
Abstract
Background
Diseases on Prunus spp. have been associated with a large number of phylogenetically different pathovars and species within the P. syringae species complex. Despite their economic significance, there is a severe lack of genomic information of these pathogens. The high phylogenetic diversity observed within strains causing disease on Prunus spp. in nature, raised the question whether other strains or species within the P. syringae species complex were potentially pathogenic on Prunus spp.
Results
To gain insight into the genomic potential of adaptation and virulence in Prunus spp., a total of twelve de novo whole genome sequences of P. syringae pathovars and species found in association with diseases on cherry (sweet, sour and ornamental-cherry) and peach were sequenced. Strains sequenced in this study covered three phylogroups and four clades. These strains were screened in vitro for pathogenicity on Prunus spp. together with additional genome sequenced strains thus covering nine out of thirteen of the currently defined P. syringae phylogroups. Pathogenicity tests revealed that most of the strains caused symptoms in vitro and no obvious link was found between presence of known virulence factors and the observed pathogenicity pattern based on comparative genomics. Non-pathogenic strains were displaying a two to three times higher generation time when grown in rich medium.
Conclusion
In this study, the first set of complete genomes of cherry associated P. syringae strains as well as the draft genome of the quarantine peach pathogen P. syringae pv. persicae were generated. The obtained genomic data were matched with phenotypic data in order to determine factors related to pathogenicity to Prunus spp. Results of this study suggest that the inability to cause disease on Prunus spp. in vitro is not the result of host specialization but rather linked to metabolic impairments of individual strains.
Similar content being viewed by others


Background
Members of the Pseudomonas syringae species complex are hemibiotrophic plant pathogenic bacteria responsible for the development of diseases on a wide range of plant species [1,2,3] but have also been isolated from non-agricultural habitats such as rivers and snow [4, 5].
Within the P. syringae species complex, more than 60 pathovars (pv.) have been defined based on the host range [6,7,8,9] whereas nine different genomospecies were identified by DNA:DNA hybridization experiments [10], which were later reflected by phylogroups (PGs) based on similarity of housekeeping genes [11, 12]. To date, a total of thirteen PGs have been defined within the P. syringae species complex [13]. As revealed by previous studies, many strains isolated from non-agricultural environments were phylogenetically closely related to plant associated strains and were also shown to be pathogenic on plants such as kiwifruit and tomato [13,14,15].
The pathogenicity and virulence of strains belonging to the P. syringae species complex has been shown to mainly rely on the presence of a gene cluster called hrp/hrc (hypersensitive reaction and pathogenicity / hypersensitive reaction and conserved) [16, 17] which was found also in many other plant pathogenic bacteria such as Xanthomonas and Erwinia [18, 19]. This gene cluster encodes for a type III secretion system (T3SS) which delivers so-called type III effectors (T3E) into the host cell, where they act by suppressing the plant immune defences and promoting virulence by different and mostly unknown mechanisms [20,21,22,23,24,25]. In addition to the hrp/hrc cluster, a second cluster encoding for a T3SS orthologous to the T3SS of rhizobia was found to be present within the P. syringae species complex [26]. However, the role of this T3SS2 in pathogenicity in P. syringae is still unknown [26].
The production of phytotoxins by members of the P. syringae species complex has also been shown to play a relevant role in virulence [27]. Beside cytotoxic phytotoxins like syringomycin and syringopeptin [28,29,30], other phytotoxins like phaseolotoxin, mangotoxin and tabtoxin have been identified within the P. syringae group able to specifically interfere with the plant amino-acid biosynthesis pathways [31,32,33,34]. Syringolin is another phytotoxin produced by strains of P. syringae that acts as an irreversible proteasome inhibitor and promotes bacterial colonization in the apoplast by inhibiting stomatal closure [35]. Moreover, members of the P. syringae species complex are also able to produce chemical compounds such as auxin, cytokinins and coronatine able to mimic plant hormones and therefore specifically interfere with the regulation of plant immune response [36,37,38,39].
With the advent of affordable next-generation sequencing technologies, hundreds of whole-genome sequence assemblies belonging to P. syringae strains became available in the public databases. Based on this data, many comparative genomic studies have been performed among strains of different pathovars with the aim to define host-specificity related factors [11, 40, 41]. Despite being relevant to investigate host-pathogen co-evolution, such studies are rarely accompanied by a proper host-range determination of the analysed strains and therefore are less suitable for investigations of pathogenicity-related elements.
To date, the Prunus genus comprises the group of hosts associated with the major number of different and polyphyletic pathovars and species within the P. syringae species complex if compared to all other known P. syringae host plants. In fact, a total of six pathovars and two independent species distributed throughout PG1, PG2 and PG3 of the P. syringae species complex have been associated with diseases on Prunus spp. [1, 42,43,44,45,46,47,48]. Distantly related strains are found naturally associated with disease on the same host (or group of hosts), raising the question whether other strains or species within the P. syringae species complex, including strains isolated from non-agricultural habitats, are also potentially pathogenic on Prunus spp. Despite their economic significance, there is a lack of genomic information on members of the P. syringae species complex pathogenic to Prunus spp. that constitutes a real obstacle to an accurate taxonomical definition and generally to a better comprehension of these pathogens.
In this study, we generated twelve complete genomes of strains belonging to the main pathovars associated with diseases on cherry trees as well as two draft genomes of the peach quarantine pathogen P. syringae pv. persicae. Together with 39 additional genome sequenced strains isolated from other hosts and from non-agricultural environments, the strains used for genome sequencing in this study were tested in vitro for pathogenicity towards Prunus spp. Based on the pathogenicity tests results, a comparative genomics approach was applied in order to define the sets of known pathogenicity related factors such as T3E and phytotoxins present in pathogenic and non-pathogenic strains.
Methods
Bacterial strains and culture conditions
A set of twelve strains from the P. syringae species complex and isolated from diseased Prunus spp. hosts was selected for whole genome sequencing using PacBio RSII or Illumina MiSeq (Table 1). For comparative genomics purpose, the whole genome data obtained in this study were combined with the publicly available genome data of 39 additional strains from the P. syringae species complex including strains isolated from Prunus spp. (n = 3), other woody plants (n = 16), herbaceous plants (n = 13) as well as strains isolated from non-agricultural environments (n = 7) and were covering nine of the thirteen PGs defined by Berge et al. [13]. The 39 additional strains were selected based on 1) their availability for testing in the laboratory, 2) having a genome with less than 1000 contigs, and 3) a set of diverse hosts and PG were covered.
All P. syringae strains used in this study were routinely grown at 28 °C on lysogenic broth (LB) agar or in LB liquid medium while shaking at 220 rpm. Most of the strains were received from collaborators as stabs or on plates. A total of 21 strains was obtained as freeze-dried samples from culture collections such as CFBP or NCPPB (Table 1) and revived according to the protocol suggested by the culture collection. The identity of strains was confirmed by cts amplification and Sanger-sequencing using the forward primer cts Fp 5′-AGTTGATCATCGAGGGCGCWGCC-3′ and the reverse primer cts Rp 5′-TGATCGGTTTGATCTCGCACGG-3′ published by Sarkar and Guttman [49]. Sequencing was performed at Microsynth AG (Balgach, Switzerland).
Whole-genome sequencing and assembly
Genomic DNA for PacBio whole genome sequencing was extracted from the selected strains following the protocol described elsewhere [50]. PacBio library preparation and sequencing were performed at the Functional Genomic Center Zurich. SMRTbells were prepared using the DNA Template Prep Kit 2.0 (3 kb to 10 kb) (Pacific Biosciences, Menlo Park, CA) and sequencing was performed on a PacBio RSII system (Pacific Biosciences) run with a P4/C2 chemistry using five to six SMRTcells per strain. Reads were assembled on the SMRT analysis software platform version 2.3.0 using the Hierarchical Genome Assembly Process (HGAP3) protocol followed by manual assembly using BLAST or the SeqMan Pro subroutine of the Lasergene Package (DNASTAR, Madison, WI). Genomic DNA for whole genome shotgun sequencing using Illumina MiSeq (Illumina, San Diego, CA) was extracted with the NucleoSpin Tissue Kit (Macherey-Nagel AG, Düren, DE) following the manufacturer’s protocol. The library preparation was then performed on an Illumina NeoPrep System (Illumina) with a TruSeq Nano DNA kit (Illumina) according to manufacturer’s instructions with six PCR cycles. Paired-end sequencing of 300 bp was performed using MiSeq Reagent Kit v.3 (Illumina) following manufacturer’s instructions. Automatic assemblies were performed using SPAdes Genome Assembler v.3.5.0 [51] on a BaseSpace Onsite v.2.1.2 (Illumina). Putative plasmids were identified by the presence of self-closing molecules during assemblies and/or of genes involved in plasmid replication or mobilization.
