fully expanded and assessed for their infection types 12–14 days post inoculation according to Steffenson et al. [69]. Infection type (IT) were based on a 0–4 Stakman IT (Infection Type) scale used for barley [17, 70]. Where 0 is highly resistant and 4 is highly susceptible with the middle range numbers representing intermediate reactions, which are further modified by + or – and a fleck (;) indicating a small necrotic area. IT1 indicates minute uredinia; IT2 small uredinia with chlorosis; IT3 medium uredinia often with chlorosis; and IT4 indicates large uredinia with chlorosis. Barley often exhibits mesothetic reactions with two or more ITs on a single leaf therefore ITs observed are recorded in order of their prevalence and this categorical disease rating was converted to a single numerical value as described previously [38]. When segregation for stem rust reactions were observed within an individual M2 spike, single heads were harvested and phenotyped with pathotype Pgt-QCCJB at the M3 generation. A consistent mutant was identified and designated as rpr9.
Segregation analysis and RIL population development
The rpr9 mutant generated in the Q21861 background was backcrossed to Q21861 for four generations and fifteen BCF2 individuals were phenotyped at each back cross (BC) generation to identify homozygous rpr9 mutant individuals. Pollen from the rpr9 mutant with the background mutations cleaned up with four generations of backcrossing was crossed with the Swiss landrace Hv584 (UMN Pathology breeding program) [70] as the female parent to generate the Hv584/rpr9 population. The Swiss landrace Hv584, is resistant to Pgt races QCCJB and TTKSK because it carries RMRL [70] but contains polymorphism across the genome compared to Q21861. Seeds of all the barley genotypes used in the study are available at Barley Pathology, NDSU. For inheritance studies 54 Hv584/rpr9 F2 individuals were phenotyped at the seedling stage with Pgt race QCCJ in the growth chamber as described in Mirlohi et al., (2008) [37]. The single gene segregation analysis was calculated utilizing a χ2 test with the null hypothesis that the rpr9 phenotype was contributed by a single recessive gene. A Hv584/rpr9 RIL population consisting of 95 individuals was developed through single seed decent till the F5:6 generation and phenotyped for QCCJ seedling disease assay.
Rag-doll test for root length analysis
Two seed germination papers/brown paper towels were placed together, and a horizontal line was drawn using a pencil at the center of the paper. The paper towels were moistened with water. Twenty rpr9 and wt Q21861 seed were selected randomly and placed separately on one half of the moist germination papers keeping them on the drawn horizontal line. The germination paper was carefully rolled vertically from the drawn line avoiding seed movement into a moderately tight tube and secured with duct tape. All the tubes were labelled and kept in a warm place at ~ 25 °C for four days before taking the root length readings in mm from seed base to the end of the longest root. An unpaired t-test was run to determine if the differences were statistically different. Similar method was followed using the 95 individual Hv584/rpr9 RILs, Q21861, Hv584 and the rpr9 mutant and root length was recorded for 3 germinated seeds and data was used for QTL mapping.
Genotyping and genetic map construction
The Hv584/rpr9 RIL population was genotyped using the 9 k Illumina Infinium iSelect assay [71] at the USDA cereal genotyping lab, Fargo ND. Markers containing greater than 30% missing data were removed from the data set. Linkage map for the Hv584/rpr9 RIL population generated using MapDisto 2.1.1 [72]. The command ‘Find linkage groups’ was used to make markers linkage group with a logarithm of the odds (LOD) value of 3.0 and rmax of 0.3. A minimum cutoff score of 10% was used to filter loci with missing data in 2-point matrices. The ‘AutoOrder’, ‘AutoCheckInversions’, and ‘AutoRipple’ commands were utilized to generate the linkage map at a LOD of 3.0 and Kosambi mapping function was used to calculate the genetic distances. The final linkage maps were drawn using ‘Draw all sequences’ command. Co-segregating markers were identified and removed, leaving a single marker within each block of co-segregating loci with the least amount of missing data at each position. For stem rust and seedling root length QTL analysis, the whole map was regenerated without using rpr9 phenotype as a marker. Non-redundant loci and their corresponding de novo genetic positions and trait data was exported to utilized in QGene 4.4 [73].
Genetic map concordance
Genetic positions based on POPSEQ [74] were also obtained for each iSelect marker using the POPSEQ position data sheet for iSelect markers downloaded from the Barlex web server (barlex.barleysequence.org). iSelect marker names for the 2701 polymorphic markers for the rpr9 mapping was imported in the Microsoft Excel and ‘vlookup’ function was used to identify POPSEQ positions. If no POPSEQ position was assigned, then IPK barley BLAST server (https://webblast.ipk-gatersleben.de/barley_ibsc/viroblast.php) based sequence and position search was conducted. Further, Barleymap online tool [75] was used to cross check the iSelect marker anchoring in the POPSEQ positions in the seven barley chromosome.
