
Abstract
Background
Bovine viral diarrhea virus (BVDV) is an economically important viral pathogen of domestic and wild ruminants. Apart from cattle, small ruminants (goats and sheep) are also the susceptible hosts for BVDV. BVDV infection could interfere both of the innate and adaptive immunity of the host, while the genes and mechanisms responsible for these effects have not yet been fully understood. Peripheral blood mononuclear cells (PBMCs) play a pivotal role in the immune responses to viral infection, and these cells were the target of BVDV infection. In the present study, the transcriptome of goat peripheral blood mononuclear cells (PBMCs) infected with BVDV-2 was explored by using RNA-Seq technology.
Results
Goat PBMCs were successfully infected by BVDV-2, as determined by RT-PCR and quantitative real-time RT-PCR (qRT-PCR). RNA-Seq analysis results at 12 h post-infection (hpi) revealed 499 differentially expressed genes (DEGs, fold-change ≥ ± 2, p < 0.05) between infected and mock-infected PBMCs. Of these genes, 97 were up-regulated and the remaining 352 genes were down-regulated. The identified DEGs were found to be significantly enriched for locomotion/ localization, immune response, inflammatory response, defense response, regulation of cytokine production, etc., under GO enrichment analysis. Cytokine-cytokine receptor interaction, TNF signaling pathway, chemokine signaling pathway, etc., were found to be significantly enriched in KEGG pathway database. Protein-protein interaction (PPI) network analysis indicated most of the DEGs related to innate or adaptive immune responses, inflammatory response, and cytokine/chemokine-mediated signaling pathway. TNF, IL-6, IL-10, IL-12B, GM-CSF, ICAM1, EDN1, CCL5, CCL20, CXCL10, CCL2, MAPK11, MAPK13, CSF1R and LRRK1 were located in the core of the network and highly connected with other DGEs.
Conclusions
BVDV-2 infection of goat PBMCs causes the transcription changes of a series of DEGs related to host immune responses, including inflammation, defense response, cell locomotion, cytokine/chemokine-mediated signaling, etc. The results will be useful for exploring and further understanding the host responses to BVDV-2 infection in goats.
Similar content being viewed by others


Background
Bovine viral diarrhea virus (BVDV) is the prototypic member of the genus Pestivirus in the family Flaviviridae, and two main different BVDV species have been recognized as BVDV-1 and BVDV-2 [1]. BVDV infection decreases productive performance and causes considerable economic losses in cattle industry worldwide [2]. Infections with both species of BVDV can induce similar diseases, from subclinical infections to severe clinical diseases including acute diarrhea, respiratory diseases, reproductive failures, congenital defects, and increased mortality due to immunosuppression [2,3,4]. Persistent infection is the common type of infection in cattle and the persistently infected (PI) animals are considered the main source of BVDV transmission [2, 5]. PI animals had also been detected in heterologous species, which amplify and facilitate the reservoirs for BVDV [6, 7]. Evidence of BVDV infection exists in 7 families (over 50 species) of Artiodactyla including Antilocapridae, Bovidae, Camelidae, Cervidae, Giraffidae, Suidae, and Tragulidae [6]. The circulation of BVDV-1 and BVDV-2 in cattle, pigs had been identified in China [8,9,10]. Our previous study has identified the prevalence of BVDV-1 in Chinese goat herds [11]. The infection of BVDV-2 in goat or sheep has been confirmed in India, Korea [12,13,14]. Therefore, the risk and prevalence of BVDV-1 and BVDV-2 in goat/sheep herds needs urgent attention.
Recently, high-throughput RNA-Sequencing (RNA-Seq) technologies give the opportunity to produce large numbers of sequence data in non-model organisms, and this method is better than the traditional microarray analysis [15, 16]. It provides a thorough understanding of the host defense mechanisms and immune evasion strategies of viral infection [17]. The transcriptional landscape in the host upon virus infection facilitates the understanding of host immune responses and defense mechanisms upon the pathogenic microorganism infection at whole mRNA level, and provides new approaches to the potential control of virus infections.
Two biotypes of BVDV are recognized: cytopathic (cp) and non-cytopathic (ncp) strains. In experimental infected calves, BVDV-specific antibody is first detected shortly after viral clearance for both biotypes. T cell proliferative responses are detectable by 3–4 weeks post-infection with cpBVDV; while delayed to about 6–8 weeks after ncpBVDV infection [18]. The primary site of BVDV replication is immune tissue, viral replication results in altered cell function or cell death in different lymphoid populations. The resulting immune suppression occurs in all acute BVDV infections [19]. Peripheral blood mononuclear cells (PBMCs), including lymphocytes, monocytes and macrophages, play a pivotal role in the host innate or adaptive immune responses to viral infection. PBMCs were the main target of BVDV infection, infection of lymphocytes and monocytes by BVDV resulted in lymphoid depletion of B cells, T helper cells, cytotoxic T cells and γ-δ T cells [20, 21]. PBMCs have been proved to be a suitable model for characterizing the host immune responses to virus infection and have been utilized for the evaluation of immune responses to animal viruses [17, 22, 23]. Global transcriptome analysis has been employed to explore the molecular events of host interaction with BVDV in bovine originated cells [24,25,26,27]. However, no report on BVDV-goat interactome is available to date. In this study, Illumina sequencing method was used to identify the transcriptome changes in BVDV-2 infected goat PBMCs. For the first time, we obtained the differentially expressed transcriptome profile in the goat PBMCs during BVDV-2 infection. The results will be helpful for better understanding the host responses to BVDV-2 infection and its relationship to viral pathogenesis in goats.
Results
Determination of BVDV-2 replication in goat PBMCs
To confirm the replication of BVDV-2 in goat PBMCs, RT-PCR and qRT-PCR were performed. As shown in Fig. 1a, 5’-UTR fragment with ~ 290 bp was amplified in infected goat PBMCs from 6 to 24 h post- infection (hpi). qRT-PCR detection showed similar results, the BVDV genome copy numbers increased from 6hpi and reached high levels at 12 and 24 hpi (Fig. 1b). These results confirmed the BVDV-2 infection in goat PBMCs. To explore the effect of early BVDV replication on gene expression, 12 hpi was selected for sampling and RNA-Seq. In addition, BVDV nucleotide was not detected in the mock infected PBMCs at any of the experimental time points.

