
Abstract
Background
Mud crabs, Scylla spp., are commercially important large-size marine crustaceans in the Indo-West Pacific region. As females have the higher growth rate and economic value, the production of all female stocks is extremely essential in aquaculture. However, the sex determination mechanism is still unclear. Development of sex-specific genetic markers based on next-generation sequencing proved to be an effective tool for discovering sex determination system in various animals.
Results
Restriction-site associated DNA sequencing (RAD-seq) was employed to isolate sex-specific SNP markers for S. paramamosain. A total of 335.6 million raw reads were obtained from 20 individuals, of which 204.7 million were from 10 females and 130.9 million from 10 males. After sequence assembly and female-male comparison, 20 SNP markers were identified to be sex-specific. Furthermore, ten SNPs in a short sequence (285 bp) were confirmed heterozygous in females and homozygous in males in a large population by PCR amplification and sequencing. Subsequently, a female-specific primer was successfully designed according to the female-specific nucleotide which could amplify an expected band from females but not from males. Thus, a rapid and effective method for molecular sexing in S. paramamosain was developed, meanwhile, this method could successfully identify the sex of S. tranquebarica and S. serrata. Finally, nine and four female-specific SNP markers were detected in S. tranquebarica and S. serrata, respectively.
Conclusions
Sex-specific SNP markers were firstly identified in crab species and showed female heterogamety and male homogamety, which provided strong genetic evidence for a WZ/ZZ sex determination system in mud crabs S. paramamosain, S. tranquebarica and S. serrata. These findings will lay a solid foundation for the study of sex determination mechanism, sex chromosome evolution, and the development of mono-sex population in crustaceans.
Similar content being viewed by others

Background
Sex determination mechanisms of animals are remarkably diverse and complicated, and they are attracting considerable interest due to its great implication in both theory and practice [1,2,3]. Unlike vertebrates, the sex determination mechanisms of crustaceans are more varied and frequently affected by genetic and/or environmental factors [4, 5]. Among crustaceans, several crab species, including Plagusia dentipes, Eriocheir japonicus and Hemigrapsus sanguineus, are thought to have a XY/XX sex determination system based on karyotype studies [6,7,8]. However, a recent study suggested that Chinese mitten crab (Eriocheir sinensis) exhibited a WZ/ZZ sex determination system according to quantitative trait locus location of the gender phenotype [5]. Meanwhile, the WZ/ZZ sex determination system has also been observed in other crustaceans, such as kuruma prawn (Penaeus japonicus) [9], Pacific white shrimp (Litopenaeus vannamei) [10], and giant freshwater prawn (Macrobrachium rosenbergii) [11]. Importantly, studies on the genetic basis of sex determination mechanism are the foundation towards future sex manipulation biotechnologies, including the development of mono-sex population [2, 12], especially for those crabs with significant sexual dimorphism, such as caribbean king crab (Mithrax spinosissimus) [13] and Japanese mitten crab (Eriocheir japonica) [14]. So far, the sex determination mechanism remains unclear in most aquaculture crustacean species, which have therefore obviously limited its potential application in the aquaculture sector.
Generally, there are three popular types of techniques for studying sex determination mechanism, i.e. cytogenetic approaches, breeding experiments, and sex-specific molecular markers [15]. The application of cytogenetic analysis is limited because some species lack visually heteromorphic sex chromosomes, while breeding experiments are mainly focused on several common species, and therefore impracticable for many species. The use of sex-linked or sex-specific markers is regarded as a powerful tool for well-understanding sex determination system in most species [2, 12, 16].
Over the last few decades, various genetic approaches have been successfully applied to identify sex-specific DNA sequences or markers in a range of aquaculture fish and crustacean species. For example, random amplified polymorphic DNA (RAPD) for turbot (Scophthalmus maximus) [17], amplified fragment length polymorphism (AFLP) for swimming crab (Portunus trituberculatus) [18] and Pacific bluefin tuna (Thunnus orientalis) [19], as well as simple sequence repeat (SSR) markers for half-smooth tongue sole (Cynoglossus semilaevis) [20, 21] and rock bream (Oplegnathus fasciatus) [22]. Currently, with the rapid development of next-generation sequencing (NGS) technologies, some novel methods have been developed for exploring sex-associated DNA markers [3, 23]. NGS-based marker systems allow highly efficient DNA markers development within a short period [24]. Restriction-site associated DNA sequencing (RAD-seq) is based on NGS technologies and can discover massive single nucleotide polymorphisms (SNPs) in various species by sequencing parts of the genome at high depth, without a reference genome [23, 25,26,27]. Recently, RAD-seq was successfully used to develop sex-specific markers in aquaculture species, such as Atlantic halibut (Hippoglossus hippoglossus) [28], European sea bass (Dicentrarchus labrax) [25], and silver carp (Hypophthalmichthys molitrix) [3].