Phylogenomics
Automatic genome annotation of the sequenced strains was performed using the GenDB platform v.2.4 [52]. The core genome phylogenetic relationships were obtained using EDGAR v.2.2 [53]. Briefly, the core genome was defined by iterative pairwise comparison of the gene content of each of the selected genomes using the bidirectional best hits (BBH) as orthology criterion. For all calculations, protein BLAST (BLASTp) was used with BLOSUM62 as similarity matrix [54, 55]. Genes were considered orthologous when a reciprocal best BLAST hit was found between two genes, and when both BLAST hits were based on alignments exceeding 70% sequence identity spanning over at least 70% of the query gene length [56]. Multiple alignments of each orthologous gene set of the core genome were calculated using the MUSCLE software [57] and non-matching parts of the alignments were removed based on GBLOCKS [58]. The resulting alignments were concatenated and used to construct a Neighbour Joining (NJ) phylogeny as implemented in the PHYLIP package [59]. Non-annotated genomes retrieved from the NCBI database were annotated using a command line annotation pipeline based on HMMer against an EDGAR based database of Pseudomonas ortholog groups followed by reference genome annotation and a comparison to the Swiss-Prot and RefSeq databases for genes that had no high quality hit in previous steps [60]. In addition to the core-genome phylogeny, the average nucleotide identity based on BLASTn (ANIb) values were calculated between each genome using EDGAR v2.2 [60].
Pathogenicity tests on immature cherry fruitlets
Pathogenicity tests on immature cherry fruitlets were performed following the protocol described elsewhere [61]. Freshly collected immature sweet cherry fruitlets (cv. Christiana × Gisela5) were dipped in 50% ethanol for 3 min and rinsed three times with sterile distilled water. All tested strains (n = 51, Table 1) were grown overnight in liquid LB medium at 28 °C while shaking at 220 rpm. Bacteria were collected by centrifugation and washed twice with sterile distilled water. Final bacterial concentration was adjusted to OD600 = 0.5 (corresponding to around 108 CFU/ml) with sterile distilled water. For each strain, ten fruitlets were inoculated by pricking in two places on the fruitlet with a sterile needle previously immersed in the bacterial suspension. Sterile distilled water was used as negative control. After inoculation, the fruitlets were put on a moist sterile filter paper into a Petri dish, sealed with parafilm and incubated at 22 °C for four days in the dark. Pathogenicity was assessed visually looking at the symptoms developed at the pricking sites.
Detached leaf bioassay
The detached leaf bioassay was performed as described elsewhere [62] with some slight modifications. Leaves from Prunus persica cv. Red Haven and from Prunus dulcis cv. Dürkheimer Riesenmandel were freshly collected and washed for 5 min under running tap water, dipped into 70% ethanol for 1 min and then into a 6.5% sodium hypochlorite solution for 5 min. After disinfection, leaves were rinsed three times in sterile distilled water and air-dried under a sterile flow bench. All tested strains (n = 24) were grown overnight in liquid LB medium at 28 °C while shaking at 220 rpm. Bacteria were collected by centrifugation and washed twice with sterile 0.8% KCl. Final concentration was adjusted to OD600 = 0.5 with sterile 0.8% KCl.
Leaves were infiltrated from the abaxial leaf side with the bacterial suspension using a sterile disposable 3 ml syringe without needle applying a gentle pressure until the mesophyll tissue became water soaked. Each leaf was infiltrated with eight to ten different strains including the positive and the negative control (i.e. P. syringae pv. syringae strain CFBP 2118 and 0.8% KCl, respectively). Every strain was infiltrated once into three different leaves. Each inoculated leaf was placed into a Petri dish containing water agar (10 g/L) sealed with parafilm and incubated for one week at 25 °C under daylight photoperiod. A strain causing the formation of a clear brownish necrotic spot on the site of infiltration for all three infiltrated leaves was considered as pathogenic.
Bacterial growth assays
All growth curves were obtained using the Bioscreen C Microbiology Analyser (Oy Growth Curves AB Ltd., Helsinki, Finland). For this purpose, bacteria were grown overnight in liquid LB medium at 28 °C while shaking at 220 rpm. Bacterial cells were then collected by centrifugation (10 min at 3220 x g), washed three times with sterile 0.8% KCl and finally diluted to an OD600 = 0.01 with LB. Each strain was tested in triplicates.
Comparative genomics of known virulence related factors
In order to determine the virulence factors profile of the selected strains, the locus tags of the corresponding amino acid sequences were obtained from the NCBI database (Additional file 1: Table S1) and used as query to screen the remaining genomes for orthologous proteins using EDGAR v2.2 [53]. For the T3E screening, the amino acid sequence of a total of 80 T3E was obtained from the Hop database available at the Pseudomonas syringae Genome Resources website (www.pseudomonas-syringae.org) and used as query in a tBLASTn analysis to retrieve the corresponding locus tags to be used in EDGAR v2.2 [53] to search for the reciprocal best hit on the selected genomes (n = 51) (Additional file 1: Table S2).
Results
Genome sequencing and assembly
De novo assembly of PacBio reads yielded a total of contigs ranging from one to seven with a mean coverage of over 100× for each of the genomes (Additional file 1: Table S3). The size of the chromosome ranged from 5.8 Mb to 6.4 Mb and with an average G + C content of 58.6% ± 0.5% for the sequenced chromosomes whereas putative plasmids ranged from 20 kb to 140 kb and generally displayed a lower G + C content (~ 55%) (Additional file 1: Table S3). Automatic genome annotation predicted a total number of coding sequences (CDS) varying between 5118 and 5995 (Additional file 1: Table S3). The whole genome sequencing of the P. syringae pv. persicae strain CFBP 1573 using Illumina MiSeq yielded a total of 214 contigs and a mean coverage of 61× (Additional file 1: Table S4). Similar results were obtained for the P. syringae pv. persicae strain NCPPB 2254 with a total 246 contigs and mean coverage of 43×. Both genomes had a total size of 6.4 Mb and a G + C content of 58% (Additional file 1: Table S4). The number of CDS predicted using GenDB was 6079 and 5990 for strains CFBP 1573 and NCPPB 2254, respectively.
Phylogenomics
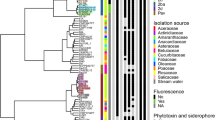
In order to clarify the exact phylogenetic position of the sequenced Prunus associated strains within the P. syringae species complex a core genome based phylogeny was generated using EDGAR v.2.2 [53]. The obtained tree was generated based on the concatenated and aligned amino acid sequences of 2085 proteins consisting of a total length of 840,202 amino acids (Fig. 1). The main clustering obtained from the core genome phylogeny reflected the PGs previously defined by Multi Locus Sequence Analysis (MLSA) [11, 49, 63] and single locus phylogeny [12, 13]. The sequenced Prunus associated strains fell into three different PGs namely PG1 (P. syringae pv. morsprunorum race 2, P. syringae pv. avii, P. syringae pv. persicae), PG2 (P. syringae pv. syringae and P. cerasi) and PG3 (P. syringae pv. morsprunorum race 1 and P. syringae pv. cerasicola). However, strains of different Prunus associated pathovars from the same PG did not form a monophyletic group (Fig. 1). Within PG1, Prunus associated strains were found in two separate clades: one with strains of the P. syringae pv. morsprunorum race 2 (PG1b) and one with P. syringae pv. persicae and P. syringae pv. avii (PG1a). Strains of P. syringae pv. syringae and P. cerasi were both belonging to the PG2 but clustered within PG2d and PG2a, respectively (Fig. 1). Sequenced strains of the same pathovar mostly tightly clustered with exception of the two P. syringae pv. syringae strains CFBP 2118 and CFBP 4215 which clustered closer to strains isolated from other hosts than to each other. The core-genome phylogeny was supported by ANIb results which revealed additionally that PGs boundaries within the P. syringae species complex, with ANIb values < 95%, actually represent species boundaries [64] (Additional file 1: Figure S1).