Quantitative trait loci analysis
For QTL analysis de novo loci position for non-cosegregating loci was used. The average of three replication of seedling root length data and stem rust phenotyping trait data was assigned for analysis. QTL analysis was conducted using the single trait multiple interval mapping (MIM) algorithm at scanning interval of 5. A permutation test consisting of 1000 iterations was carried out to determine a LOD threshold at the p ≤ 0.05 significance level.
Statistical analysis
Mean Score of categorical disease ITs (0–4) was calculated in MS-Excel-2018. The χ2 test (goodness of fit) to determine 3:1 segregation ratio was carried out in SAS 9.4. To determine the root length phenotype, root length was measured in a continuous scale and unpaired t-test was carried out in SAS 9.4 (p ≤ 0.05).
DNA extraction, fragmentation optimization, exome capture library preparation, and sequencing
Eleven seed of wt Q21861, and the rpr9 mutant were placed on water-soaked Whatman filter paper in a disposable petri dish for 24 h. Embryos were excised using a sterilized DNase free scalpel and a total of five excised embryos were used for DNA extraction using the PowerPlant Pro DNA isolation kit (MoBIO Laboratories Inc., QIAGEN Carlsbad CA). Mechanical lysis of samples was done using a mechanical bead beater at 2000 rpm for 2 cycles of 3 min each and the manufacturer’s protocol was followed for DNA isolation. The quality of extracted DNA was checked by running an aliquot of 1 μL of gDNA on a 1% agarose gel supplemented with GelRED (Biotium) fluorescent nucleic acid dye. The DNA was determined to have good integrity when it showed a high molecular weight band ~ 15–20 kb with minimal low molecular weight smearing indicative of DNA degradation. The gDNA was quantified using the Qubit Broad Range DNA Quantification kit (Thermo Scientific). Enzymatic DNA shearing was optimized to generate desired fragment sizes of 250–450 bp by conducting a time course experiment with digestion reactions consisting of 1.5 μg of gDNA in a 20 μl reaction with NEB dsDNA Fragmentase enzyme, 1x Fragmentase reaction buffer and 10 mM MgCl2 (New England Biolabs, Ipswich MA). Digestion reactions were incubated at 37 °C for 10, 15, 20, 25 and 30 min and then inactivated by adding 5 μl of 0.5 M EDTA, followed by AMPure XP magnetic bead DNA purification (Agencourt). DNA size distribution was analyzed on the Agilent 2100 Bioanalyzer (Agilent Technologies) using a DNA 1000 kit (Agilent Technologies) following the manufacturer protocol for chip loading and data analysis. The 25-min enzymatic digestion was found to produce the optimal fragment size distribution ranging between 250 and 450 base pairs and was used to produce fragmented DNA libraries of Q21861, and rpr9 (Additional file 10: Figure S6).
Fragmented gDNA samples were used for whole exome capture using the Roche NimbleGen SeqCap EZ Developer probe pool barley exome design 120426_Barley_BEC_D04 with a total capture design size of 88.6 Mb. After exome capture the KAPA HTP gDNA library preparation kit was used for Illumina sequencing library preparation. The standard manufacturer protocol was followed for library preparation using the KAPA HTP kit, except for size selection being performed on a Pippin Prep gel purification system (Sage Science) with a 250–450 bp targeted size selection. The gDNA used to prepare the barcoded barley whole exome capture multiplexed library was developed according to seqCAP EZ Library SR user guide 4.1 protocol. Quality and size distribution of the final capture library was determined using a bioanalyzer as previously described. A Qubit fluorometer was used to quantify the library for final dilution and sequencing on an Illumina NextSeq flow cell generating 150 base pair single end reads. Qubit readings were used since the bioanalyzer tends to underestimate the quantity for size selected libraries due to presence of DNA fragments not falling within the desired range (150–450). Thus, posing a possible chance of over-flooding of the flow cells due to the under-diluted library.