Mud crabs (Scylla spp.) are highly valuable commercial aquaculture species and fishery resources in the Indo-West Pacific region, such as China [29], Thailand [30], Vietnam [31], Philippines [32] and Malaysia [33]. The aquaculture output of mud crab in China reached approximately 148,977 tons, the top among all marine commercial crabs [34]. Although the farming and fishing output of mud crab increased tremendously in recent years, the current scale of production is still too inadequate to meet the market demand [35]. A major challenge in mud crab aquaculture industry presently is how to develop a set of fast and viable seed production techniques so as to improve supply [36]. Additionally, Scylla paramamosain, one of mud crab species, exhibits significant sexual dimorphism in growth rate and body size, with females growing faster and having higher nutritive value than males [37]. Thus, it is essential to develop sex-linked markers for production of mono-sex breeding, shortening farming duration, as well as understanding the genetic basis of sex determination system [3, 28]. However, there is currently no available genetic information on sex determination system for these economically important crustacean species.
In the present study, RAD-seq technology was employed to identify and characterize sex-specific SNP markers in mud crab S. paramamosain. A rapid and reliable molecular method based on the female-specific SNP markers was developed to identify the genetic sex of individuals at early developmental stages. Further, the developed PCR-based genetic sex identification method was applied in two other mud crab species, S. tranquebarica and S. serrata. The results suggest a WZ/ZZ sex determination system in S. paramamosain, S. tranquebarica and S. serrata. These findings will provide new insights into the mechanism of sex determination in brachyuran crabs, as well as facilitate the development of mono-sex population of mud crab and related crustacean species.
Results
RAD sequencing
A total of 335,600,178 raw reads were generated after RAD-seq, with 204,685,780 for females and 130,914,398 for males (Table 1). After filtering low quality reads, 172,889,526 clean reads for females and 111,335,216 clean reads for males were obtained, with an average of 14,211,237 per sample. The average read length of each sample ranged from 126.2 to 134.1 bp, while the average clean reads rate of all samples was 83.75%. Based on the read quality and quantity, female sample named “F2A” was selected as a reference, and its clean reads were clustered and assembled (Additional file 1). After filtering, 272,347 tags were used for assembly, retrieving 69,799,531 bp data and 242,137 contigs. The contig N50 was 300 bp and GC content was 40.50% (Additional file 1).
Identification of sex-specific SNP markers
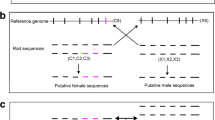
The reads of 20 individuals were mapped to the reference sample “F2A” (Additional file 2), with the mapping rate for each individual varying from 49.73 to 80.34%. After alignment analysis, 1,780,706 polymorphic SNP markers across the 20 individuals were obtained. These SNP markers were subjected to sex-specific marker identification, and as a result, a total of 20 sex-specific SNP markers (Table 3) were identified in 11 contigs by comparing the SNPs between sexes. Next, 10 primer pairs were designed to amplify the corresponding sequences (Table 2). After testing, primer 11,508 (Table 2) could successfully amplify Cluster_384014 with 285 bp in length (Table 3), which was regarded as a sex-related sequence. PCR amplification and sequencing analysis showed that this sequence was completely identical between sexes except 10 SNPs. These 10 SNPs were heterozygous (including C/T, G/A, A/T, T/G) in all females, but homozygous in all males (Fig. 1), indicating that the sex determination system in S. paramamosain is WZ/ZZ with females being heterogametic. These SNP markers were then successfully validated by PCR assay and sequencing using additional 195 specimens (106 females and 89 males).

Ten female-specific SNP markers in the sequencing chromatograms of the sex-related sequence of Scylla paramamosain. SNP1: C/T; SNP2: C/T; SNP3: C/T; SNP4: T/C; SNP5: G/A; SNP6: C/T; SNP7: A/T; SNP8: T/G; SNP9: A/T; SNP10: A/T
Development of the PCR-based genetic sex identification method
First, the sequence (Cluster_384014) which contained the 10 female-specific SNP markers was lengthened to 2315 bp by comparing with the whole genome sequences of S. paramamosain (unpublished data) and molecular cloning. This sequence was submitted to GenBank database under the accession number of MH133208. Subsequently, the PCR-based genetic sex identification method was successfully developed based on the lengthened sequence and the female-specific SNP markers. A pair of female-specific primers (SPFS, Table 2, Fig. 2) produced a stable female-specific band with 320 bp in length in females, but not in males. Meanwhile, a pair of control primers (SPC, Table 2, Fig. 2) produced a 282 bp long band in both females and males. Four nucleotides mismatches were artificially created in the female-specific primer, of which three were at the female-specific SNPs, SNP 7, SNP 8 and SNP 9, and one was at the 5′ end of the primer (Table 2, Fig. 2). A total of 96 specimens (48 females and 48 males) were used to determine the accuracy and precision of the PCR-based genetic sex identification method. The results showed a 100% accuracy for identification of genetic sex of S. paramamosain. (Fig. 3 and Additional file 3).