Pathogenicity tests results and virulence factors profile of the analyzed Pseudomonas syringae strains. Strains sequenced in this study are indicated in bold. Strains are ordered based on the core-genome. Approximately Maximum-Likelihood phylogenetic tree constructed on the similarity of 2085 protein sequences corresponding to a total alignment length of 840,202 amino acids per genome. The local support values computed using the Shimodaira-Hasegawa test are indicated close to the nodes. The tree was generated using EDGAR v.2.2 [53]. The strain names refer to the code field from Table 1. Phylogroups (PGs) are reported on the left and are separated by horizontal dashed lines whereas clades are reported on the right and are separated by horizontal dotted lines. Results of the pathogenicity tests performed on immature cherry fruitlets, peach and almond detached leaves are reported in the first three columns (see graphical legend). No pathogenicity test was performed for strains displaying a X sign in a white square. A strain was defined as possessing T3SS2, a second cluster encoding for a type III 3 secretion system (T3SS) homologous to the one found in rhizobia, if at least 22 out of the 27 genes constituting this system were retrieved. Presence (black) and absence (white) of clusters for biosynthesis and regulations of the known phytohormones (pink) and phytotoxins (blue) is also reported. The generation time in hours was derived from the slope of the logarithmic (log10) growth curve. IaaM (tryptophan monooxygenase) and IaaH (indoleacetamide hydrolase) are responsible for the synthesis of auxin whereas IaaL (indole-acetic acid-lysine synthase) is conjugating auxin to lysine decreasing the concentration of the active form of auxin. Locus tags used for the genotypic screening are reported in Additional file 1: Tables S1 and S2. N.D.: not determined
In vitro pathogenicity tests of members of the P. syringae species complex on Prunus spp.
Results of the cherry immature fruitlets revealed a high pathogenicity potential towards Prunus spp. within the P. syringae species complex. In particular, of the 51 strains tested on immature cherry fruitlets, 32 strains were pathogenic, ten were classified as non-pathogenic whereas nine strains were associated with unclear phenotypes, i.e. neither necrotic nor water soaked (Fig. 1). Most of the pathogenic strains (n = 23) caused brownish, water-soaked superficial lesions similar to those caused by P. syringae pv. morsprunorum race 1 and race 2 whereas the remaining strains (n = 9) caused formation of black-brown sunken necrotic lesions which are typically caused by P. syringae pv. syringae (Fig. 2A). Most of the non-pathogenic strains belonged to PG1 (n = 5) and PG3 (n = 4) and included six strains originally isolated from Prunus spp. like the peach pathogen P. syringae pv. persicae NCPPB 2254 and the P. syringae pv. morsprunorum race 2 pathotype strain M302280 (Fig. 1). Results obtained from the detached leaf assays (Fig. 2B) on peach and almond were mostly congruent with the virulence profile obtained from the cherry immature fruitlets inoculation but additionally revealed few strains possessing a narrower host range. In fact, P. syringae pv. persicae NCPPB 2254 and P. syringae pv. actinidifoliorum ICMP 18883 were both non-pathogenic on cherry fruitlets but were clearly pathogenic if inoculated on peach and almond leaves. Additionally, P. cerasi PL58 was non-pathogenic on peach leaves but showed symptoms on cherry immature fruitlets and almond leaves as well (Fig. 1). Furthermore, the P. avellanae strain PaVt10 was not pathogenic on cherry fruitlets and peach leaves but was symptomatic on almond leaves. Only five strains resulted to be non-pathogenic in all three in vitro pathogenicity tests, namely P. syringae pv. morsprunorum race 2 strain M302280 (PG1), P. amygdali pv. dendropanacis CFBP 3226 (PG3), P. amygdali CFBP 3205 (PG3) and both P. syringae pv. cerasicola strains CFBP 6109 and CFBP 6110 (PG3).

Representative results of the pathogenicity tests on cherry immature fruitlets (A) and on peach detached leaf (B). (A) Typical symptoms observed two days post inoculation with suspensions of (A1) Pseudomonas syringae pv. morsprunorum race 2 CFBP 2116, (A2) P. syringae pv. syringae CFBP 2118 and (A3) with sterile distilled water. (B) Typical results obtained at (B1) 0 days post inoculation (dpi), (B2) 2 dpi and (B3) 7 dpi during the detached leaves assays performed on peach (Prunus persica cv. Red Haven) infiltrated with suspensions of “10”: P. syringae pv. morsprunorum race 2 M302280; “41”: P. syringae pv. cerasicola CFBP 6110; “36”: P. syringae pv. morsprunorum race 1 CFBP 3840; “11”: P. syringae pv. morsprunorum race 1 CFBP 6411; “37”: P. syringae pv. morsprunorum race 1 CFBP 2116; “44”: P. syringae pv. phaseolicola 1448a; “Pos.”: P. syringae pv. syringae CFBP 2118 and “Neg.”: 0.8% KCl
Distribution of known virulence-related factors
In order to investigate a potential link between known virulence-related factors and the observed pattern of pathogenicity, the publicly available genomes of the strains selected for this study (Table 1) were screened for the presence of clusters of genes known to be involved in pathogenicity of P. syringae.
All strains possessed a complete hrp/hrc cluster (Fig. 1), with exception of P. viridiflava CFBP 1590, which lacked most of the genes within this cluster. The distribution of the T3SS2 among the strains considered in this study was not consistent with the PG defined based on core-genome phylogeny and even varied among strains of the same pathovar (Fig. 1). Moreover, the presence of the T3SS2 could not explain the pathogenicity profiles obtained in this study (Fig. 1).
Using in silico screening for 80 known T3E (Additional file 1: Table S2), the total number of T3E retrieved per strain range from one in the T3SS-impaired P. viridiflava CFBP 1590 to 45 found in the genome of the P. syringae pv. tomato DC3000. The overall T3E presence/absence distribution profile mostly reflected the core-genome phylogeny: closely related strains possessed similar T3E repertoires with some rearrangements. Also here, the T3E profiles could not explain the pathogenicity results. It was noticed that the T3E HopAA, which is located in the conserved effector locus (CEL) was absent in the genomes of the five strains that were non-pathogenic on cherry, peach and almond. However, HopAA was also missing in the genomes of P. syringae pv. phaseolicola 1448a and in P. syringae pv. aesculi 0893_23, which, in contrast, were pathogenic. As already noticed by Lindeberg et al. [65], the number of T3E present in strains from the PG2 is generally lower in comparison to strains of PG1 or PG3 (Fig. 3). However, both P. cerasi strains, belonging to PG2a, possessed almost the double number of T3E when compared to all other members of the PG2 while most of the T3E in P. cerasi were located on plasmids (Fig. 3). On the other hand, the presence of clusters for the synthesis of the necrosis-inducing phytotoxins syringomycin and syringopeptin co-occurred with the phenotype obtained from the immature cherry fruitlets assay: with exception of P. syringae CC1583 (PG10b), the strains causing necrotic lesions (Fig. 1) possessed clusters related to the production and regulation of syringomycin, syringopeptin or both. These clusters were generally found in strains of PG2, which were also shown to possess a lower amount of T3E (~ 20 T3E per strain). However, P. syringae strain CC1557, belonging to the quite distantly related PG10a, also possessed the syringomycin cluster. The syringolin cluster was exclusively found in strains from the PG2 and mostly within the PG2d clade whereas the phaseolotoxin cluster was only present in P. syringae pv. phaseolicola 1448a (PG3) and two strains of P. syringae pv. actinidiae (PG1). The mangotoxin cluster was restricted to strains from the PG2 and specifically found within the clades PG2a and PG2b (Fig. 1). The genes involved in the synthesis of the plant hormone auxin (indoleacetic acid, IAA), iaaM and iaaH, encoding respectively the tryptophane monooxygenase and IAA hydrolase were found in strains belonging to PG2d and PG3, but as well as in some strains within PG1 (n = 3). In contrast, the iaaL gene encoding the IAA-lysine synthase responsible for the reversible inactivation of IAA was found throughout the entire phylogenetic tree. The cluster for the biosynthesis of coronatine was found only in six distantly related strains whereas only three closely related strains within PG3 were potentially able to produce cytokinins.