Identification of viral infection in goat PBMCs. a Amplification of 5’UTR by RT-PCR at 6 h, 12 h and 24 hpi in BVDV-2 infected goat PBMCs. 1: Mock-infected PBMC at 12hpi; 2: Infected PBMC at 6 hpi; 3: Infected PBMC at 12 hpi; 4: Infected PBMC at 24 hpi; 5: DNA Marker DL-2000 plus. b Detection of viral genome copy numbers in goat PBMCs by qRT-PCR at different time points
Transcriptome quantification
After RNA-seq, a total of 122,154,110 raw reads (B2: 62679448; N: 59474662) were obtained. After removing low-quality reads and reads with adaptor sequences, 113,957,630 clean reads (B2: 58748222; N: 55209408) were obtained (Table 1). We then queried the clean reads against the latest reference genome (Gallus_gallus-5.0, https://www.ncbi.nlm.nih.gov/assembly/GCF_001704415.1) and mapped using TopHat (http://tophat.cbcb.umd.edu/). For B2 and N samples, 50,308,617 and 47,349,951 reads were mapped to the reference genome with mapped rate of 85.63 and 85.76%, respectively. Among the matched 13,974 target genes for B2, 9883 with RPKM≥1; For N, 13905 target genes were matched and 9952 with RPKM≥1 (Table 1).
DEGs analysis and functional annotation
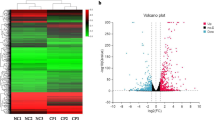
After the gene mapping and the Cuffdiff analyses in terms of FRKM, a total of 449 genes were identified as significantly differentially expressed for infected group (B2), when comparing with the mock-infected control group (N, fold change (FC) ≥ ± 2, p < 0.05). Among the 449 genes, 97 were up-regulated and 352 genes were down-regulated (Fig. 2, Additional file 4: Figure S1, Additional file 1: Table S1).

Summary of the differentially expressed genes between mock and BVDV-2 infected samples at 12hpi
The 449 DEGs were annotated to 54 different GO terms. The up-regulated DEGs were annotated to 38 GO terms and the down-regulated DEGs were annotated to 53 GO terms (Fig. 3). The most annotated GO terms were metabolic process (BP), cellular process (BP), response to stimulus (BP), biological regulation (BP), localization (BP), cell (CC), cell part (CC), membrane (CC), membrane part (CC), extracellular region (CC), organelle (CC) and binding (MF), etc. (Fig. 3).

GO annotation for the DEGs between mock and BVDV-2 infected goat PBMCs
Functional and PPI analysis of DEGs
After GO enrichment analysis, DEGs were enriched into different GO terms. For immune related DEGs, significant enrichment was observed in regulation of locomotion/ localization, immune response, inflammatory response, immune system process, defense response, regulation of cytokine production of BP group and in cytokine activity, chemokine activity, receptor binding of MF group. (Fig. 4 and Additional file 2: Table S2).

GO enrichment analysis for the DEGs between mock and BVDV-2 infected goat PBMCs. Circles indicate numbers of enriched genes and colors depict the P value
To further define DEGs function, KEGG pathway/enrichment analysis was performed. Among the fifteen significantly enriched pathways, cytokine-cytokine receptor interaction, TNF signaling pathway, chemokine signaling pathway, complement and coagulation cascades and NOD-like receptor signaling pathway were found to be enriched to canonical pathways (Fig. 5, Table 2 and Additional file 3: Table S3). The cytokine-cytokine receptor interaction pathway was the pathway enriched with most number of DEGs (n = 29). For the 29 DEGs, CCL4, CCL3, CXCL10, CCL5, CCL22, CCL20, GM-CSF, TNF, IL-6, IL-17A, IL-12B, IL-19, IL-10, TNFRSF13C, TNFRSF8, TNFRSF9 and XCL1 were up-regulated; while TNFSF12, CSF1R, TNFRSF21, CSF3R, regakine-1(LOC102170772), CCL2, CCL24, CCL17, CCL14, CCL25, IL-5RA and PPBP were down-regulated. In addition, TNF, IL-6, CXCL10, CCL4, CCL3, CCL5, CCL20, CCL2 and regakine-1 were enriched in at least three of the six pathways mentioned above (Table 2).

Top 30 pathways enriched in KEGG database for the DEGs between mock and BVDV-2 infected goat PBMCs. Circles indicate numbers of enriched genes and colors depict the P value
STRING analysis was used to explore the potential interaction network of the DEGs. As shown in Fig. 6, most of the DEGs related to innate or adaptive immune responses, inflammatory response, cytokine/chemokine-mediated signaling pathway, etc. Among the up-regulated genes, TNF, IL-6, IL-10, IL-12B, GM-CSF, ICAM1, EDN1, CCL20, CXCL10 and CCL5 were located in the core of the network and linked to lots of other DGEs; for the down-regulated genes, the key points included CCL2, MAPK11, MAPK13, CSF1R and LRRK1, etc., which linked to more genes. In addition, not all DEGs showed connection with others because their functions were either unrelated or have not yet been clarified (Additional file 5: Figure S2). These DGEs were not included in Fig. 6 during the analysis.

PPI network of the selected DEGs based on STING analysis results and fold change information. The up-regulated genes are shown in red and down-regulated genes are shown in blue with the gradient showing the extent of expression. The size of the node indicates connectivity
Partial validation of RNA-Seq data
To further validate the RNA-Seq data, DEGs with annotations associated with immune responses from RNA-seq were selected for qRT-PCR analysis. As shown in Table 3, eighteen selected genes exhibited a concordant direction both in RNA-Seq and qRT-PCR analysis. The correlation coefficient between RNA-Seq and qRT-PCR results was high (R2 = 0.91). Some of the DEGs were further determined by Western blot or ELISA, as shown in Fig. 7, the expression of Annexin A2 decreased obviously (Fig. 7a) and the expression of TNF-α, GM-CSF and IL-6 were increased significantly (Fig. 7b). In addition, Viperin (used as a control) expression showed no change between infected and mock-infected group, which was consistent with the RNA-Seq result. These results confirmed that the differential expression genes identified by RNA-Seq is reliable.