Part of female-related DNA sequence cloned based on the genome sequence of Scylla paramamosain. The red color letters in the sequence indicate the ten female-specific SNPs, where SNP1: C/T; SNP2: C/T; SNP3: C/T; SNP4: T/C; SNP5: G/A; SNP6: C/T; SNP7: A/T; SNP8: T/G; SNP9: A/T; SNP10: A/T. The underlined text indicated the primer binding sites in the PCR-based genetic sex identification method, where Control-F and Control-R were the control primer (SPC) binding sites, and Female-specific-F and Female-specific-R were the female-specific primer (SPFS) binding sites

Application of the PCR-based genetic sex identification method in Scylla paramamosain. Female-specific band (320 bp): PCR products amplified with the female-specific primer (SPFS); Control band (282 bp): PCR products amplified with control primer (SPC); M: marker; a: the agarose gel electrophoresis results for 12 females and 12 males cultured in a pond at Shantou, China. b: the agarose gel electrophoresis results for another 12 females and 12 males cultured in a pond at Shantou, China
Application of the molecular genetic sex identification method to individuals at early developmental stages
The PCR-based genetic sex identification method was applied to determine the sex and sex ratio of 180 offspring (unknown sex) of S. paramamosain from a full-sib family at three different early developmental stages, i.e. megalopa stage (M), the first crablet stage (C1), and the second crablet stage (C2). At these three developmental stages, the number of females were 30, 24, 25, and the number of males were 30, 36, 35, respectively (Table 4). The female/male ratio at each developmental stage was 1.00, 0.67 and 0.71, respectively, with the total sex ratio being 0.78 (Table 4). There was a tendency toward a lower female/male ratio with the growth and development of S. paramamosain. Chi-square tests showed that the sex ratio at each developmental stage and the total sex ratio did not exhibit significant differences with the 1:1 separation ratio (P > 0.05) (Table 4).
Cross-species application of the molecular genetic sex identification method and identification of sex-specific SNP markers
To test the feasibility of the newly developed molecular sexing method in three other species of genus Scylla, the female-specific primer (SPFS, Table 2) was used to amplify the potential bands in S. tranquebarica, S. serrata and S. olivacea. Interestingly, the results showed that this method worked well in S. tranquebarica and S. serrata despite an extremely weak band exhibited in the males of S. serrata (Fig. 4). To investigate whether the sex-specific SNP markers exist in these three species, primer 11,508 (Table 2) was employed to amplify the corresponding fragment and then sent for sequencing. After sequence comparison, nine (SNP2-SNP10, Table 5) and four (SNP2, SNP3, SNP9, SNP10, Table 5) sex-specific SNP markers were identified from S. tranquebarica and S. serrata, respecitively. The genotypes of these sex-specific markers were identical to those detected in S. paramamosain, being heterozygous in all females, but homozygous in all males. In addition, no sex-specific SNP markers were observed in S. olivacea (Table 5).

Application of the PCR-based genetic sex identification method in three other species of genus Scylla. Female-specific band (320 bp): PCR products amplified with female-specific primer (SPFS); The PCR-based genetic sex identification method was effective for S. tranquebarica and S. serrata, but not for S. olivacea, even though an extremely faint band exhibited in male individuals of S. serrata
Discussion
Studies on sex determination system of crabs are much limited. In the latter part of the last century, the XY/XX sex determination system was suggested for crabs [8, 38], but recently, the WZ/ZZ sex determination system was found in Chinese mitten crab [5]. To the best of our knowledge, this study is the first to focus on the sex determination system of genus Scylla. Here, 10 female-specific SNP markers were verified from DNA sequence in 195 individuals of S. paramamosain. These identified SNP markers were heterozygous in all females but homozygous in all males, suggesting a WZ/ZZ female heterogametic sex determination system in S. paramamosain. Additionally, the female-specific SNP markers were detected in a short DNA sequence of both females and males, which indicated the W and Z chromosomes of S. paramamosain are not fully differentiated from each other at DNA level and there are still many homologous loci, as suggested by Cui et al. [5]. The stable sex-specific genetic markers deteced in this study may due to the meiotic recombination suppression between sex chromosomes [39,40,41].