Type III effector (T3E) profile of the 52 Pseudomonas syringae strains used in this study. Strains sequenced in this study are indicated in bold. The amino acid sequence of a total of 80 T3E (Additional file 1: Table S2) was obtained from the Hop database available at the P. syringae Genome Resources website (www.pseudomonas-syringae.org) and used as query in a tBLASTn analysis to retrieve the corresponding locus tags to be used in EDGAR v.2.2 [53] for search of the reciprocal best hit on the selected genomes. Black squares indicate presence whereas white squares indicate absence of the T3E. For strains sequenced in this study using PacBio RSII as well as for the complete genomes P. syringae pv. tomato DC3000 and P. syringae pv. phaseolicola 1448a, grey squared indicates T3E located on plasmids. Strains are ordered based on the core-genome phylogeny constructed in Fig. 1 together with phenotypical analysis whereas T3E are ordered based on their abundance from left to right in descending order. The strain names refer to the code field from Table 1. Phylogroups are reported on the left and are separated by horizontal dashed lines whereas clades are reported on the right and are separated by horizontal dotted lines. The last column indicates the total number of T3E per strain. CEL: the T3E located in the conserved effector locus
Again, the presence of known pathogenicity factors was not related to the differences in virulence on cherry, peach and almond. Indeed, most of the analysed genes or gene clusters mainly reflected the core-genome phylogeny and could not reveal why closely related strains differed in their pathogenicity towards the tested hosts.
Divergence of the HrpA protein among the P. syringae species complex
The hrpA gene within the hrp/hrc cluster encodes for the extracellular pilus of the T3SS, which is essential for a functional T3SS and has been shown to be under diversifying selection [66]. Two homologous HrpA proteins were found within the P. syringae species complex: one variant was found in strains of PG1, PG6, PG9 and PG10 and named HrpA1 (for HrpA like PG1) whereas the other variant was present in strains belonging to PG2, PG3 and PG5 and named HrpA2 (for HrpA like PG2) (Fig. 4). The pattern of distribution of these two HrpA variants did not reflect the core genome phylogeny. In fact, the genome of PG6 strain P. syringae pv. tagetis ICMP 4091 contained the HrpA1 variant gene, but the strain was phylogenetically positioned equidistantly to PG2 and PG3 strains, which both have the HrpA2 variant. The same situation was observed for the PG5 strain P. cannabina pv. alisalensis ES4326 (Fig. 1). The sequence analysis of HrpA1 revealed a higher level of polymorphism within strains of the same PG if compared to HrpA2 (Fig. 4). Polymorphisms of HrpA1 and HrpA2 mostly reflected the PGs but HrpA1 within strains of the PG1 displayed some more specific polymorphisms, which were generally shared among strains of the same pathovar. However, the HrpA1 protein from P. syringae pv. tomato NCPPB 1108 (PG1a) was identical to the HrpA1 protein of strains belonging to PG1b including strains isolated from water and snow (Fig. 4). Strains of the PG2 possessed a 100% identical HrpA2 protein and within PG3, the HrpA2 sequence was almost identical independent of the pathovar (Fig. 4).

Alignment of the HrpA1 and HrpA2 proteins retrieved from the 51 genomes of members of the Pseudomonas syringae species complex analyses in this study. Red lines indicate the phylogroup (PG) borders, whereas red dashed lines indicate clade borders. Amino acids are only reported if different from the reference sequences (GenBank Accession no. AKT31917 and CFBP2118_03968, respectively) which are entirely displayed in the top line of each alignment
Potential link between pathogenicity and growth rate
The results obtained from the comparative genomics of known virulence related factors did not reveal any direct link with the results obtained from the pathogenicity tests. However, it was noticed that non-pathogenic strains usually grew at a lower growth rate in rich medium (LB) if compared to their closely related pathogenic strains (Fig. 1). For example, the non-pathogenic strain P. syringae pv. morsprunorum race 2 M302280 displayed a generation time of 100 min which is three times higher than what was observed for the two pathogenic P. syringae pv. morsprunorum race 2 strains analysed in this study. The same trend was observed with the pathogenic and non-pathogenic strains of the PG3 (Fig. 1). This suggested that a metabolic impairment could be a potential reason why those strains were not pathogenic within the timeframe of the experiments. Comparative genomics between closely related pathogenic and non-pathogenic strains revealed some mutations affecting genes involved in metabolic pathways in non-pathogenic strains, which were previously shown to be related to virulence of plant pathogenic bacteria (Table 2).
Discussion
A prerequisite for the development of effective and targeted control measures against plant diseases is the comprehension of the mechanisms adopted by the pathogen for successful host infection.
Bacterial canker caused by members of the P. syringae species complex on Prunus spp. is responsible for relevant yield losses in both fruit and wood production worldwide [67, 68]. However, with the exception of few comparative genomics studies of pathogens on Prunus spp. [40, 69, 70] the repertoire of pathogenicity related factors in Prunus spp. associated strains remains largely unstudied. Taking advantage of the complete as well as the draft genomes generated in this study and combining them with a consistent set of publicly available genomes, we generated a whole genome based phylogeny of the P. syringae species complex comprising all known pathovars and species that have ever been associated with diseases in Prunus spp. (status April 2017), including the newly described P. cerasi species [46] and the quarantine peach pathogen P. syringae pv. persicae.
The methodology used in this study to test pathogenicity relied on two different in vitro assays, i.e. the use of detached immature fruitlet and detached leaf assays, which were previously shown to be reliable for cherry [61, 62, 70] but also for other woody hosts [70, 71]. While the use of detached organs instead of the whole plant could potentially affect the results of pathogenicity tests, the pattern of pathogenicity retrieved from this study is largely congruent with the patterns obtained from the inoculation of whole plants (C.E. Morris, personal communication), therefore supporting the veracity of the results. Nevertheless, it might be necessary to repeat the pathogenicity tests to further validate the results. Furthermore, the possibility to co-inoculate different strains together with the positive and negative controls was crucial to reduce the potential effect of physiological variation of leaves. The large number of potentially pathogenic stains retrieved from this study and the fact that strains belonging to the same pathovar varied in their pathogenicity towards Prunus spp. highlighted the importance of a proper host range determination in order to perform reasonable comparative genomics studies, especially if intended to investigate factors involved in host-specificity. Indeed, it is important to consider that a strain never isolated from a particular host could still be pathogenic on that host, as also previously shown for the P. syringae-kiwifruit and P. syringae-tomato pathosystems [14, 15]. At the same time, these findings revealed the weakness of the pathovar designation system for P. syringae taxonomy. A clear example is constituted by the two races of P. syringae pv. morsprunorum, whose ANIb values (~ 88%) are clearly below the species boundaries of 95% [64]. This indicates that they rather should be considered as separate species. Therefore, the genomic data supports the claims to revise the taxonomic position of the P. syringae species complex [72].
The results from this study also revealed that strains isolated from water reservoirs such as stream water and snow could potentially constitute a threat for Prunus spp. plantations, supporting the direct link between the agricultural and non-agricultural habitats occupied by P. syringae as already reported [4, 14, 15, 73,74,75]. These findings also provides some important hints for cultural practices implementation especially regarding the maintenance and hygiene of water-irrigation systems. In fact, due to the persistence of potentially pathogenic P. syringae strains in water basins, the use of closed (i.e. recirculating) irrigation systems should be avoided and if possible the water should be disinfected or sterilized prior to use to prevent the spread of this pathogen within plantations [76].
A first systematic screen and comparison of known virulence related factors in strains associated with Prunus spp. was performed in this study, revealing a high variability in the set of virulence factors comprising both T3E set as well as phytotoxins and phytohormons production. This observation led to the conclusion that pathogenicity on Prunus spp. may be achieved by different and currently unknown mechanisms that could not be detected in this study as we only used already known virulence related factors. However, the high level of susceptibility observed for this group of hosts to members of the P. syringae species complex could reflect the lack of proper defense mechanisms in the host rather than the evolution of specific virulence strategies in the pathogen. In fact, the results of our comparative genomics approach did not show an obvious match with the results obtained from the pathogenicity tests even though the description of potential link to woody compounds in the past [70]. The only exception was P. viridiflava strain CFBP 1590 where the absence of pathogenicity can be related to the lack of a complete T3SS combined with an extremely reduced T3E repertoire [77, 78].