Partial validation of RNA-Seq data by Western blot and ELISA. a The expression of Annexin A2 and Viperin in the samples was determined by Western blot using rabbit anti-Viperin polyclonal antibody (Abcam) and mouse anti-AnnexinA2 antibody (Santa Cruz). b The concentration of GM-CSF, TNF- and IL-6 present in the samples were determined by commercial ELISA kit and calculated with the formula derived from the standard curve. Data was shown as the mean ± S.D. Columns marked with * (P < 0.05) are significantly different from each other
Discussions
BVDV is one of the most important viral diseases of various species of domestic animals. Regardless of clinical presentation, all BVDV infections result in significant loss of immune tissue, the resulting immune suppression and increased severity of subsequent infections [28]. The different genotypes and biotypes of BVDV, wide spectrum of susceptible host, ability to induce persistently infection, as well as its ability to interfere both innate and adaptive immunity of the host, make it difficult for prevention and control.
Microarray and RNA-Seq analysis are the two main techniques for transcriptome analysis. There were many reports about transcriptome changes upon animal virus infection by microarray analysis [29,30,31]. As a revolutionary tool, RNA-Seq has been widely used in recent years, such as BTV, PRRSV, PPRV and NDV [22, 32,33,34]. Several studies had explored the effect of BVDV infection on mRNA expression changes in different bovine cells [25, 27, 35], but understanding of BVDV-host interaction is still far from complete. Comparing to the studies in cattle little is known regarding the cellular impact of BVDV-2 infection in goats. Herein, in the present study, goat PBMCs were infected with a ncp BVDV-2 strain, and the transcriptome was evaluated at 12 hpi to identify the global gene expression changes, to understand and delineate the mechanism of early responses induced by virus infection. Transcriptional changes of the DEGs involved in immunological processes were analyzed specially. The results from GO, KEGG and PPI analysis indicated that various numbers of DEGs were involved in different biological processes of host immune responses.
The innate immunity is the first defense line against infectious disease. The PBMCs were the gatekeeper of virus spread during systemic viral infections and include several subpopulations that may cooperate in the activation processes [36]. These cells play important role in innate and adaptive immune responses. The pathogen-associated molecular pattern receptors (PAMPs) such as Toll-like receptors (TLRs) or RIG-I-like receptors (RLRs) recognize pathogens, then released a series of related antiviral cytokines. In this study, under BVDV-2 infection, the expression of TLRs, RLRs, interferon (IFN) and the ISGs were not significant induced. Contrarily, the mRNA levels of lysozyme, beta-defense, competent of complement system (C1q, C1r, C1s, CFD) and IFITM3 (one of the ISGs) was found to be down-regulated. A previous study showed that TLR3, type I IFN gene was up-regulated in ncp BVDV-infected monocytes at 1hpi but not at 24hpi [37]. It has also been reported that ncpBVDV dose not induce type I IFN in vitro and block the induction of type I IFN by dsRNA or other viruses and interferon tau-stimulated ISGs expression [18, 38]. The interaction of ncp BVDV with its host cells impairs both of the innate and adaptive immunity [39]. During BVDV infection, viral RNA firstly triggers IFN synthesis, while the viral RNase Erns protein inhibits IFN expression; in addition, Npro promotes the degradation of IRF-3, which effectively blocks IFN expression in BVDV-infected cells [39,40,41]. So, the inhibition of immune genes mentioned above might result from the interference of BVDV-2 infection on the related immune pathways. The inhibition of IFN synthesis and other competent of innate immunity might play an important role in escaping innate immunity and the establishment of effective infection for BVDV in host cells.
The complement system, which consists of both soluble factors and cell surface receptors, is one of the major innate defense systems. The main role of complement system is to protect against infections, it also links the innate and adaptive immune responses [42]. In the present study, C1q, C1r, C1s, CFD were down-regulated, while C3 was up-regulated; imply that BVDV-2 infection in goat PBMCs may inhibit the classical or alternative pathways of the complement system activation.
Inflammation was one of the important anti-microorganism responses upon infection. Once exposed to infectious agents, host cells produced certain cytokines, such as TNF-α, IL-1 and IL-6, resulting in the development of inflammation. TNF-α is an essential mediator of inflammation and also facilitates the transition from innate to adaptive immunity. IL-6 affects both inflammation and adaptive immunity. It promotes some aspects of inflammation, especially in response to tissue damage and severe infections since it is a major mediator of the acute phase reaction and of septic shock. Many viruses, including BVDV, could induce inflammation after infection. Studies in cattle and sheep have shown that the pathology of BVDV associated with the replication of virus and the production of pro-inflammatory cytokines and induced inflammatory responses [43,44,45]. One study examined the mRNA expression in tracheo-bronchial lymph nodes of BVDV infected beef calves and found that high virulence BVDV-2 strain induced pro-inflammatory (TNF-α, IL-12, IL-1β, IL-2, IFN-γ) and anti-inflammatory (IL-4 and IL-10) cytokines, while low virulence BVDV-1a strain only up-regulated IL-12 and IL- 15 gene expression [46]. In an other study, both cp and ncp BVDV biotypes suppressed pro-inflammatory cytokines TNF-α, IL-1β, and IL-6, but did not change IL-12 and INF-γ gene expression in bovine PBMCs [37]. As a member of pestivirus, virulent CSFV infection in pigs resulted in the secretion of cytokines associated with inflammation or apoptosis such as TNF-α, IL-2, IL-4, IL-6, and IL-10 [47]. The BVDV-2 strain used in this study induced fever, viremia and lymphopenia in experimentally infected goats (unpublished data). The clinical signs, together with the pattern of increased TNF-α, IL-6, IL-12, IL-10 and IL-17 mRNA level observed in BVDV2-infected goat PBMCs in this study, suggested that BVDV-2 induced an acute inflammatory response in the early stage of infection. The enriched pathways related to cytokine/chemokine signaling provide obvious evidence that specific immune responses are activated during that acute phase of BVDV infection.