Exome capture data analysis3
The rpr9 mutant and wildtype Q21861 sequencing reads were parsed by their specific barcodes added during library preparation. The quality scores of the raw sequencing reads were determined using the FQC dashboard [76]. The Illumina reads were imported into CLC Genomics Workbench v8.0 in FASTQ format and trimmed for the presence of adapter sequences. Mutant and wildtype reads were aligned to the barley reference genome and the barley reference gene set using the BWA ‘mem’ algorithm [77] with default settings. The alignments were used to identify deleted region utilizing two separate data analysis pipelines: (1) Small deletions (less than 100 bp) were identified using SAMtools ‘mpileup’ with default settings [78], the identified variants were filtered for a minimum read depth of three and a minimum individual genotype quality of 10 using VCFtools [79], 2) As fast neutron mutagenesis may induce large chromosomal deletions, sequencing coverage was calculated from the reads aligned to the reference gene set using SAMtools ‘idxstats’ to identify full gene deletions. To eliminate reads that map to paralogous genes and therefore may give rise to the identification of false negative deletions, only uniquely mapping reads were extracted using SAMtools ‘view’ and used in coverage analysis. Since we mapped the rpr9 QTL and determined that ~ 610 high confidence genes are present in between the flanking markers, a ratio of captured reads between rpr9 and Q21861 was calculated with a cutoff score of 0.05 to identify genes which have very low or no reads in the rpr9 exome capture. Physical positions of fully deleted exome capture targets were obtained from the barley physical map [39], allowing for the identification of candidate genes within the delimited region and characterization of the larger chromosomal deletions typically containing multiple genes generated with the high energy fast neutron irradiation.
Availability of data and materials
All the material and data can be obtained from the authors upon request through the Barley Pathology lab, Department of Plant Pathology at North Dakota State University.
Abbreviations
- ABA:
-
Abscisic acid
- bp:
-
Base pair
- CDS:
-
Coding determining sequence
- ch.:
-
Chromosome
- cM:
-
Centi Morgan
- CNL:
-
Coiled coil-NB-LRR
- ETI:
-
Effector triggered immunity
- FN:
-
Fast neutron
- HPI:
-
Hours post inoculation
- HR:
-
Hypersensitive response
- IT:
-
Infection type
- LRK:
-
Receptor like kinase
- Mbp:
-
Million base pair
- NbSGT1 :
-
Nicotiana benthamiana suppressor of G2 allele of SKP1
- NLR:
-
Nucleotide-binding domain and leucine-rich repeat
- NQO:
-
NAD(P)H-quinone oxidoreductase
- PAMP:
-
Pathogen associated molecular pattern
- PCD:
-
Programmed cell death
- Pgt :
-
Puccinia graminis f. sp. tritici
- PR:
-
Pathogen related
- PRRs:
-
Pattern recognition receptors
- PTI:
-
PAMP triggered immunity
- QTL:
-
Quantitative trait loci
- RIL:
-
Recombinant inbred line
- RLKs:
-
Receptor-like kinases
- Rme1 :
-
rpg4 modifier element 1
- RMRL:
-
rpg4-mediated resistance locus
- RMRL2:
-
rpg4-mediated resistance locus 2
- rpr9 :
-
Required for P. graminis resistance 9
- SCF:
-
SKP/Cullin/F-box and ring finger protein Rbx1
- SKP1:
-
S-phase kinase- associated protein
- STPK:
-
Serine / threonine protein kinase
- TNL:
-
Toll/interlukin 1 receptor-Nucleotide binding-Leucine rich repeats
- UTR:
-
Untranslated region
- wt:
-
Wild type
References
Leonard KJ, Szabo LJ. Stem rust of small grains and grasses caused by Puccinia graminis. Mol Plant Pathol. 2005;6:99–111. https://doi.org/10.1111/j.1364-3703.2005.00273.x.
Steffenson BJ. Analysis of durable resistance to stem rust in barley. Breeding for disease resistance; 1992. p. 153–67.
Brueggeman R, Rostoks N, Kudrna D, Kilian A, Han F, Chen J, et al. The barley stem rust-resistance gene Rpg1 is a novel disease-resistance gene with homology to receptor kinases. Proc Natl Acad Sci U S A. 2002;99:9328–33. https://doi.org/10.1073/pnas.142284999.
Nirmala J, Brueggeman R, Maier C, Clay C, Rostoks N, Kannangara CG, et al. Subcellular localization and functions of the barley stem rust resistance receptor-like serine/threonine-specific protein kinase Rpg1. Proc Natl Acad Sci U S A. 2006;103:7518–23. https://doi.org/10.1073/pnas.0602379103.
Nirmala J, Drader T, Chen X, Steffenson B, Kleinhofs A. Stem rust spores elicit rapid RPG1 phosphorylation. Mol Plant-Microbe Interact. 2010;23:1635–42. https://doi.org/10.1094/MPMI-06-10-0136.