Sex determination system is essential not only for the study of reproductive biology and the process of genome evolution [5, 42], but also for the development of sex manipulation techniques, especially in economically important species with obvious sexual dimorphic characters such as mud crab. Given the large number of chromosomes in crustaceans, to elucidate the sex determination system using cytogenetic investigations and high-resolution linkage map is challenging [5]. Fortunately, sex-linked DNA markers provides an alternative tool to study sex determination system and it has shown great potential application in various aquaculture species [3, 27, 43]. In this study, the sex-linked SNPs provide a strong evidence for chromosomal sex determination system and serve as a crucial foundation for development of mono-sex population of S. paramamosain [2, 12]. Additionally, these sex-linked markers will be helpful for identifying sex-related contigs from whole genome assemblies, thereby broadening our current knowledge of sex chromosome genes and evolution [15].
The ability to correctly identify sex of animals is of critical importance in ecological and evolutionary studies, as well as for artificial breeding and farming purposes [44, 45]. Several approaches have been developed for sex identification in animals, such as behavior observation [46], morphological and external characteristics [47], ultrasonography [48] and hormone levels [49]. However, these methods are prone to high error rates and sometimes technically complex [45]. Genetic sex identification is an essential sex determination tool when the sex of an organism can not be discriminated morphologically [12]. In the last decade, sex-specific DNA markers have been successfully used to distinguish sex of different species, especially in animals at their early developmental stages without obvious sexual traits [12, 21, 43, 50]. The PCR-based method for genetic sex identification is believed to be expedient, time-saving and cost-effective [12, 45, 51].
The sex of S. paramamosain is indistinguishable by morphological traits at their early developmental stages, especially younger than one-month old. The lack of sex identification methods have tremendously limited the study on sex determination and differentiation between males and females. Thus, the molecular sex identification method for S. paramamosain is essential for these reasons. Among the PCR-based sex identification methods, sex-specific primers were usually designed with different strategies, for example, sex-specific DNA sequence [3, 50], two mismatched nucleotides in the forward primer [12], and DNA sequence deletion between sexes [43]. In the present study, four nucleotide mismatches containing three female-specific SNP markers were artificially created that gave positive PCR amplification in females but not in males. Importantly, the mismatch in the 3’ end of the female-specific primer played a vital role in this method because the initial binding site with DNA template is at the 3’ end of the primer. Thus, in addition to providing a novel method for determining the sex of S. paramamosain, especially during larval and juvenile stages, this method also provided new insights into the application of sex-specific SNP markers to genetic sex identification of other brachyuran species in the future, and it should be beneficial for the marker-assisted sex control breeding and aquaculture [21, 50, 52].
Further, the investigation of sex and sex ratio of individuals at three early developmental stages (M, C1 and C2) from a full-sib family showed no significant deviation from the expected ratio of 1:1, which was consistent with the genetic sex determination mechanism suggested in this study. However, the number of females decreased with the growth and development of S. paramamosain. In our previous study, the number of female S. paramamosain at two-month old (older than C2) was also lower than males, but the sex ratio significantly skewed towards females from three-month old to four-month old (unpublished data). The variation of the sex ratio during the process of growth and development could be due to the known aggressive and cannibalistic behavior of Scylla spp., and the growing competition for food and spouses, especially for male crabs [33], which leads to injury and death of males.
Sex-specific markers can also provide insights into sex chromosome conservation and evolution [15, 27]. The same sex-specific markers and sex-determining DNA sequence were identified and verified in bighead carp, silver carp and grass carp by 2b-RAD sequencing, suggesting that the same pathways might be involved in sex determination systems [3]. In this study, S. tranquebarica and S. serrata were proved to have nine and four identical female-specific SNP markers with S. paramamosain, respectively, suggesting that these three crab species may have a homologous nascent W chromosome and share the most recent common ancestor [3]. Therefore, the WZ/ZZ sex determination system is suggested for S. paramamosain, S. tranquebarica and S. serrata. While the data obtained in the present study was unable to draw conclusion on the genetic sex determination system of S. olivacea because no sex-specific markers were observed. However, the sex-specific SNP markers may exist in other genome regions of S. olivacea, hence, further studies should be carried out in the future to explore this.
Although mud crabs have been captured and cultured for over 110 years, the taxonomy of genus Scylla was controversial until the revision by Keenan et al. in 1998 [53], based on genetic variations, external morphology and morphometric characters analysis [54]. Phylogenetic study on genus Scylla based on COI sequence of mtDNA showed that S. paramamosain is genetically closest to S. tranquebarica, and then to S. serrata, while, the largest genetic distance was found with S. olivacea [55, 56]. In the present study, the number of identical female-specific SNP markers were found to be nine between S. paramamosain and S. tranquebarica, four between S. paramamosain and S. serrata, but none between S. paramamosain and S. olivacea, an observation which is supported by the phylogenetic relationships of genus Scylla as determined by previous studies [55, 56]. Nevertheless, we suggest that for deeply understanding the evolution and phylogenetic relationships of genus Scylla, more research should be carried out.