The serum amyloid A (SAA) family comprises a number of apolipoproteins, which include acute-phase SAAs (A-SAAs) and constitutive SAAs (C-SAAs). A-SAAs have been identified in all vertebrates and could be induced as much as 1000-fold during inflammation [48]. Transcription of SAAs members LOC102168428 (SAA like) and LOC100860781 (SAA3) were found to be up-regulated in this study. Members of A-SAAs have been identified to be activated upon infection and proven useful as an inflammatory marker for many viral diseases of animals [49,50,51]. Similarly, Ganheim C. et al. reported a significant acute phase response with elevated values of SAA and other acute phase proteins in calves experimentally infected with BVDV [52]. So, SAA might be considered as a diagnostic marker for BVDV induced inflammation.
Viral infection usually induces the recruitment of inflammatory cells to the infection site, and this activity is regulated by various cytokines and chemokines. Chemokines are a great superfamily of at least 50 small (8- to 10-kDa) structurally related chemoattractant proteins, which play a key role in initiating the innate and subsequently adaptive immune responses. Chemokines have many roles in the regulation of leukocyte development, angiogenesis, tumor growth and metastasis [53]. They coordinate the migration of leukocytes and hence dictate the course of many inflammatory and immune responses by recruiting different immune cells toward sites of infection [54]. Monocytes/macrophages are part of the first line of defense and have been shown to release a variety of chemokines in response to infection [31]. Studies have determined chemokines expression upon the infection of several viruses, such as PRRSV, PEDV and PCV2 [30, 32, 55]. Study by Helal et al. showed that low expression of CXCR4 and high expression of IL-10 is associated with the production of PI calves in a herd level [56]. Trzeciak-Ryczek A. et al. reported that ncpBVDV infection increases CXCR4, CXCL12 mRNA expression in bovine PBMCs [57]. In another study, detection of chemokine profile in cp and ncp BVDV infected bovine monocytes/macrophages demonstrated up-regulation of several key chemokines of the CCL and CXCL families to cpBVDV, but not ncpBVDV [31]. In our study, BVDV-2 infected goat PBMCs showed increased transcription of CCL3, CCL4, CCL5, CCL20, CXCL10 and decrease of CCL2, suggesting that such chemokines may contribute to the recruitment and regulation of macrophages or other inflammatory cells to the infected sites after infection.
GM-CSF was another up-regulated DEG found in our study. It is not only an inducer of differentiation and proliferation of granulocytes/macrophages but also involved in a wide range of biological processes in both innate and adaptive immunity [58]. GM-CSF has been widely used as adjuvant for vaccines and has been shown powerful as an important therapeutic target in several autoimmune and inflammatory diseases [58, 59]. The induction of GM-CSF expression has been identified in several viruses and shown to alter pathogenic “M1-like” macrophage inflammation after influenza A virus infection [60,61,62]. The high expression of GM-CSF in goat PBMCs indicated the regulation role of it in host immune responses after BVDV-2 infection.
Conclusions
In conclusion, the changes of a series of immunity related genes in BVDV-2 infected goat PBMCs were observed by RNA-Seq analysis. The results will be useful for exploring and further understanding host responses to BVDV-2 infection in goats.
Methods
Virus and cells
MDBK cells were (purchased from the China Institute of Veterinary Drug Control) cultured in DMEM (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Transgen, Bio, Inc.,China) and kept at 37 °C. For the production of stock virus, MDBK cell monolayer was inoculated with the ncpBVDV2 strain C201604 (GenBank No. MG420995, identified and kept in our lab) with multiplicity of infection (MOI) of 0.1. At 3 days post infection, the infected cells were harvested, lysed by three cycles of freeze-thaw, centrifuged at 8000×g and 4 °C for 10 min and then aliquoted and stored at − 70 °C until use.
Goat, PBMCs culture and virus infection
Goats used for blood collection were purchased from a goat farm (private farm, 210 goats) located in Jurong county, Jiangsu province. The goats were vaccinated against goat pox virus, foot and mouth disease virus, peste des petits ruminants virus and Mycoplasma mycoides subsp.capri. The animal was further screened for BVDV and viral specific neutralizing antibodies using RT-PCR and virus neutralization test, respectively. EDTA anticoagulant whole blood were collected from the goats (n = 3) and PBMCs were separated using Histopaque-1077 (Sigma) by density gradient centrifugation at 500×g for 20 min, then washed three times with RPMI-1640 medium at 500×g for 10 min. Cells from each animal were suspended to 1× 106 cells/mL with complete RPMI 1640 medium (RPMI 1640 containing 10% FBS) and seeded in six well plates (n = 4, 8 wells/animal). Two plates were infected with BVDV2 (2 × 105 TCID50/mL, MOI = 0.1) and the other two plates were served as mock-infected control. Samples from one infected plate and one mock-infected plate were collected at 6, 12 and 24 hpi for virus replication detection. At 12 hpi, medium was discarded and the cells of the left two plates (infected and mock-infected) were washed twice with PBS, mixed and pelleted for RNA-Seq.
RNA extraction, library preparation and RNA sequencing (RNA-seq)
Total RNA was extracted from the infected (B2) and mock-infected (N) PBMCs using TRIzol reagent (Invitrogen) according the manufacturer’s instructions and genomic DNA was removed using DNase I (TaKara). RNA quality (RNA integrity number, RIN) was determined using Agilent 2100 Bioanalyser and quantified using the ND-2000 (NanoDrop). High-quality RNA sample (OD260/280 = 1.8~2.2, OD260/230 ≥ 2.0, RIN ≥ 6.5, 28S:18S ≥ 1.0, > 10 μg) was used to construct sequencing library.