We confirmed that strains possessing a small T3E repertoire were potential producers of necrosis-inducing phytotoxins like syringomycin and/or syringopeptin [40, 65] and belonged mostly to PG2 [65]. In addition, strains of PG9, PG10a and PG10b were found to possess the genes necessary for the production of at least one of those phytotoxins [13]. Nevertheless, only strains of PG2 possessed both syringomycin and syringopeptin clusters, which were previously shown to be physically linked and located on a 155-kb genomic island [79, 80]. The observed mutually exclusive presence of clusters for production of necrosis-inducing phytotoxins versus the evolution of large T3E repertoires reveals a potential trend of pathogenesis subgroup specialization within the P. syringae species complex with strains of the PG2 adopting a more generalist pathogenicity strategy and most of the remaining PGs relying on a specific host-targeted pathogenicity mechanism. This specialization is reflected by the broad vs. narrow host range observed within the P. syringae species complex [81, 82]. In contrast to the T3E, phytotoxins were never recognized by the host resulting in a non-compatible interaction. Therefore, in a specific environment, a pathogenicity mechanism relying on necrosis-inducing phytotoxins would theoretically be selectively more advantageous than the production of specialized T3E, which could lead to the induction of plant immunity [83, 84]. However T3E, phytohormons and other non-necrosis inducing phytotoxins are related to a more precise modulation of the host-physiology [31, 85] and could promote the long-term survival of bacterial populations in the host-plant which can serve as source of inoculum for further infection [86, 87].
Nevertheless, syringomycin and syringopeptin were shown to be the major virulence determinant for P. syringae pv. syringae strain B301-D in vitro [88], highlighting the fact that for strains possessing necrosis-inducing phytotoxins, the T3SS/T3E strategy may be of secondary importance. This would also be congruent with the observation that the HrpA protein among PG2 strains is 100% identical suggesting that low diversifying selection acts on that gene in contrast to the HrpA found in PG1 strains [66]. In addition, the HrpA protein is conserved also within strains of PG3, but strains from this PG have a narrower host range when compared to P. syringae pv. syringae strains [45, 47, 89].
A positive trend was observed between the necrotic phenotype on cherry immature fruitlets and presence of clusters for the production of syringomycin and syringopeptin [88]. However, while the PG10b strain P. syringae CC1583 was lacking the syringomycin and syringopeptin clusters, it was associated with necrotic lesions in cherry fruitlet tests, suggesting that this strain is probably able to produce another necrosis inducing phytotoxin, which is still not characterized. Although strains within PG2 usually possessed a smaller set of T3E (~ 15), both P. cerasi strains constituted an exception having almost the double of T3E. As we obtained high quality genomes for those strains, it was possible to determine that around half of those T3E were located on plasmids (Fig. 3) thereby highlighting the importance of horizontal gene transfer in P. syringae [82, 90].
Strains of the quarantine peach pathogen P. syringae pv. persicae are known to produce a necrosis inducing phytotoxin called persicomycin [91]. However, no necrotic phenotype was observed on cherry fruitlets inoculated with P. syringae pv. persicae NCPPB 2254. As persicomycin production was shown to be thermoregulated [91] it is possible that it was not induced under the used assay conditions. Moreover P. syringae pv. persicae NCPPB 2254 was never tested previously for production of persicomycin. On the other hand, the P. syringae pv. persicae pathotype strain CFBP 1573 which was shown to produce persicomycin under in vitro conditions [91] did not cause necrotic lesions on cherry immature fruitlets as well (M. Kałuźna, personal communication), leaving the role of this phytotoxin in pathogenicity open.
The positive trend between the reduced growth rate in rich medium and the pathogenicity led to the hypothesis that the inability of the identified P. syringae strains to cause disease was rather related to a metabolic impairment of those strains which does not allow them to reach population densities able to trigger disease [86]. The T3E screening revealed that all strains that resulted in a non-pathogenic phenotype on all three hosts were lacking a single T3E, namely HopAA, which has been shown to contribute to efficient formation of bacterial colonies in planta [92]. However, as P. syringae pv. phaseolicola strain 1448a is lacking this T3E as well, but was still growing at a higher growth rate and was pathogenic to cherry, peach and almond, this hypothesis can be rejected. Comparative genomics between closely related pathogenic and non-pathogenic strains revealed mutations within genes of metabolic pathways previously shown to be involved in virulence of plant pathogenic bacteria [93,94,95,96,97]. However, the role of those pathways in the pathogenicity of Prunus spp. is still unclear.
Conclusion
Based on the obtained results, it is clear that the ability of P. syringae strains to cause disease on Prunus spp. is not the result of a common evolutionary event but is most probably due to an independent loss or gain of different factors in individual strains, not necessarily related to virulence. Moreover, the large number of strains found to be pathogenic on the tested hosts revealed that the Prunus spp. – P. syringae pathosystem does not represent the most suitable case for the investigation of virulence-related factors. A more comprehensive phenotyping and genome comparisons of both pathogen and host would provide more indications in order to reveal key factors in the pathogenicity of P. syringae on cherry, peach and almond.
Abbreviations
- ANIb:
-
Average nucleotide identity based on BLASTN searches
- CDS:
-
Coding sequences
- IAA:
-
Indole acetic acid
- MLSA:
-
Multilocus sequence analysis
- OD:
-
Optical density
- PG:
-
Phylogroup
- pv:
-
Pathovar
- T3E:
-
Type III effectors
- T3SS:
-
Type III secretion system
References
Crosse JE. Bacterial canker of stone-fruits: IV. Investigation of a method for measuring the inoculum potential of cherry trees. Ann Appl Biol. 1959;47(2):306–17.
Hirano SS, Upper CD. Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae—a pathogen, ice nucleus, and epiphyte. Microbiol Mol Biol Rev. 2000;64(3):624–53.
Pieterse CMJ, Leon-Reyes A, Van der Ent S, Van Wees SCM. Networking by small-molecule hormones in plant immunity. Nat Chem Biol. 2009;5(5):308–16.
Morris CE, Sands DC, Vinatzer BA, Glaux C, Guilbaud C, Buffière A, Yan S, Dominguez H, Thompson BM. The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J. 2008;2(3):321–34.
Monteil CL, Cai R, Liu H, Llontop ME, Leman S, Studholme DJ, Morris CE, Vinatzer BA. Nonagricultural reservoirs contribute to emergence and evolution of Pseudomonas syringae crop pathogens. New Phytol. 2013;199(3):800–11.
Young JM, Dye DW, Bradbury JF, Panagopoulos CG, Robbs CF. A proposed nomenclature and classification for plant pathogenic bacteria. N Z J Agric Res. 1978;21(1):153–77.
Dye D, Bradbury J, Goto M, Hayward A, Lelliott R, Schroth MN. International standards for naming pathovars of phytopathogenic bacteria and a list of pathovar names and pathotype strains. Rev Plant Pathol. 1980;59(4):153–68.
Palleroni NJ, Genus I. Pseudomonas. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey's Manual of Systematic Bacteriology The Proteobacteria, Part C: The Gammaproteobacteria, vol. Volume 2. Dordrecht: Springer; 2005. p. 323–79.
Young JM. Taxonomy of Pseudomonas syringae. J Plant Pathol. 2010;92(1sup):S1.5–S1.14.
Gardan L, Shafik H, Belouin S, Broch R, Grimont F, Grimont PA. DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Dowson 1959). Int J Syst Bacteriol. 1999;49:469–78.
Sarkar SF, Gordon JS, Martin GB, Guttman DS. Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics. 2006;174(2):1041–56.
Parkinson N, Bryant R, Bew J, Elphinstone J. Rapid phylogenetic identification of members of the Pseudomonas syringae species complex using the rpoD locus. Plant Pathol. 2011;60(2):338–44.
Berge O, Monteil CL, Bartoli C, Chandeysson C, Guilbaud C, Sands DC, Morris CE. A user's guide to a data base of the diversity of Pseudomonas syringae and its application to classifying strains in this phylogenetic complex. PLoS One. 2014;9(9):e105547.