Conclusions
RAD-seq technology has proven to be a good tool for the identification of sex-specific markers in non-model organisms. To the best of our knowledge, this is the first study to identify female-specific SNP markers in mud crab (S. paramamosain) that showed heterozygous in females but homozygous in males. Subsequently, a rapid and effective method for molecular genetic sex identification was developed, by which, the sex of S. paramamosain, S. tranquebarica and S. serrata could be distinguished with a 100% accuracy. Moreover, nine and four identical female-specific SNP markers similar to those in S. paramamosain were identified in S. tranquebarica and S. serrata, respectively. These findings provided a solid evidence for a WZ/ZZ sex determination system in these three mud crab species. This study will not only further our understanding of sex determination mechanism and genome evolution of genus Scylla, but also facilitate mono-sex breeding and aquaculture of these crab species.
Methods
Samples collection and DNA extraction
Twenty wild S. paramamosain (10 males and 10 females), randomly collected from the inshore of Hainan Province, China, were used to determine the potential sex-specific SNP markers by RAD-seq. Another 195 specimens (106 females and 89 males) were collected and used to confirm the sex-specific SNP markers, of which 115 were from the inshore of Shantou and 80 were from a culture pond in Hainan. In addition, 96 specimens (48 females and 48 males) were collected from two culture ponds in Shantou and Raoping for verification of the molecular sex identification method developed based on the sex-specific SNP markers. Another 180 young progenies with unknown sex from a full-sib family were sampled and used to distinguish the sex and sex ratio using the developed molecular sex identification method. Among these progenies, 60 individuals were at megalopa stage (M), 60 individuals were at the first crablet stage (C1), and 60 individuals were at the second crablet stage (C2). In order to test the feasibility of the sex-specific SNP markers and the developed sex identification method in other Scylla species, S. tranquebarica (N = 20, 10 males and 10 females) and S. olivacea (N = 6, 3 males and 3 females) were collected from Setiu Wetlands, Terengganu, Malaysia, whereas S. serrata (N = 6, 3 males and 3 females) was caught from Iloilo, Philippines. The sex of these crabs was identified based on the shape of the abdomen. Females have wider and more globular abdomens, whereas males have narrow and straight abdomens [57].
Before sampling, the crabs were placed on ice for anesthetization (about 10 min). Muscle tissues were frozen in liquid nitrogen and preserved at − 80 °C until DNA extraction. The whole body of the individuals at early developmental stages (M, C1 and C2) were used for DNA extraction due to their small size. Genomic DNA was extracted using TIANamp Marine Animals DNA kit (Tiangen Biotech Co. Ltd., Beijing, China) and treated with RNase A to remove residual RNA. The DNA quality was assessed by agarose gel electrophoresis and quantified using a Qubit fluorometer (Life Technologies).
RAD library preparation and sequencing
The RAD library was prepared following previous methods [58,59,60] with slight modifications. Briefly, approximately 1 μg of genomic DNA from each individual was digested with EcoRI-HF. The P1 adapter which contains individual-specific index sequence of 5 bp long barcode (Table 1) was ligated to the purified products of restriction reaction. The ligated samples were then pooled, purified and randomly sheared to short fragments with an average size of 350 bp using Bioruptor (Diagenode, Liège, Belgium). The P2 adapter which contains a 3′ dT overhang was ligated to the sheared DNA fragments. Furthermore, the ligation mix with 300 to 500 bp was purified using the MinElute Gel Extraction Kit (Qiagen). The library was further enriched by high-fidelity PCR using P1 and P2 adapter primers. Finally, the PCR products comprising between 300 and 500 bp were purified and sequenced (2 × 150-bp paired-end reads) on Illumina Hiseq 3000 platform. Raw data generated in this study has been submitted to the NCBI Short Read Archive (SRA) under the accession number SRP135178.
Sequence analysis and sex-specific SNPs discovery
The raw reads from the Illumina Hiseq were first sorted according to the individual-specific index sequence, and then the indexes and low-quality reads were removed by Trim Galore software (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Quality control analysis was performed to assess the length distribution and GC content of the clean reads using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Individual which named “F2A” has the best sequencing quality and quantity, so clean reads from it were clustered for reference using the software CD-HIT-EST (http://weizhongli-lab.org/cd-hit/) under the following thresholds: sequence identity ≥97%, minimum coverage 10, and maximum coverage 400, with the cluster analysis results shown in Additional file 1. After the cluster analysis, clean reads were assembled to contigs using the Spades software (http://cab.spbu.ru/software/spades/) with k-mer size of 21, 33 and 55. The contigs shorter than 150 bp were excluded from assembly. The assembled sequences of the sample “F2A” were regarded as a reference, with reads from each sample aligned to the assembled reference of “F2A” using Burrows-Wheeler Alignment Tool (BWA) [61]. The alignment results were processed using SAMtools [62] to identify candidate genetic variants among all sequenced samples, including both SNPs and INDELs. Bcftools and Vcfutils.pl were used to filter variants with the default parameters, such as the minimum root-mean-square read mapping quality of 10, a minimum depth of 2 and the minimum number of read supporting the variant of 2. The SNPs of males and females were summarized respectively, and then the sex-specific SNP markers were identified based on the SNP comparison between males and females. Only SNP markers with a polymorphic genotype associated with the sex were regarded as sex-specific markers.