RNA-Seq transcriptome libraries were prepared by TruSeqTM RNA sample preparation Kit from Illumina (San Diego, CA), using 1 μg of total RNA. Shortly, mRNA was isolated with polyA selection by magnetic oligo (dT) beads and fragmented into small pieces using fragmentation buffer. cDNA synthesis, end repair, A-base addition and ligation of the Illumina-indexed adaptors were performed according to Illumina’s protocol. Libraries were then selected for cDNA target fragments of 200–300 bp on 2% Low Range Ultra Agarose followed by PCR amplified for 15 cycles using Phusion DNA polymerase (NEB). After quantified by TBS380, paired-end libraries prepared for both the infected and mock-infected samples (B2 and N) were sequenced with the Illumina HiSeq PE 2X151bp read length. RNA-Seq was performed by Shanghai Biozeron Biotech Co. Ltd.
Reads quality control and mapping
The raw paired end reads were trimmed and quality controlled by Trimmomatic with default parameters (http://www.usadellab.org/cms/index.php?page=trimmomatic). Clean reads were then separately aligned to the reference caprine genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_001704415.1) with orientation mode using TopHat software (http://tophat.cbcb.umd.edu/), which can align RNA-Seq reads to a genome in order to identify gene expression and exon-exon splice junctions. It is built on the ultrafast short read mapping program Bowtie2 to map with default parameters.
Differential expression genes (DEGs) analysis and annotation
To identify DEGs between the two samples, the expression level for each transcript was calculated using the fragments per kilobase of exon per million mapped reads (FPKM) method. Cuffdiff (http://cufflinks.cbcb.umd.edu/) was used for differential expression analysis and the DEGs were selected using the following criteria: the logarithmic of fold change was greater than 2 and the p-fdr should be less than 0.05. All DEGs were subjected to Gene Ontology (GO) annotation based on GO database.
Functional and protein-protein interaction (PPI) analysis of DEGs
To understand the functions of the DEGs, GO functional enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were carried out by Goatools (https://pypi.org/project/goatools/) and KOBAS (http://kobas.cbi.pku.edu.cn/). DEGs were significantly enriched in GO terms and KEGG pathways when their p-value was less than 0.05.
PPI network among the DEGs was analyzed using the STRING (http://string-db.org/) database, which included direct and indirect associations of proteins. After analyzing the result from STRING analysis and expression change information for each DEG, the network figure was drawn for the selected DEGs (connected with one or more DEGs) by using Cytoscape software.
RT-PCR and quantitative real-time RT-PCR (qRT-PCR)
Total RNAs were extracted from infected and mock-infected PBMCs using TransZol UP reagent (Transgen, Bio, Inc.,China) according to the manufacturer’s instruction.
The RT-PCR was performed to identify the replication of BVDV and carried out with EasyScript one-step RT-PCR supermix (Transgen, Bio, Inc., China) in a 20 μl reaction mixture containing 10 μl of 2 × R-Mix, 20 pM of each primer (F: 5'-ATGCCCWTAGTAGGACTAGCA-3', R: 5'-TCAACTCCATGTGCCATGTAC-3'), 0.4 μl of E-Mix, 2 μl extracted RNA and 6.6 μl ddH2O. The reaction was run in a thermocycler (Mjmini, BIO-RAD) with the following program: reverse transcription at 45 °C for 30 min; denaturation at 94 °C for 5 min, 35 cycles composed of denaturation at 94 °C for 30 s, annealing for 30 s at 54 °C and extension at 72 °C for 30 s; and was terminated with a final extension of 10 min at 72 °C. Amplification products were detected by electrophoresis in 1.2% agarose gels.
The qRT-PCR amplification was carried out with TransScript one-step qRT- PCR supermix (Transgen, Bio, Inc., China) in a 20 μl reaction mixture containing 10 μl of 2 × Supermix, 20 pM of each primer (Table 4), 0.5 μl of E-Mix, 0.4 μl of passive reference Dye and 2 μl extracted RNA. The reaction was run in ABI Step One instrument as the following procedure: samples were incubated at 45 °C for 5 min firstly; then heated at 94 °C for 30 s and a two-step cycle (5 s at 94 °C, 30 s at 60 °C) was repeated for 40 cycles. GAPDH was used as the internal control and relative quantification of target gene expression was the target transcript in infected group to that of mock-infected group and expressed as –ΔΔCt.
Virus titration
For titration of virus stock, four replicates of 10-fold serially diluted virus (starting from 1/10) were inoculated on MDBK cell monolayer in 96-well culture plates. After 48 h incubation, the culture plates were fixed at 4 °C for 30 min with ice cold absolute ethyl alcohol and subjected to immunofluorescence staining with BVDV-specific mouse monoclonal antibody Mix (RAE2020, AHVLA, UK; 1:200 diluted in PBS) and FITC-conjugated goat anti-mouse IgG (BOSTER, Wuhan, China; 1:200 diluted in PBS). The fluorescence signals were observed undera fluorescence microscopy (ZEISS) and viral titers was expressed as the 50% tissue culture infective dose (TCID50)/mL by Reed-Muench method.
Western blot
Cell lysates samples were separated by 12% SDS-PAGE and transferred onto nitrocellulose membranes (Pall) using a semi-dry transfer cell (Bio-Rad) at 1 V/cm2 for 40 min. The membrane was treated sequentially with 5% skimmed milk in PBST (PBS containing 0.05% Tween-20) at 37 °C for 2 h, with different primary antibodies (1/200 diluted rabbit anti-Viperin polyclonal antibody (Abcam), 1/400 diluted mouse anti-AnnexinA2 antibody (Santa Cruz), 1/1000 diluted anti-β-actin/GAPDH monoclonal antibody (Transgen, Bio, Inc., China)) at 37 °C for 2 h, and with different secondary antibodies (1/1000 diluted rabbit anti-mouse or goat anti-rabbit IgG antibody conjugated to HRP (Transgen, Bio, Inc., China)). After three washes with PBST, the color development was performed using enhanced chemiluminescence luminal reagent (Thermo Scientific Pierce).
Elisa
The concentration of GM-CSF, TNF-α and IL-6 present in the infected and mock-infected PBMCs samples were determined by commercial ELISA kit (Jiangsu Yutong Biotech Co., Ltd., China) according to the manufacturer’s instructions and calculated with the formula derived from the standard curve.