Nirmala J, Dahl S, Steffenson BJ, Kannangara CG, von Wettstein D, Chen X, et al. Proteolysis of the barley receptor-like protein kinase RPG1 by a proteasome pathway is correlated with Rpg1-mediated stem rust resistance. Proc Natl Acad Sci U S A. 2007;104:10276–81. https://doi.org/10.1073/pnas.0703758104.
Roelfs AP. Epidemiology of the cereal rusts in North America. Can J Plant Pathol. 1989;11:86–90.
Roelfs AP, Long DL, Roberts JJ. Races of Puccinia graminis in the United States during 1991. Plant Dis. 1993;77:129–32.
Jin Y, Steffenson BJ, Fetch TG. Sources of resistance to pathotype QCC of Puccinia graminis f. sp. tritici in barley. Crop Sci. 1994;34:285–8.
Jin Y, Steffenson BJ, Miller JD. Inheritance of resistance to pathotypes QCC and MCC of Puccinia graminis f. sp. tritici in barley line Q21861 and temperature effects on the expression of resistance. Pathology. 1994;84:452–5.
Borovkova IG, Steffenson BJ, Jin Y, Rasmussen JB, Kilian A, Kleinhofs A, et al. Identification of molecular markers linked to the stem rust resistance gene rpg4 in barley. Phytopathology. 1995;85:181–5.
Pretorius ZA, Singh RP, Wagoire WW, Payne TS. Detection of virulence to wheat stem rust resistance gene Sr31 in Puccinia graminis. f. Sp. tritici in Uganda. Plant Dis 2000;84:203–203.
Jin Y, Singh RP, Ward RW, Wanyera R, Kinyua M, Njau P, et al. Characterization of seedling infection types and adult plant infection responses of monogenic Sr gene lines to race TTKS of Puccinia graminis f. sp. tritici. Plant Dis. 2007;91:1096–9.
Jin Y, Szabo LJ, Pretorius ZA, Singh RP, Ward R Jr. TF. Detection of virulence to resistance gene Sr24 within race TTKS of Puccinia graminis f. sp. tritici. Plant Dis. 2008;92:923–6.
Steffenson BJ, Zhou H, Chai Y, Grando S. Vulnerability of cultivated and wild barley to african stem rust race TTKSK. In: Zhang G, Li C, Liu X, editors. Advance in barley sciences. Dordrecht: Springer Netherlands; 2013. p. 243–55. https://doi.org/10.1007/978-94-007-4682-4_21.
Stokstad E. Deadly wheat fungus threatens world’s breadbaskets. Science. 2007;315:1786–7.
Steffenson BJ, Jin Y, Brueggeman RS, Kleinhofs A, Sun Y. Resistance to stem rust race TTKSK maps to the rpg4/Rpg5 complex of chromosome 5H of barley. Phytopathology. 2009;99:1135–41. https://doi.org/10.1094/PHYTO-99-10-1135.
Druka A, Kudrna D, Han F, Kilian A, Steffenson B, Frisch D, et al. Physical mapping of the barley stem rust resistance gene rpg4. Mol Gen Genet. 2000;264:283–90. https://doi.org/10.1007/s004380000320.
Kleinhofs A, Brueggeman R, Nirmala J, Zhang L, Mirlohi A, Druka A, et al. Barley stem rust resistance genes: structure and function. The Plant Genome. 2009;2:109–20.
Arora D, Gross T, Brueggeman R. Allele characterization of genes required for rpg4-mediated wheat stem rust resistance identifies Rpg5 as the R gene. Phytopathology. 2013;103:1153–61. https://doi.org/10.1094/PHYTO-01-13-0030-R.
Wang X, Richards J, Gross T, Druka A, Kleinhofs A, Steffenson B, et al. The rpg4-mediated resistance to wheat stem rust (Puccinia graminis) in barley (Hordeum vulgare) requires Rpg5, a second NBS-LRR gene, and an actin depolymerization factor. Mol Plant-Microbe Interact. 2013;26:407–18. https://doi.org/10.1094/MPMI-06-12-0146-R.
Brueggeman R, Druka A, Nirmala J, Cavileer T, Drader T, Rostoks N, et al. The stem rust resistance gene Rpg5 encodes a protein with nucleotide-binding-site, leucine-rich, and protein kinase domains. Proc Natl Acad Sci U S A. 2008;105:14970–5. https://doi.org/10.1073/pnas.0807270105.