Development and verification of sex-specific SNP markers
Based on the results of RAD-seq, 11 contigs were found to contain sex-specific SNPs. A total of ten pairs of primers (Table 2) were designed by the Primer Premier 5.0 software, with the exclusion of one contig (cluster 40,991, Table 3) due to the near-end position of the sex-specific SNP marker. PCR amplification was then used to validate the specificity of each sex-specific SNP marker. The PCR reactions were carried out with a total volume of 25 μl, containing 50 ng of template DNA, 2.5 μl of 10 × PCR buffer, 2.0 μl of dNTPs (2.5 mM each), 0.5 μl of each primer (10 μM), 0.5 μl of Taq DNA polymerase (5 U/μl, TransGen Biotech, Beijing, China) and water to the final volume. PCR programs were as follows: an initial denaturation step at 94 °C for 5 min; then 34 cycles of 94 °C for 30 s, annealing temperature of primer pair for 30 s, and 72 °C for 40 s; and a final extension step at 72 °C for 7 min. The amplified products were separated by 1% agarose gel electrophoresis, and the expected PCR fragments were directly sequenced in both directions by Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China). The sex-specific SNP markers were determined manually by the peaks and their colors in the chromatogram. Among the sequences, one containing sex-specific SNPs with the best quality and quantity was regarded as sex-related sequence, and was used for further analysis. Furthermore, the sex-related sequence was amplified and the sex-specific SNP markers were verified using 195 other specimens (106 females and 89 males) with known sex. The PCR products were sequenced and the sex-specific SNPs were genotyped as described above.
Development of a rapid method for genetic sex identification
First, the sex-related sequence was aligned to the genome of S. paramamosain (unpublished data), obtaining a longer sequence. The obtained sequence was then verified through molecular cloning and sequencing. The primers (FLFE-1 and FLFE-2, Table 2) used for DNA cloning were designed based on the genome sequences. Next, in order to develop a rapid PCR-based method to identify genetic sex of S. paramamosain, a pair of female-specific primers (SPFS, Table 2) was designed based on the sex-specific SNP markers, which is expected to amplify the sex-specific SNP regions only in females and show female-specific band on agarose gel electrophoresis. Meanwhile, a pair of control primers (SPC, Table 2) were designed based on the longer sex-related sequence and regarded as a control to avoid the presence of false-negative results, which can amplify the same size fragment in both males and females. The optimized PCR parameters of the PCR-based genetic sex identification method are as follows: total PCR reaction volume of 20 μl, containing 75 ng of template DNA, 2.5 μl of 10 × PCR buffer, 1.0 μl of dNTPs (2.5 mM each), 0.25 μl of each primer (10 μM), 0.4 μl of Taq DNA polymerase (5 U/μl, TransGen Biotech, Beijing, China) and water to the final volume. The PCR program was as follows: an initial denaturation step at 94 °C for 5 min; then 35 cycles of 94 °C for 30 s, 65 °C for 30 s, and 72 °C for 40 s; and a final extension step at 72 °C for 7 min. PCR amplification was conducted in 96 specimens (48 females and 48 males) to verify the PCR-based genetic sex identification method.
Application of the PCR-based genetic sex identification method for determining the sex of individuals at early developmental stages
The sex of 180 individuals of S. paramamosain from a full-sib family at three different early developmental stages, i.e. M, C1 and C2, was identified by the PCR-based genetic sex identification method. The sex ratio at each developmental stage was estimated according to the identification result. Chi-square tests were used to estimate the fitness of sex ratio (female/male) to the expected 1:1.
Cross-species application and verification of sex-specific SNP markers
The sex-specific SNP markers identified from S. paramamosain were cross-species tested in three other Scylla species, i.e. S. tranquebarica, S. serrata and S. olivacea, using PCR amplification and sequencing methods as described above. To test the feasibility of the newly developed molecular sex identification method, PCR amplifications were also carried out (Table 2) in these three other Scylla species.
Data analysis
Chi-square tests were performed using the SPSS 21.0 for Windows (SPSS, Michigan Avenue, Chicago, IL, USA), and differences were considered statistically significant at P < 0.05.