Monteil CL, Lafolie F, Laurent J, Clement J-C, Simler R, Travi Y, Morris CE. Soil water flow is a source of the plant pathogen Pseudomonas syringae in subalpine headwaters. Environ Microbiol. 2014;16(7):2038–52.
Bartoli C, Lamichhane JR, Berge O, Guilbaud C, Varvaro L, Balestra GM, Vinatzer BA, Morris CE. A framework to gauge the epidemic potential of plant pathogens in environmental reservoirs: the example of kiwifruit canker. Mol Plant Pathol. 2015;16(2):137–49.
Lindgren PB, Peet RC, Panopoulos NJ. Gene cluster of Pseudomonas syringae pv. "phaseolicola" controls pathogenicity of bean plants and hypersensitivity of nonhost plants. J Bacteriol. 1986;168(2):512–22.
Lindgren PB, Panopoulos NJ, Staskawicz BJ, Dahlbeck D. Genes required for pathogenicity and hypersensitivity are conserved and interchangeable among pathovars of Pseudomonas syringae. Mol Gen Genet. 1988;211(3):499–506.
Beer SV, Bauer DW, Jiang XH, Laby RJ, Sneath BJ, Wei Z-M, Wilcox DA, Zumoff CH. The hrp gene cluster of Erwinia amylovora. In: Hennecke H, DPS V, editors. Advances in Molecular Genetics of Plant-Microbe Interactions Vol 1: Proceedings of the 5th International Symposium on the Molecular Genetics of Plant-Microbe Interactions, Interlaken, Switzerland, September 9–14, 1990. Dordrecht: Springer Netherlands; 1991. p. 53–60.
Bonas U, Schulte R, Fenselau F, Minsavage GV, Staskawicz BJ, Stall RE. Isolation of a gene cluster from Xanthomonas campestris pv. Vesicatoria that determines pathogenicity and the hypersensitive response on pepper and tomato. Mol Plant Microbe Interact. 1991;4(1):81–8.
Lindgren PB. The role of hrp genes during plant-bacterial interactions. Annu Rev Phytopathol. 1997;35(1):129–52.
Wei W, Plovanich-Jones A, Deng W, Jin Q, Collmer A, Huang H, He S. The gene coding for the Hrp pilus structural protein is required for type III secretion of Hrp and Avr proteins in Pseudomonas syringae pv. Tomato. Proc Natl Acad Sci U S A. 2000;97(5):2247–52.
Jin Q, He S-Y. Role of the Hrp pilus in type III protein secretion in Pseudomonas syringae. Science. 2001;294(5551):2556–8.
Mackey D, Holt Iii BF, Wiig A, Dangl JL. RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell. 2002;108(6):743–54.
Shao F, Golstein C, Ade J, Stoutemyer M, Dixon JE, Innes RW. Cleavage of Arabidopsis PBS1 by a bacterial type III effector. Science. 2003;301(5637):1230–3.
Xiang T, Zong N, Zou Y, Wu Y, Zhang J, Xing W, Li Y, Tang X, Zhu L, Chai J, et al. Pseudomonas syringae effector AvrPto blocks innate immunity by targeting receptor kinases. Curr Biol. 2008;18(1):74–80.
Gazi AD, Sarris PF, Fadouloglou VE, Charova SN, Mathioudakis N, Panopoulos NJ, Kokkinidis M. Phylogenetic analysis of a gene cluster encoding an additional, rhizobial-like type III secretion system that is narrowly distributed among Pseudomonas syringae strains. BMC Microbiol. 2012;12(1):188.
Bender C, Palmer D, Peñaloza-Vázquez A, Rangaswamy V, Ullrich M. Biosynthesis of coronatine, a thermoregulated phytotoxin produced by the phytopathogen Pseudomonas syringae. Arch Microbiol. 1996;166(2):71–5.
Iacobellis NS, Lavermicocca P, Grgurina I, Simmaco M, Ballio A. Phytotoxic properties of Pseudomonas syringae pv. Syringae toxins. Physiol Mol Plant Pathol. 1992;40(2):107–16.
Di Giorgio D, Camoni L, Mott KA, Takemoto JY, Ballio A. Syringopeptins, Pseudomonas syringae pv. Syringae phytotoxins, resemble syringomycin in closing stomata. Plant Pathol. 1996;45(3):564–71.
Hutchison ML, Gross DC. Lipopeptide phytotoxins produced by Pseudomonas syringae pv. Syringae: comparison of the biosurfactant and ion channel-forming activities of syringopeptin and syringomycin. Mol Plant-Microbe Interact. 1997;10(3):347–54.
Patil SS, Tam LQ. Mode of action of the toxin from Pseudomonas phaseolicola: I. Toxin specificity, chlorosis, and ornithine accumulation. Plant Physiol. 1972;49(5):803–7.
Turner JG. Tabtoxin, produced by Pseudomonas tabaci, decreases Nicotiana tabacum glutamine synthetase in vivo and causes accumulation of ammonia. Physiol Plant Pathol. 1981;19(1):57–67.
Arrebola E, Cazorla FM, Durán VE, Rivera E, Olea F, Codina JC, Pérez-García A, de Vicente A. Mangotoxin: a novel antimetabolite toxin produced by Pseudomonas syringae inhibiting ornithine/arginine biosynthesis. Physiol Mol Plant Pathol. 2003;63(3):117–27.
Arai T, Kino K. A novel L-amino acid ligase is encoded by a gene in the phaseolotoxin biosynthetic gene cluster from Pseudomonas syringae pv. Phaseolicola 1448A. Biosci Biotechnol Biochem. 2008;72(11):3048–50.
Schellenberg B, Ramel C, Dudler R. Pseudomonas syringae virulence factor syringolin a counteracts stomatal immunity by proteasome inhibition. Mol Plant-Microbe Interact. 2010;23(10):1287–93.
Kosuge T, Heskett MG, Wilson EE. Microbial synthesis and degradation of indole-3-acetic acid. I. The conversion of L-tryptophan to indole-3-acetamide by an enzyme system from Pseudomonas savastanoi. J Biol Chem. 1966;241(16):3738–44.
Surico G, Iacobellis NS, Sisto A. Studies on the role of indole-3-acetic acid and cytokinins in the formation of knots on olive and oleander plants by Pseudomonas syringae pv. Savastanoi. Physiol Plant Pathol. 1985;26(3):309–20.
Glass NL, Kosuge T. Cloning of the gene for indoleacetic acid-lysine synthetase from Pseudomonas syringae subsp. savastanoi. J Bacteriol. 1986;166(2):598–603.
Geng X, Jin L, Shimada M, Kim MG, Mackey D. The phytotoxin coronatine is a multifunctional component of the virulence armament of Pseudomonas syringae. Planta. 2014;240(6):1149–65.
Baltrus DA, Nishimura MT, Romanchuk A, Chang JH, Mukhtar MS, Cherkis K, Roach J, Grant SR, Jones CD, Dangl JL. Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Path. 2011;7(7):e1002132.
Dudnik A, Dudler R. Genomics-based exploration of virulence determinants and host-specific adaptations of Pseudomonas syringae strains isolated from grasses. Pathogens. 2014;3(1):121–48.
Crosse JE, Garrett CME. Studies on the bacteriophagy of Pseudomonas morsprunorum, Ps. syringae and related organisms. J Appl Microbiol. 1963;26(2):159–77.
Freigoun SO, Crosse JE. Host relations and distribution of a physiological and pathological variant of Pseudomonas morsprunorum. Ann Appl Biol. 1975;81(3):317–30.
Young JM. New plant disease record in New Zealand: Pseudomonas syringae pv. Persicae from nectarine, peach, and Japanese plum. N Z J Agric Res. 1987;30(2):235–47.
Kamiunten H, Nakao T, Oshida S. Pseudomonas syringae pv. Cerasicola, pv. Nov., the causal agent of bacterial gall of cherry tree. J Gen Plant Pathol. 2000;66(3):219–24.
Kałużna M, Willems A, Pothier JF, Ruinelli M, Sobiczewski P, Puławska J. Pseudomonas cerasi sp. nov. (non Griffin, 1911) isolated from diseased tissue of cherry. Syst Appl Microbiol. 2016;39(6):370–7.