Brueggeman RS, Solanki S. Barley Stem Rust resistance mechanisms: diversity, Genestructure, and function suggest a recently evolved host-pathogen relationship. In: Singh DP, editor. Management of wheat and barley diseases. Boca Raton: CRC Press; 2017. p. 579–604. https://doi.org/10.1201/9781315207537-26.
Sharma Poudel R, Al-Hashel AF, Gross T, Gross P, Brueggeman R. Pyramiding rpg4- and Rpg1-mediated Stem rust resistance in barley requires the Rrr1 gene for both to function. Front Plant Sci. 2018;9:1789. https://doi.org/10.3389/fpls.2018.01789.
Solanki S, Ameen G, Borowicz P, Brueggeman RS. Shedding light on penetration of cereal host stomata by wheat stem rust using improved methodology. Sci Rep. 2019;9:7939. https://doi.org/10.1038/s41598-019-44280-6.
Solanki S, Ameen G, Richards JS, Brueggeman R. Modulation of integrated decoy R-genes/transcription factor assembly elicits wheat stem rust resistance responses in barley: rpg4/Rpg5-mediated Ug99 resistance. In: Smith K, Dill-Macky R, editors. International Barley Genetics Symposium. Minneapolis-St. Paul: IBGS; 2016.
Orozco-Cárdenas ML, Narváez-Vásquez J, Ryan CA. Hydrogen peroxide acts as a second messenger for the induction of defense genes in tomato plants in response to wounding, systemin, and methyl jasmonate. Plant Cell. 2001;13:179–91.
van Esse HP, Bolton MD, Stergiopoulos I, de Wit PJGM, Thomma BPHJ. The chitin-binding Cladosporium fulvum effector protein Avr4 is a virulence factor. Mol Plant-Microbe Interact. 2007;20:1092–101. https://doi.org/10.1094/MPMI-20-9-1092.
Sánchez-Vallet A, Saleem-Batcha R, Kombrink A, Hansen G, Valkenburg D-J, Thomma BPHJ, et al. Fungal effector Ecp6 outcompetes host immune receptor for chitin binding through intrachain LysM dimerization. Elife. 2013;2:e00790. https://doi.org/10.7554/eLife.00790.
Bigeard J, Colcombet J, Hirt H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol Plant. 2015;8:521–39. https://doi.org/10.1016/j.molp.2014.12.022.
Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–9. https://doi.org/10.1038/nature05286.
Glazebrook J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol. 2005;43:205–27. https://doi.org/10.1146/annurev.phyto.43.040204.135923.
Li X, Lassner M, Zhang Y. Deleteagene: a fast neutron deletion mutagenesis-based gene knockout system for plants. Comp Funct Genomics. 2002;3:158–60. https://doi.org/10.1002/cfg.148.
Li X, Song Y, Century K, Straight S, Ronald P, Dong X, et al. A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J. 2001;27:235–42. https://doi.org/10.1046/j.1365-313x.2001.01084.x.
Koornneef M, Dellaert LW, van der Veen JH. EMS- and radiation-induced mutation frequencies at individual loci in Arabidopsis thaliana (L.) Heynh. Mutat Res. 1982;93:109–23.
Zhang L, Fetch T, Nirmala J, Schmierer D, Brueggeman R, Steffenson B, et al. Rpr1, a gene required for Rpg1-dependent resistance to stem rust in barley. Theor Appl Genet. 2006;113:847–55. https://doi.org/10.1007/s00122-006-0342-y.
Mirlohi A, Brueggeman R, Drader T, Nirmala J, Steffenson BJ, Kleinhofs A. Allele sequencing of the barley stem rust resistance gene Rpg1 identifies regions relevant to disease resistance. Phytopathology. 2008;98:910–8. https://doi.org/10.1094/PHYTO-98-8-0910.
Zhou H, Steffenson BJ, Muehlbauer G, Wanyera R, Njau P, Ndeda S. Association mapping of stem rust race TTKSK resistance in US barley breeding germplasm. Theor Appl Genet. 2014;127:1293–304. https://doi.org/10.1007/s00122-014-2297-8.
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, et al. A chromosome conformation capture ordered sequence of the barley genome. Nature. 2017;544:427–33. https://doi.org/10.1038/nature22043.
Sallam AH, Tyagi P, Brown-Guedira G, Muehlbauer GJ, Hulse A, Steffenson BJ. Genome-wide association mapping of Stem rust resistance in Hordeum vulgare subsp. spontaneum. G3 (Bethesda). 2017;7:3491–507. https://doi.org/10.1534/g3.117.300222.