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Abbreviations
- BVDV:
-
Bovine viral diarrhea virus
- DEGs:
-
Differentially expressed genes
- FPKM:
-
Fragments per kilobase of exon per million mapped reads
- GO:
-
Gene Ontology
- Hpi:
-
Hours post infection
- IFN:
-
Interferon
- ISGs:
-
Interferon stimulated genes
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MOI:
-
Multiplicity of infection
- PAMPs:
-
Pathogen-associated molecular pattern receptors
- PBMCs:
-
Peripheral blood mononuclear cells
- PBST:
-
PBS containing 0.05% Tween-20
- RLRs:
-
RIG-I-like receptors
- RNA-Seq:
-
RNA sequencing
- SAA:
-
serum amyloid A
- TCID50 :
-
Median tissue culture infective dose
- TLRs:
-
Toll-like receptors
References
Smith DB, Meyers G, Bukh J, Gould EA, Monath T, Scott Muerhoff A, Pletnev A, Rico-Hesse R, Stapleton JT, Simmonds P, et al. Proposed revision to the taxonomy of the genus Pestivirus, family Flaviviridae. J Gen Virol. 2017;98(8):2106–12.
Pinior B, Firth CL, Richter V, Lebl K, Trauffler M, Dzieciol M, Hutter SE, Burgstaller J, Obritzhauser W, Winter P, et al. A systematic review of financial and economic assessments of bovine viral diarrhea virus (BVDV) prevention and mitigation activities worldwide. Pre Vet Med. 2017;137(Pt A):77–92.
Walz PH, Grooms DL, Passler T, Ridpath JF, Tremblay R, Step DL, Callan RJ, Givens MD. American College of Veterinary Internal M: control of bovine viral diarrhea virus in ruminants. J Vet Intern Med. 2010;24(3):476–86.
Wolff PL, Schroeder C, McAdoo C, Cox M, Nelson DD, Evermann JF, Ridpath JF. Evidence of bovine viral diarrhea virus bnfection in three species of sympatric wild ungulates in Nevada: life history strategies may maintain endemic infections in wild populations. Front Microbiol. 2016;7:292.
Ostachuk A. Bovine viral diarrhea virus structural protein E2 as a complement regulatory protein. Arch Virol. 2016;161(7):1769–82.
Passler T, Walz PH. Bovine viral diarrhea virus infections in heterologous species. Anim Health Res Rev. 2010;11(2):191–205.
Griebel PJ. BVDV vaccination in North America: risks versus benefits. Anim Health Res Rev. 2015;16(1):27–32.
Deng Y, Sun CQ, Cao SJ, Lin T, Yuan SS, Zhang HB, Zhai SL, Huang L, Shan TL, Zheng H, et al. High prevalence of bovine viral diarrhea virus 1 in Chinese swine herds. Vet Microbiol. 2012;159(3–4):490–3.
Ran X, Chen X, Ma L, Wen X, Zhai J, Wang M, Tong X, Hou G, Ni H. A systematic review and meta-analysis of the epidemiology of bovine viral diarrhea virus (BVDV) infection in dairy cattle in China. Acta Trop. 2019;190:296–303.
Tao J, Liao J, Wang Y, Zhang X, Wang J, Zhu G. Bovine viral diarrhea virus (BVDV) infections in pigs. Vet Microbiol. 2013;165(3–4):185–9.
Mao L, Li W, Yang L, Wang J, Cheng S, Wei Y, Wang Q, Zhang W, Hao F, Ding Y, et al. Primary surveys on molecular epidemiology of bovine viral diarrhea virus 1 infecting goats in Jiangsu province. China BMC Vet Res. 2016;12(1):181.
Han YJ, Chae JB, Chae JS, Yu DH, Park J, Park BK, Kim HC, Yoo JG, Choi KS. Identification of bovine viral diarrhea virus infection in Saanen goats in the Republic of Korea. Trop Anim Health Prod. 2016;48(5):1079–82.
Mishra N, Dubey R, Rajukumar K, Tosh C, Tiwari A, Pitale SS, Pradhan HK. Genetic and antigenic characterization of bovine viral diarrhea virus type 2 isolated from Indian goats (Capra hircus). Vet Microbiol. 2007;124(3–4):340–7.
Mishra N, Rajukumar K, Tiwari A, Nema RK, Behera SP, Satav JS, Dubey SC. Prevalence of bovine viral diarrhoea virus (BVDV) antibodies among sheep and goats in India. Trop Anim Health Prod. 2009;41(7):1231–9.
Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8(6):469–77.
Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107(1):1–15.
Zhao FR, Xie YL, Liu ZZ, Shao JJ, Li SF, Zhang YG, Chang HY. Transcriptomic analysis of porcine PBMCs in response to FMDV infection. Acta Trop. 2017;173:69–75.
Brackenbury LS, Carr BV, Charleston B. Aspects of the innate and adaptive immune responses to acute infections with BVDV. Vet Microbiol. 2003;96(4):337–44.
Ridpath JF, Neill JD. Pestiviruses: old enemies and new challenges. Anim Health Res Rev. 2015;16(1):1–3.
Chase CC, Thakur N, Darweesh MF, Morarie-Kane SE, Rajput MK. Immune response to bovine viral diarrhea virus--looking at newly defined targets. Anim Health Res Rev. 2015;16(1):4–14.
Song QJ, Weng XG, Cai DJ, Zhang W, Wang JF. Forsythoside a inhibits BVDV replication via TRAF2-dependent CD28-4-1BB signaling in bovine PBMCs. PLoS One. 2016;11(9):e0162791.
Singh A, Prasad M, Mishra B, Manjunath S, Sahu AR, Bhuvana Priya G, Wani SA, Sahoo AP, Kumar A, Balodi S, et al. Transcriptome analysis reveals common differential and global gene expression profiles in bluetongue virus serotype 16 (BTV-16) infected peripheral blood mononuclear cells (PBMCs) in sheep and goats. Genomics data. 2017;11:62–72.