Abbreviations
- AFLP:
-
Amplified fragment length polymorphism
- NGS:
-
Next-generation sequencing
- RAD-seq:
-
Restriction-site associated DNA sequencing
- RAPD:
-
Random amplified polymorphic DNA
- SNPs:
-
Single nucleotide polymorphisms
- SSR:
-
Simple sequence repeat
References
Pennell MW, Mank JE, Peichel CL. Transitions in sex determination and sex chromosomes across vertebrate species. Mol Ecol. 2018; https://doi.org/10.1111/mec.14540.
Pan ZJ, Li XY, Zhou FJ, Qiang XG, Gui JF. Identification of sex-specific markers reveals male heterogametic sex determination in Pseudobagrus ussuriensis. Mar Biotechnol. 2015;17:441–51.
Liu H, Pang M, Yu X, Zhou Y, Tong J, Fu B. Sex-specific markers developed by next-generation sequencing confirmed an XX/XY sex determination system in bighead carp (Hypophthalmichehys nobilis) and silver carp (Hypophthalmichthys molitrix). DNA Res. 2018. https://doi.org/10.1093/dnares/dsy009.
Ford AT. Can you feminise a crustacean? Aquat Toxicol. 2008;88:316–21.
Cui Z, Hui M, Liu Y, Song C, Li X, Li Y, et al. High-density linkage mapping aided by transcriptomics documents ZW sex determination system in the Chinese mitten crab Eriocheir sinensis. Heredity. 2015;115:206–15.
Niiyama H. The problem of male heterogamety in the decapod Crustacea, with special reference to the sex-chromosomes in Plagusia dentipes de Haan and Eriocheir japonicus de Haan. J Fac Sci Hokkaido imp Univ Ser VI. Zoology. 1937;5:283–95.
Niiyama H. The XY chromosomes of the shore-crab, Hemigrapsus sanguineus (de Haan). Jpn J Genet. 1938;14:34–8.
Lécher P, Defaye D, Noel P. Chromosomes and nuclear DNA of Crustacea. Invertebr Reprod Dev. 1995;27:85–114.
Li Y, Byrne K, Miggiano E, Whan V, Moore S, Keys S, et al. Genetic mapping of the kuruma prawn Penaeus japonicus using AFLP markers. Aquaculture. 2003;219:143–56.
Zhang L, Yang C, Zhang Y, Li L, Zhang X, Zhang Q, et al. A genetic linkage map of Pacific white shrimp (Litopenaeus vannamei): sex-linked microsatellite markers and high recombination rates. Genetica. 2007;131:37–49.
Jiang XH, Qiu GF. Female-only sex-linked amplified fragment length polymorphism markers support ZW/ZZ sex determination in the giant freshwater prawn Macrobrachium rosenbergii. Anim Genet. 2013;44:782–5.
Xu D, Lou B, Xu H, Li S, Geng Z. Isolation and characterization of male-specific DNA markers in the rock bream Oplegnathus fasciatus. Mar Biotechnol. 2013;15:221–9.
Baeza JA, Anderson JR, Spadaro AJ, Behringer DC. Sexual dimorphism, allometry, and size at first maturity of the caribbean king crab, Mithrax spinosissimus, in the Florida Keys. J Shellfish Res. 2014;31:909–16.
Kobayashi S, Satake K. Difference in body size and sexual dimorphism in the ogasawara mitten crab, Eriocheir ogasawaraensis, and the Japanese mitten crab, Eriocheir japonica. Pac Sci. 2017;71:171–88.
Gamble T, Zarkower D. Identification of sex-specific molecular markers using restriction site-associated DNA sequencing. Mol Ecol Resour. 2014;14:902–13.
Charlesworth D, Mank JE. The birds and the bees and the flowers and the trees: lessons from genetic mapping of sex determination in plants and animals. Genetics. 2010;186:9–31.
Vale L, Dieguez R, Sánchez L, Martínez P, Viñas A. A sex-associated sequence identified by RAPD screening in gynogenetic individuals of turbot (Scophthalmus maximus). Mol Biol Rep. 2014;41:1501–9.
Lai X, Gao H, Yan B, Yan W, Shen S. Identification of sex-linked DNA markers by AFLP in Portunus trituberculatus. Gene. 2014;544:216–9.
Agawa Y, Iwaki M, Komiya T, Honryo T, Tamura K, Okada T, et al. Identification of male sex-linked DNA sequence of the cultured Pacific bluefin tuna Thunnus orientalis. Fish Sci. 2015;81:113–21.
Chen SL, Ji XS, Shao CW, Li WL, Yang JF, Liang Z, et al. Induction of mitogynogenetic diploids and identification of WW super-female using sex-specific SSR markers in half-smooth tongue sole (Cynoglossus semilaevis). Mar Biotechnol. 2012;14:120–8.