Dodds PN, Lawrence GJ, Catanzariti A-M, Teh T, Wang C-IA, Ayliffe MA, et al. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc Natl Acad Sci U S A. 2006;103:8888–93. https://doi.org/10.1073/pnas.0602577103.
Slovak R, Ogura T, Satbhai SB, Ristova D, Busch W. Genetic control of root growth: from genes to networks. Ann Bot. 2016;117:9–24. https://doi.org/10.1093/aob/mcv160.
Ameen G, Drader T, Sager L, Steffenson BJ, Kleinhofs A, Brueggeman RS. Rcs5 is a wall associated kinase gene that putatively functions as a dominant susceptibility factor in the barley–Cochliobolus sativus interaction: The 12th international barley genetics symposium, Minneapolis, MN USA. 2016. pp. 26–30.
Parker MA. The pleiotropy theory for polymorphism of disease resistance genes in plants. Evolution. 1990;44:1872–5. https://doi.org/10.1111/j.1558-5646.1990.tb05257.x.
Burstin J, Marget P, Huart M, Moessner A, Mangin B, Duchene C, et al. Developmental genes have pleiotropic effects on plant morphology and source capacity, eventually impacting on seed protein content and productivity in pea. Plant Physiol. 2007;144:768–81. https://doi.org/10.1104/pp.107.096966.
Willems AR, Schwab M, Tyers M. A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta. 2004;1695:133–70. https://doi.org/10.1016/j.bbamcr.2004.09.027.
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–9. https://doi.org/10.1038/416703a.
Xu G, Ma H, Nei M, Kong H. Evolution of F-box genes in plants: different modes of sequence divergence and their relationships with functional diversification. Proc Natl Acad Sci U S A. 2009;106:835–40. https://doi.org/10.1073/pnas.0812043106.
Farrás R, Ferrando A, Jásik J, Kleinow T, Okrész L, Tiburcio A, et al. SKP1-SnRK protein kinase interactions mediate proteasomal binding of a plant SCF ubiquitin ligase. EMBO J. 2001;20:2742–56. https://doi.org/10.1093/emboj/20.11.2742.
Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A. 1998;95:11324–9. https://doi.org/10.1073/pnas.95.19.11324.
Dong OX, Ao K, Xu F, Johnson KCM, Wu Y, Li L, et al. Individual components of paired typical NLR immune receptors are regulated by distinct E3 ligases. Nature plants. 2018;4(9):699.
Liu Y, Schiff M, Marathe R, Dinesh-Kumar SP. Tobacco Rar1, EDS1 and NPR1/NIM1 like genes are required for N-mediated resistance to tobacco mosaic virus. Plant J. 2002;30:415–29. https://doi.org/10.1046/j.1365-313X.2002.01297.x.
Liu Y, Schiff M, Serino G, Deng X-W, Dinesh-Kumar SP. Role of SCF ubiquitin-ligase and the COP9 signalosome in the N gene-mediated resistance response to tobacco mosaic virus. Plant Cell. 2002;14:1483–96.
Shirasu K, Lahaye T, Tan MW, Zhou F, Azevedo C, Schulze-Lefert P. A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell. 1999;99:355–66.
Li C, Liu Z, Zhang Q, Wang R, Xiao L, Ma H, et al. SKP1 is involved in abscisic acid signalling to regulate seed germination, stomatal opening and root growth in Arabidopsis thaliana. Plant Cell Environ. 2012;35:952–65. https://doi.org/10.1111/j.1365-3040.2011.02464.x.
Zhang Y, Xu W, Li Z, Deng XW, Wu W, Xue Y. F-box protein DOR functions as a novel inhibitory factor for abscisic acid-induced stomatal closure under drought stress in Arabidopsis. Plant Physiol. 2008;148:2121–33. https://doi.org/10.1104/pp.108.126912.
Stefanowicz K, Lannoo N, Zhao Y, Eggermont L, Van Hove J, Al Atalah B, et al. Glycan-binding F-box protein from Arabidopsis thaliana protects plants from Pseudomonas syringae infection. BMC Plant Biol. 2016;16:213. https://doi.org/10.1186/s12870-016-0905-2.
van den Burg HA, Tsitsigiannis DI, Rowland O, Lo J, Rallapalli G, Maclean D, et al. The F-box protein ACRE189/ACIF1 regulates cell death and defense responses activated during pathogen recognition in tobacco and tomato. Plant Cell. 2008;20:697–719. https://doi.org/10.1105/tpc.107.056978.