Manjunath S, Mishra BP, Mishra B, Sahoo AP, Tiwari AK, Rajak KK, Muthuchelvan D, Saxena S, Santra L, Sahu AR, et al. Comparative and temporal transcriptome analysis of peste des petits ruminants virus infected goat peripheral blood mononuclear cells. Virus Res. 2017;229:28–40.
Oguejiofor CF, Cheng Z, Abudureyimu A, Anstaett OL, Brownlie J, Fouladi-Nashta AA, Wathes DC. Global transcriptomic profiling of bovine endometrial immune response in vitro. II. Effect of bovine viral diarrhea virus on the endometrial response to lipopolysaccharide. Biol Reprod. 2015;93(4):101.
Yamane D, Zahoor MA, Mohamed YM, Azab W, Kato K, Tohya Y, Akashi H. Microarray analysis reveals distinct signaling pathways transcriptionally activated by infection with bovine viral diarrhea virus in different cell types. Virus Res. 2009;142(1–2):188–99.
Demasius W, Weikard R, Hadlich F, Muller KE, Kuhn C. Monitoring the immune response to vaccination with an inactivated vaccine associated to bovine neonatal pancytopenia by deep sequencing transcriptome analysis in cattle. Vet Res. 2013;44:93.
Villalba M, Fredericksen F, Otth C, Olavarria V. Transcriptomic analysis of responses to cytopathic bovine viral diarrhea virus-1 (BVDV-1) infection in MDBK cells. Mol Immunol. 2016;71:192–202.
Taxis TM, Bauermann FV, Ridpath JF, Casas E. Circulating MicroRNAs in serum from cattle challenged with bovine viral diarrhea virus. Front Genet. 2017;8:91.
Zhang H, Li Y, Huang X, Zheng C. Global transcriptional analysis of model of persistent FMDV infection reveals critical role of host cells in persistence. Vet Microbiol. 2013;162(2–4):321–9.
Li W, Liu S, Wang Y, Deng F, Yan W, Yang K, Chen H, He Q, Charreyre C, Audoneet JC. Transcription analysis of the porcine alveolar macrophage response to porcine circovirus type 2. BMC Genomics. 2013;14:353.
Burr S, Thomas C, Brownlie J, Offord V, Coffey TJ, Werling D. Potential evidence for biotype-specific chemokine profile following BVDV infection of bovine macrophages. Vet Immunol Immunopathol. 2012;150(1–2):123–7.
Badaoui B, Rutigliano T, Anselmo A, Vanhee M, Nauwynck H, Giuffra E, Botti S. RNA-sequence analysis of primary alveolar macrophages after in vitro infection with porcine reproductive and respiratory syndrome virus strains of differing virulence. PLoS One. 2014;9(3):e91918.
Liu W, Qiu X, Song C, Sun Y, Meng C, Liao Y, Tan L, Ding Z, Liu X, Ding C. Deep sequencing-based transcriptome profiling reveals avian interferon-stimulated genes and provides comprehensive insight into Newcastle disease virus-induced host responses. Viruses. 2018;10(4). https://doi.org/10.3390/v10040162.
Manjunath S, Kumar GR, Mishra BP, Mishra B, Sahoo AP, Joshi CG, Tiwari AK, Rajak KK, Janga SC. Genomic analysis of host - Peste des petits ruminants vaccine viral transcriptome uncovers transcription factors modulating immune regulatory pathways. Vet Res. 2015;46:15.
Neill JD, Ridpath JF. Gene expression changes in MDBK cells infected with genotype 2 bovine viral diarrhoea virus. Vet Microbiol. 2003;96(4):301–12.
Colavita F, Bordoni V, Caglioti C, Biava M, Castilletti C, Bordi L, Quartu S, Iannetta M, Ippolito G, Agrati C, et al. ZIKV infection induces an inflammatory response but fails to activate types I, II, and III IFN response in human PBMC. Mediat Inflamm. 2018;2018:2450540.
Lee SR, Pharr GT, Boyd BL, Pinchuk LM. Bovine viral diarrhea viruses modulate toll-like receptors, cytokines and co-stimulatory molecules genes expression in bovine peripheral blood monocytes. Comp Immunol Microb. 2008;31(5):403–18.
Cheng Z, Chauhan L, Barry AT, Abudureyimu A, Oguejiofor CF, Chen X, Wathes DC. Acute bovine viral diarrhea virus infection inhibits expression of interferon tau-stimulated genes in bovine endometrium. Biol Reprod. 2017;96(6):1142–53.
Peterhans E, Schweizer M. BVDV: a pestivirus inducing tolerance of the innate immune response. Biologicals. 2013;41(1):39–51.
Hilton L, Moganeradj K, Zhang G, Chen YH, Randall RE, McCauley JW, Goodbourn S. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J Virol. 2006;80(23):11723–32.
Matzener P, Magkouras I, Rumenapf T, Peterhans E, Schweizer M. The viral RNase E (rns) prevents IFN type-I triggering by pestiviral single- and double-stranded RNAs. Virus Res. 2009;140(1–2):15–23.
Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology. 2011;411(2):362–73.
Bielefeldt-Ohmann H. The pathologies of bovine viral diarrhea virus infection. A window on the pathogenesis. Vet Clin N Am-Food A. 1995;11(3):447–76.
Hewicker-Trautwein M, Trautwein G, Frey HR, Liess B. Variation in neuropathogenicity in sheep fetuses transplacentally infected with non-cytopathogenic and cytopathogenic biotypes of bovine-virus diarrhoea virus. Zentralbl Veterinarmed B. 1995;42(9):557–67.
Ellis JA, West KH, Cortese VS, Myers SL, Carman S, Martin KM, Haines DM. Lesions and distribution of viral antigen following an experimental infection of young seronegative calves with virulent bovine virus diarrhea virus-type II. Can J Vet Res. 1998;62(3):161–9.
Palomares RA, Brock KV, Walz PH. Differential expression of pro-inflammatory and anti-inflammatory cytokines during experimental infection with low or high virulence bovine viral diarrhea virus in beef calves. Vet Immunol Immunopathol. 2014;157(3–4):149–54.