Psallidas PG. Hyperplastic canker – a perennial disease of almond caused by Pseudomonas amygdali. EPPO Bull. 1997;27(4):511–7.
Ménard M, Sutra L, Luisetti J, Prunier JP, Gardan L. Pseudomonas syringae pv. Avii (pv. Nov.), the causal agent of bacterial canker of wild cherries (Prunus avium) in France. Eur J Plant Pathol. 2003;109(6):565–76.
Sarkar SF, Guttman DS. Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl Environ Microbiol. 2004;70(4):1999–2012.
Pitcher DG, Saunders NA, Owen RJ. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett Appl Microbiol. 1989;8(4):151–6.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, et al. GenDB--an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31(8):2187–95.
Blom J, Kreis J, Spänig S, Juhre T, Bertelli C, Ernst C, Goesmann A. EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016;44:W22–8.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89(22):10915–9.
Smits THM, Rezzonico F, Kamber T, Blom J, Goesmann A, Frey JE, Duffy B. Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol Plant-Microbe Interact. 2010;23(4):384–93.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–77.
Felsenstein J. PHYLIP (phylogeny inference package), version 3.57 c. Seattle: University of Washington; 1995.
Linke B, Giegerich R, Goesmann A. Conveyor: a workflow engine for bioinformatic analyses. Bioinformatics. 2011;27(7):903–11.
Kałużna M, Sobiczewski P. Virulence of Pseudomonas syringae pathovars and races originating from stone fruit trees. Phytopathologia. 2009;54:71–4.
Bedford KE, Sholberg PL, Kappel F. Use of a detached leaf bioassay for screening sweet cherry cultivars for bacterial canker resistance. Acta Hort. 2003;622:365–8.
Hwang MSH, Morgan RL, Sarkar SF, Wang PW, Guttman DS. Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl Environ Microbiol. 2005;71(9):5182–91.
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(1):81–91.
Lindeberg M, Cunnac S, Collmer A. Pseudomonas syringae type III effector repertoires: last words in endless arguments. Trends Microbiol. 2012;20(4):199–208.
Guttman DS, Gropp SJ, Morgan RL, Wang PW. Diversifying selection drives the evolution of the type III secretion system pilus of Pseudomonas syringae. Mol Biol Evol. 2006;23(12):2342–54.
Vicente JG, Alves JP, Russell K, Roberts SJ. Identification and discrimination of Pseudomonas syringae isolates from wild cherry in England. Eur J Plant Pathol. 2004;110(4):337–51.
Bultreys A, Kałużna M. Bacterial cankers caused by Pseudomonas syringae on stone fruits species with special emphasis on the pathovars syringae and morsprunorum race 1 and race 2. J Plant Pathol. 2010;92(1):S21–33.
Hulin MT, Armitage AD, Vicente JG, Holub EB, Baxter L, Bates HJ, Mansfield JW, Jackson RW, Harrison RJ. Comparative genomics of Pseudomonas syringae reveals convergent gene gain and loss associated with specialization onto cherry (Prunus avium). New Phytol. 2018;219(2):672–96.
Hulin MT, Mansfield JW, Brain P, Xu X, Jackson RW, Harrison RJ. Characterization of the pathogenicity of strains of Pseudomonas syringae towards cherry and plum. Plant Pathol. 2018;67(5):1177–93.
Moragrega C, Llorente I, Manceau C, Montesinos E. Susceptibility of European pear cultivars to Pseudomonas syringae pv. Syringae using immature fruit and detached leaf assays. Eur J Plant Pathol. 2003;109(4):319–26.
Gomila M, Busquets A, Mulet M, García-Valdés E, Lalucat J. Clarification of taxonomic status within the Pseudomonas syringae species group based on a phylogenomic analysis. Front Microbiol. 2017;8:2422.
Morris CE, Bardin M, Kinkel LL, Moury B, Nicot PC, Sands DC. Expanding the paradigms of plant pathogen life history and evolution of parasitic fitness beyond agricultural boundaries. PLoS Path. 2009;5(12):e1000693.
Morris CE, Sands DC, Vanneste JL, Montarry J, Oakley B, Guilbaud C, Glaux C. Inferring the evolutionary history of the plant pathogen Pseudomonas syringae from its biogeography in headwaters of rivers in North America, Europe, and New Zealand. mBio. 2010;1(3):e00107–10.
Morris CE, Monteil CL, Berge O. The life history of Pseudomonas syringae: linking agriculture to earth system processes. Annu Rev Phytopathol. 2013;51(1):85–104.
Halstead AJ, Scrace JM. Chapter 11: Pests and diseases of outdoor ornamentals, including hardy nursery stock. In: Alford DV, editor. Pests and disease managment handbook. New York: John Wiley and Sons; 2008.
Araki H, Tian D, Goss EM, Jakob K, Halldorsdottir SS, Kreitman M, Bergelson J. Presence/absence polymorphism for alternative pathogenicity islands in Pseudomonas viridiflava, a pathogen of Arabidopsis. Proc Natl Acad Sci U S A. 2006;103(15):5887–92.
Ruinelli M, Blom J, Pothier JF. Complete genome sequence of Pseudomonas viridiflava CFBP 1590, isolated from diseased cherry in France. Genome Announc. 2017; in press.
Scholz-Schroeder BK, Soule JD, Lu SE, Grgurina I, Gross DC. A physical map of the syringomycin and syringopeptin gene clusters localized to an approximately 145-kb DNA region of Pseudomonas syringae pv. Syringae strain B301D. Mol Plant-Microbe Interact. 2001;14(12):1426–35.
Gross DC, Grgurina I, Scholz-Schroeder BK, Lu S-E. Characteristics of the syr-syp genomic island of Pseudomonas syringae pv. syringae strain B301D. In: Iacobellis NS, Collmer A, Hutcheson SW, Mansfield JW, Morris CE, Murillo J, Schaad NW, Stead DE, Surico G, Ullrich MS, editors. Pseudomonas syringae and related pathogens: Biology and Genetic. Dordrecht: Springer Netherlands; 2003. p. 137–45.
KWE R. Chapter 3: Pseudomonas syringae pathovars. In: Singh US, Singh RP, Kohomoto K, editors. Pathogenesis and host specificity in plant diseases Histopathological, biochemical and molecular bases. vol. Volume I: Prokaryotes. Oxford: Elsevier Science Ltd; 1995.
Kvitko BH, Park DH, Velásquez AC, Wei C-F, Russell AB, Martin GB, Schneider DJ, Collmer A. Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Path. 2009;5(4):e1000388.
Block A, Alfano JR. Plant targets for Pseudomonas syringae type III effectors: virulence targets or guarded decoys? Curr Opin Microbiol. 2011;14(1):39–46.
Qi D, Dubiella U, Kim SH, Sloss DI, Dowen RH, Dixon JE, Innes RW. Recognition of the protein kinase AVRPPHB SUSCEPTIBLE1 by the disease RESISTANCE protein RESISTANCE TO PSEUDOMONAS SYRINGAE5 is dependent on S-acylation and an exposed loop in AVRPPHB SUSCEPTIBLE1. Plant Physiol. 2014;164(1):340–51.
Arrebola E, Cazorla FM, Codina JC, Gutiérrez-Barranquero JA, Pérez-García A, Vicente A. Contribution of mangotoxin to the virulence and epiphytic fitness of Pseudomonas syringae pv. Syringae. Int Microbiol. 2009;12:87–95.
Rico A, Preston GM. Pseudomonas syringae pv. Tomato DC3000 uses constitutive and apoplast-induced nutrient assimilation pathways to catabolize nutrients that are abundant in the tomato apoplast. Mol Plant-Microbe Interact. 2008;21(2):269–82.
Rico A, McCraw SL, Preston GM. The metabolic interface between Pseudomonas syringae and plant cells. Curr Opin Microbiol. 2011;14(1):31–8.
Scholz-Schroeder BK, Hutchison ML, Grgurina I, Gross DC. The contribution of syringopeptin and syringomycin to virulence of Pseudomonas syringae pv. Syringae strain B301D on the basis of sypA and syrB1 biosynthesis mutant analysis. Mol Plant Microbe Interact. 2001;14(3):336–48.