Chesis PL, Levin DE, Smith MT, Ernster L, Ames BN. Mutagenicity of quinones: pathways of metabolic activation and detoxification. Proc Natl Acad Sci U S A. 1984;81:1696–700.
Hurley B, Subramaniam R, Guttman DS, Desveaux D. Proteomics of effector-triggered immunity (ETI) in plants. Virulence. 2014;5:752–60. https://doi.org/10.4161/viru.36329.
Kawalleck P, Keller H, Hahlbrock K, Scheel D, Somssich IE. A pathogen-responsive gene of parsley encodes tyrosine decarboxylase. J Biol Chem. 1993;268:2189–94.
Guillet G, De Luca V. Wound-inducible biosynthesis of phytoalexin hydroxycinnamic acid amides of tyramine in tryptophan and tyrosine decarboxylase transgenic tobacco lines. Plant Physiol. 2005;137:692–9. https://doi.org/10.1104/pp.104.050294.
Lehmann T, Pollmann S. Gene expression and characterization of a stress-induced tyrosine decarboxylase from Arabidopsis thaliana. FEBS Lett. 2009;583:1895–900. https://doi.org/10.1016/j.febslet.2009.05.017.
Yogendra KN, Dhokane D, Kushalappa AC, Sarmiento F, Rodriguez E, Mosquera T. StWRKY8 transcription factor regulates benzylisoquinoline alkaloid pathway in potato conferring resistance to late blight. Plant Sci. 2017;256:208–16. https://doi.org/10.1016/j.plantsci.2016.12.014.
Bindschedler LV, Dewdney J, Blee KA, Stone JM, Asai T, Plotnikov J, et al. Peroxidase-dependent apoplastic oxidative burst in Arabidopsis required for pathogen resistance. Plant J. 2006;47:851–63. https://doi.org/10.1111/j.1365-313X.2006.02837.x.
Kimura M, Kawano T. Salicylic acid-induced superoxide generation catalyzed by plant peroxidase in hydrogen peroxide-independent manner. Plant Signal Behav. 2015;10:e1000145. https://doi.org/10.1080/15592324.2014.1000145.
Chasov AV, Gordon LK, Kolesnikov OP, Minibaeva FV. Cell surface peroxidase--generator of superoxide anion in wheat root cells under wound stress. Tsitologiia. 2002;44:691–6.
Sun Y, Steffenson BJ. Reaction of barley seedlings with different stem rust resistance genes toPuccinia graminis f. sp.tritici andPuccinia graminis f. sp.secalis. Can J Plant Pathol. 2005;27:80–9. https://doi.org/10.1080/07060660509507198.
Steffenson BJ, Solanki S, Brueggeman RS. Landraces from mountainous regions of Switzerland are sources of important genes for stem rust resistance in barley. Alp Bot. 2016;126:23–33. https://doi.org/10.1007/s00035-015-0161-3.
Stakman EC, Stewart DM, Loegering WQ. Identification of physiologic races of Puccinia graminis var. tritici. US Department of Agric. 1962;ARSE-617:p53.
Muñoz-Amatriaín M, Cuesta-Marcos A, Endelman JB, Comadran J, Bonman JM, Bockelman HE, et al. The USDA barley core collection: genetic diversity, population structure, and potential for genome-wide association studies. PLoS One. 2014;9:e94688. https://doi.org/10.1371/journal.pone.0094688.
Lorieux M. MapDisto: fast and efficient computation of genetic linkage maps. Mol Breeding. 2012;30:1231–5. https://doi.org/10.1007/s11032-012-9706-y.
Joehanes R, Nelson JC. QGene 4.0, an extensible Java QTL-analysis platform. Bioinformatics. 2008;24:2788–9. https://doi.org/10.1093/bioinformatics/btn523.
Mascher M, Muehlbauer GJ, Rokhsar DS, Chapman J, Schmutz J, Barry K, et al. Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). Plant J. 2013;76:718–27. https://doi.org/10.1111/tpj.12319.
Cantalapiedra CP, Boudiar R, Casas AM, Igartua E, Contreras-Moreira B. BARLEYMAP: physical and genetic mapping of nucleotide sequences and annotation of surrounding loci in barley. Mol Breeding. 2015;35:13. https://doi.org/10.1007/s11032-015-0253-1.
Brown J, Pirrung M, McCue LA. FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017. https://doi.org/10.1093/bioinformatics/btx373.
Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–95. https://doi.org/10.1093/bioinformatics/btp698.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. https://doi.org/10.1093/bioinformatics/btr330.