Wang J, Sun Y, Meng XY, Li LF, Li Y, Luo Y, Wang W, Yu S, Yin C, Li S, et al. Comprehensive evaluation of the host responses to infection with differentially virulent classical swine fever virus strains in pigs. Virus Res. 2018;255:68–76.
Uhlar CM, Whitehead AS. Serum amyloid a, the major vertebrate acute-phase reactant. EurJ Biochem. 1999;265(2):501–23.
Hulten C, Sandgren B, Skioldebrand E, Klingeborn B, Marhaug G, Forsberg M. The acute phase protein serum amyloid a (SAA) as an inflammatory marker in equine influenza virus infection. Acta Vet Scand. 1999;40(4):323–33.
Pomorska-Mol M, Markowska-Daniel I, Kwit K, Stepniewska K, Pejsak Z. C-reactive protein, haptoglobin, serum amyloid a and pig major acute phase protein response in pigs simultaneously infected with H1N1 swine influenza virus and Pasteurella multocida. BMC Vet Res. 2013;9:14.
Stenfeldt C, Heegaard PM, Stockmarr A, Tjornehoj K, Belsham GJ. Analysis of the acute phase responses of serum amyloid a, haptoglobin and type 1 interferon in cattle experimentally infected with foot-and-mouth disease virus serotype O. Vet Res. 2011;42:66.
Ganheim C, Hulten C, Carlsson U, Kindahl H, Niskanen R, Waller KP. The acute phase response in calves experimentally infected with bovine viral diarrhoea virus and/or Mannheimia haemolytica. J Vet Med B Infect Dis Vet Public Health. 2003;50(4):183–90.
Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. New Engl J Med. 2006;354(6):610–21.
Sallusto F, Lanzavecchia A. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol Rev. 2000;177:134–40.
Wang X, Ohnstad M, Nelsen A, Nelson E. Porcine epidemic diarrhea virus does not replicate in porcine monocyte-derived dendritic cells, but activates the transcription of type I interferon and chemokine. Vet Microbiol. 2017;208:77–81.
Helal MA, Hoshino Y, Takagi S, Tajima M. C-X-C chemokine receptor type 4 and cytokine expressions in cows of a dairy herd with high prevalence of calves persistently infected with bovine viral diarrhea virus. Jap J Vet Res. 2013;61(3):109–16.
Weiner CM, Smirnova NP, Webb BT, Van Campen H, Hansen TR. Interferon stimulated genes, CXCR4 and immune cell responses in peripheral blood mononuclear cells infected with bovine viral diarrhea virus. Res Vet Sci. 2012;93(2):1081–8.
Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. 2016;12(1):37–48.
Zhao W, Zhao G, Wang B. Revisiting GM-CSF as an adjuvant for therapeutic vaccines. Cell Mol Immunol. 2018;15(2):187–9.
Halstead ES, Umstead TM, Davies ML, Kawasawa YI, Silveyra P, Howyrlak J, Yang L, Guo W, Hu S, Hewage EK, et al. GM-CSF overexpression after influenza a virus infection prevents mortality and moderates M1-like airway monocyte/macrophage polarization. Respir Res. 2018;19(1):3.
Takano T, Azuma N, Satoh M, Toda A, Hashida Y, Satoh R, Hohdatsu T. Neutrophil survival factors (TNF-alpha, GM-CSF, and G-CSF) produced by macrophages in cats infected with feline infectious peritonitis virus contribute to the pathogenesis of granulomatous lesions. Arch Virol. 2009;154(5):775–81.
Trzeciak-Ryczek A, Tokarz-Deptula B, Deptula W. Expression of IL-1beta, IL-2, IL-10, TNF-beta and GM-CSF in peripheral blood leukocytes of rabbits experimentally infected with rabbit haemorrhagic disease virus. Vet Microbiol. 2016;186:71–81.
Acknowledgements
Thanks to Fuxing Zha, Liang Zeng and Yan Cheng of Shanghai Biozeron Biotech Co. Ltd. for the assistance in performing analysis of the RNA-Seq results.
Funding
This work was supported by: National Key Research and Development Program of China (2016YFD0500908) (for the isolation of BVDV-2 stain, RNA-Seq and preparing of the manuscript); Jiangsu Agriculture Science and Technology Innovation Fund (CX (18)2003) (for the characterization of BVDV-2 strain); National Natural Science Foundation of China (31402180, 31802196, 31702272) and Natural Science Foundation of Jiangsu Province, China (BK20170595) (for the design of the study, collection, analysis and interpretation of data, results validation by qRT-PCR, Western blot and ELISA).
Author information
Authors and Affiliations
Contributions
WLL designed and participated in all the experiments. FH, XS and JZL performed the animal experiments and PBMCs culture. LLY, WWZ, MS and CYZ participated in the RT-PCR and qRT-PCR detection. WLL, LM and RXL analyzed the data, wrote and revised the manuscript. MJL and JYJ help the experiment design. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval: The feeding of goats and collection of blood samples were performed in strict accordance with the guidelines of Jiangsu Province Animal Regulations (Government Decree No 45).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Differentially expressed genes between mock and BVDV-2 infected samples at 12hpi. (XLS 111 kb)
Additional file 2:
Table S2. GO enrichment results of the DEGs between mock and BVDV-2 infected samples. (XLS 159 kb)
Additional file 3:
Table S3. KEGG pathway enrichment result of the DEGs between mock and BVDV-2 infected samples. (XLS 17 kb)
Additional file 4:
Figure S1. Heat map of the DEGs between mock and BVDV-2 infected samples. Red indicates up-regulation and blue indicates down-regulation. (PDF 47 kb)
Additional file 5:
Figure S2. STRING analysis of all DEGs between mock and BVDV-2 infected samples. (SVG 8290 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Li, W., Mao, L., Shu, X. et al. Transcriptome analysis reveals differential immune related genes expression in bovine viral diarrhea virus-2 infected goat peripheral blood mononuclear cells (PBMCs). BMC Genomics 20, 516 (2019). https://doi.org/10.1186/s12864-019-5830-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5830-y