Ogimi C, Higuchi H, Takikawa Y. Bacterial gall disease of Kakuremino (Dendropanax trifidus Mak.) caused by Pseudomonas syringae pv. Dendropanacis pv. Nov. Jpn J Phytopathol. 1988;54(3):296–302.
Nowell RW, Green S, Laue BE, Sharp PM. The extent of genome flux and its role in the differentiation of bacterial lineages. Genome Biol Evol. 2014;6(6):1514–29.
Barzic MR. Persicomycin production by strains of Pseudomonas syringae pv. Persicae. Physiol Mol Plant Pathol. 1999;55(4):243–50.
Badel JL, Shimizu R, Oh HS, Collmer A. A Pseudomonas syringae pv. Tomato avrE1/hopM1 mutant is severely reduced in growth and lesion formation in tomato. Mol Plant-Microbe Interact. 2006;19(2):99–111.
Peñaloza-Vázquez A, Kidambi SP, Chakrabarty AM, Bender CL. Characterization of the alginate biosynthetic gene cluster in Pseudomonas syringae pv. Syringae. J Bacteriol. 1997;179(14):4464–72.
Yu J, Peñaloza-Vázquez A, Chakrabarty AM, Bender CL. Involvement of the exopolysaccharide alginate in the virulence and epiphytic fitness of Pseudomonas syringae pv. Syringae. Mol Microbiol. 1999;33(4):712–20.
Guo W, Cai L-L, Zou H-S, Ma W-X, Liu X-L, Zou L-F, Li Y-R, Chen X-B, Chen G-Y. Ketoglutarate transport protein KgtP is secreted through the type III secretion system and contributes to virulence in Xanthomonas oryzae pv. Oryzae. Appl Environ Microbiol. 2012;78(16):5672–81.
Aldridge P, Metzger M, Geider K. Genetics of sorbitol metabolism in Erwinia amylovora and its influence on bacterial virulence. Mol Gen Genet MGG. 1997;256(6):611–9.
Mellgren EM, Kloek AP, Kunkel BN. Mqo, a tricarboxylic acid cycle enzyme, is required for virulence of Pseudomonas syringae pv. Tomato strain DC3000 on Arabidopsis thaliana. J Bacteriol. 2009;191(9):3132–41.
Templeton MD, Warren BA, Andersen MT, Rikkerink EH, Fineran PC. Complete DNA sequence of Pseudomonas syringae pv. Actinidiae, the causal agent of kiwifruit canker disease. Genome Announc. 2015;3(5):e01054–15.
Marcelletti S, Scortichini M. Comparative genomic analyses of multiple Pseudomonas strains infecting Corylus avellana trees reveal the occurrence of two genetic clusters with both common and distinctive virulence and fitness traits. PLoS One. 2015;10(7):e0131112.
McCann HC, Rikkerink EHA, Bertels F, Fiers M, Lu A, Rees-George J, Andersen MT, Gleave AP, Haubold B, Wohlers MW, et al. Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Path. 2013;9(7):e1003503.
Marcelletti S, Ferrante P, Petriccione M, Firrao G, Scortichini M. Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain-specific features involved in adaptation and virulence to Actinidia species. PLoS One. 2011;6(11):e27297.
O'Brien H, Thakur S, Gong Y, Fung P, Zhang J, Yuan L, Wang P, Yong C, Scortichini M, Guttman D. Extensive remodeling of the Pseudomonas syringae pv. avellanae type III secretome associated with two independent host shifts onto hazelnut. BMC Microbiol. 2012;12(1):141.
Bartoli C, Carrere S, Lamichhane JR, Varvaro L, Morris CE. Whole-genome sequencing of 10 Pseudomonas syringae strains representing different host range spectra. Genome Announc. 2015;3(2):e00379–15.
Baltrus DA, Yourstone S, Lind A, Guilbaud C, Sands DC, Jones CD, Morris CE, Dangl JL. Draft genome sequences of a phylogenetically diverse suite of Pseudomonas syringae strains from multiple source populations. Genome Announc. 2014;2(1):e01195–13.
Buell CR, Joardar V, Lindeberg M, Selengut J, Paulsen IT, Gwinn ML, Dodson RJ, Deboy RT, Durkin AS, Kolonay JF, et al. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. Tomato DC3000. Proc Natl Acad Sci U S A. 2003;100(18):10181–6.
Feil H, Feil WS, Chain P, Larimer F, DiBartolo G, Copeland A, Lykidis A, Trong S, Nolan M, Goltsman E, et al. Comparison of the complete genome sequences of Pseudomonas syringae pv. Syringae B728a and pv. Tomato DC3000. Proc Natl Acad Sci U S A. 2005;102(31):11064–9.
Gardiner DM, Stiller J, Covarelli L, Lindeberg M, Shivas RG, Manners JM. Genome sequences of Pseudomonas spp. isolated from cereal crops. Genome Announc. 2013;1(3):198–204.
Moretti C, Cortese C, Passos da Silva D, Venturi V, Ramos C, Firrao G, Buonaurio R. Draft genome sequence of Pseudomonas savastanoi pv. Savastanoi strain DAPP-PG 722, isolated in Italy from an olive plant affected by knot disease. Genome Announc. 2014;2(5):e00864–14.
Joardar V, Lindeberg M, Jackson RW, Selengut J, Dodson R, Brinkac LM, Daugherty SC, Deboy R, Durkin AS, Giglio MG, et al. Whole-genome sequence analysis of Pseudomonas syringae pv. Phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J Bacteriol. 2005;187(18):6488–98.
Acknowledgements
The authors thank Marilena Palmisano and Dr. Fabio Rezzonico (ZHAW, Wädenswil, CH) for technical assistance and Dr. Jeff Dangl (The University of North Carolina, Chapel Hill, US), Prof. Dr. David Guttman (University of Toronto, CA), Dr. Cindy E. Morris (Institut National de la Recherche Agronomique, Montfavet, FR), Dr. Monika Kałużna and Dr. Joanna Puławska (Research Institute of Horticulture, Skierniewice, PL), Dr. Marco Scortichini (Research Centre for Fruit Trees, Rome, IT), Dr. Roger G. Shivas and Yu Pei Tan (Department of Agriculture, Fisheries, and Forestry, Brisbane, AU), Dr. John Stavrinides (University of Regina, Saskatchewan, CA), and Dr. Joel Vanneste (Plant and Food Research, Hamilton, NZ), for providing some of the strains used in this study.
Funding
Funding was provided by the Swiss Secretariat for Education, Research and Innovation (SBFI C12.0099) within the European research network COST Action FA1104 ‘Sustainable production of high-quality cherries for the European market’ and in part by the European Union Seventh Framework (FP7/2007–2013) under the grant agreement no. 613678 (DROPSA). THMS was supported by the Department of Life Sciences and Facility Management of ZHAW.
Availability of data and materials
The genomic dataset supporting the conclusions of this article are deposited under the accession numbers reported in Table 1 of this manuscript.
Author information
Authors and Affiliations
Contributions
MR, THMS, JFP conceived and designed the experiments; MR performed the experiments; MR, JB and JFP analyzed the data; MR, THMS and JFP interpreted the results. All of the authors contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
This file contains all supplementary tables and figures listed below. Table S1. List of phytotoxins and phytohormons screened in this study. Table S1. List of type III effectors (T3E) screened in this study and their respective locus tags in reference genomes. Table S2. Metrics of the PacBio RSII sequencing results. Table S3. Metrics of the Illumina MiSeq sequencing results for the two Pseudomonas syringae pv. persicae (Ppe) strains. Table S4. Metrics of the Illumina MiSeq sequencing results for the two Pseudomonas 11 syringae pv. persicae (Ppe) strains. Figure S1. Average nucleotide identity (ANI) matrix based on BLASTn and derived phylogeny of a set of the Pseudomonas syringae genomes used in this study. Figure S2. Pathogenicity test results performed on cherry immature fruitlets. (PDF 1680 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ruinelli, M., Blom, J., Smits, T.H.M. et al. Comparative genomics and pathogenicity potential of members of the Pseudomonas syringae species complex on Prunus spp. BMC Genomics 20, 172 (2019). https://doi.org/10.1186/s12864-019-5555-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5555-y