Acknowledgements
The acknowledge to the USDA-ARS small grains genotyping lab in Fargo, ND, USA for providing Illumina NextSeq500 sequencing platform.
Funding
This research was supported by the National Science Foundation CAREER grant No. 1253987 awarded to Dr. Robert S. Brueggeman. “Any opinions, findings, and conclusions or recommendations expressed in this project are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.”
Author information
Authors and Affiliations
Contributions
RB and SS designed the experiment. TF identified the mutants. SS, GA, AK, HA, TG, PG, XW, AS and PT performed the wet lab and greenhouse experiments. JR and SS carried out the bioinformatics analysis. RB and SS wrote the manuscript. RB, SS, JR, PT and GA proofread the manuscript. RB obtained funding for research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Pathogen inoculations and containment was carried out as per the local legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Seedling QCCJ phenotyping and root length measurement data on Hv584/rpr9 F2:6 RIL population. (XLSX 30 kb)
Additional file 2:
Table S2. List of 2701 redundant loci across the seven chromosomes of barley used for the genetic map construction. (XLSX 73 kb)
Additional file 3:
Figure S1. Linkage map of Hv584/rpr9 F2:6 recombinant inbred line population spanning the seven barley chromosomes containing 563 polymorphic non-redundant SNP markers, created using MapDisto software. (PDF 158 kb)
Additional file 4:
Figure S2. QTL map of rpr9 region on barley chromosome 3H for average diseases ratings on Hv584/rpr9 RILs inoculated with Pgt race QCCJB. X axis represents the all 90 non-redundant loci (representing 390 redundant loci) on the ch. 3H and Y axis represents the LOD score. Map was generated in QGene 4.4 using single trait MIM algorithm. (JPG 817 kb)
Additional file 5:
Figure S3. QTL mapping showed that the rpr9 region on barley chromosomes 3 is the only significant QTL detected for average seedling root length using the Hv584/rpr9 RIL population. X axis represents the non-redundant loci on the barley chromosomes and Y axis represents the LOD score. Map was generated in QGene 4.4 using single trait MIM algorithm. (JPG 372 kb)
Additional file 6:
Table S3. List of 487 exome capture target genes under the rpr9 QTL with their physical position and gene orientation on barley ch. 3H. Exome capture reads for Q21861-wt and Q21861-rpr9 mutant and their coverage ratio for each target is given. (XLSX 41 kb)
Additional file 7:
Figure S4. Possible orientation of gene cluster in the rpr9 region on barley chromosome 3H. (A) Current annotation of gene order in rpr9 region. (B) Proposed gene order orientation in Q21861-rpr9 mutant and Q21861-wt for gene HORVU3Hr1G074960 based on the exome capture and SNP marker SCRI-RS-27417 read alignment. Read alignment in Q21861-wt (4839 reads) was present across the CDS and 3′ UTR, however, in Q21861-rpr9 mutant 269 exome capture reads were aligned to 3′ coding and UTR region which includes the physical position of SNP marker SCRI-RS-27417. In figure, red arrows denote the genes present in the rpr9 region, black arrows represent the rpr9 flanking genes and asterisk with vertical black bar shows the position of SNP in marker SCRI-RS-27417 on chromosome 3H represented by horizontal white bar. (PDF 1320 kb)
Additional file 8:
Figure S5. Average root length distribution for Hv584 x rpr9 F6 RIL population (yellow bars) along with wild type Q21861 (green bar), mutant rpr9 (red bar) and Hv584 (blue bar) in increasing order. The Y-axis represents root length in millimeters and X-axis denote 95 Hv584 x rpr9 F6 RILs with parents used for root length measurement. (JPG 1239 kb)
Additional file 9:
Table S4. List of fifteen annotated high confidence SKP-1 like genes in barley whole genome assembly. (XLSX 51 kb)
Additional file 10:
Figure S6. Size determination of fractionated DNA after 25 min of barley DNA digestion with NEB DNA fragmentize enzyme on Bioanalyzer 1000 DNA chip. A 25-min enzymatic digestion produced DNA fragments in the size range of 250–450 base pairs, which was required for exome capture library preparation. The X-axis represents the fluorescence units and Y-axis represents the size of DNA fragments. The two terminal peaks represent the DNA ladder peaks with the lower marker at 25 base pairs (left) and the upper marker 1000 base pairs (right). (JPG 950 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Solanki, S., Richards, J., Ameen, G. et al. Characterization of genes required for both Rpg1 and rpg4-mediated wheat stem rust resistance in barley. BMC Genomics 20, 495 (2019). https://doi.org/10.1186/s12864-019-5858-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5858-z