Liao X, Xu G, Chen SL. Molecular method for sex identification of half-smooth tongue sole (Cynoglossus semilaevis) using a novel sex-linked microsatellite marker. Int J Mol Sci. 2014;15:12952–8.
Xu D, Lou B, Li S, Sun X, Zhan W, Chen R, et al. A novel sex-linked microsatellite marker for molecular sexing in rock bream fish Oplegnathus fasciatus. Biochem Syst Ecol. 2015;62:66–8.
Zhang Q, Liu C, Liu Y, VanBuren R, Yao X, Zhong C, et al. High-density interspecific genetic maps of kiwifruit and the identification of sex-specific markers. DNA Res. 2015;22:367–75.
Kafkas S, Khodaeiaminjan M, Güney M, Kafkas E. Identification of sex-linked SNP markers using RAD sequencing suggests ZW/ZZ sex determination in Pistacia vera L. BMC Genomics. 2015;16:98.
Palaiokostas C, Bekaert M, Taggart JB, Gharbi K, McAndrew BJ, Chatain B, et al. A new SNP-based vision of the genetics of sex determination in European sea bass (Dicentrarchus labrax). Genet Sel Evol. 2015;47:68.
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet. 2016;17:81–92.
Gamble T. Using RAD-seq to recognize sex-specific markers and sex chromosome systems. Mol Ecol. 2016;25:2114–6.
Palaiokostas C, Bekaert M, Davie A, Cowan ME, Oral M, Taggart JB, et al. Mapping the sex determination locus in the Atlantic halibut (Hippoglossus hippoglossus) using RAD sequencing. BMC Genomics. 2013;14:566.
Ma H, Ma C, Ma L, Cui H. Novel polymorphic microsatellite markers in Scylla paramamosain and cross-species amplification in related crab species. J Crustacean Biol. 2010;30:441–4.
Nooseng S. Status of mud crab industry in Thailand. In: Proceedings of the international seminar-workshop on mud crab aquaculture and fisheries management, 10–12 April 2013, Tamil Nadu, India, Rajiv Gandhi Centre for Aquaculture; 2015. p. 37–43.
Petersen EH, Glencross BD, Truong HP, Vu AT, Le Anh T. Recent changes in the bioeconomics of lobster and mud crab mariculture in Vietnam. Asian J Agric Dev. 2016;13:89–105.
Quinitio ET. Overview of the mud crab industry in the Philippines. In: Philippines: in the forefront of the mud crab industry development: proceedings of the 1st National mud Crab Congress, 16–18 November 2015, Iloilo City, Philippines, aquaculture department, southeast Asian fisheries development center; 2017. p. 1–12.
Fazhan H, Waiho K, Azri MFD, Al-Hafiz I, Norfaizza WIW, Megat FH, et al. Sympatric occurrence and population dynamics of Scylla spp. in equatorial climate: effects of rainfall, temperature and lunar phase. Estuar Coast Shelf Sci. 2017;198:299–310.
Yearbook, C.F.S. China Fishery Statistical Yearbook 2017. Beijing (in Chinese): Fisheries Bureau, Department of Agriculture of China. Beijing: 2017. p. 22.
Ma HY, Li SJ, Feng NN, Ma CY, Wang W, Chen W, et al. First genetic linkage map for the mud crab (Scylla paramamosain) constructed using microsatellite and AFLP markers. Genet Mol Res. 2016. https://doi.org/10.4238/gmr.15026929.
Gao J, Wang X, Zou Z, Jia X, Wang Y, Zhang Z. Transcriptome analysis of the differences in gene expression between testis and ovary in green mud crab (Scylla paramamosain). BMC Genomics. 2014;15:585.
Jiang W, Ma HY, Ma CY, Li SJ, Liu YX, Qiao ZG, et al. Characteristics of growth traits and their effects on body weight of G1 individuals in the mud crab (Scylla paramamosain). Genet Mol Res. 2014;13:6050–9.
Legrand JJ, Legrand-Hamelin E, Juchault P. Sex determination in Crustacea. Biol Rev. 1987;62:439–70.
Devlin RH, Nagahama Y. Sex determination and sex differentiation in fish: an overview of genetic, physiological, and environmental influences. Aquaculture. 2002;208:191–364.
Groenen MA, Wahlberg P, Foglio M, Cheng HH, Megens HJ, Crooijmans RP, et al. A high-density SNP-based linkage map of the chicken genome reveals sequence features correlated with recombination rate. Genome Res. 2009;19:510–9.
Popa A, Samollow P, Gautier C, Mouchiroud D. The sex-specific impact of meiotic recombination on nucleotide composition. Genome Biol Evol. 2012;4:412–22.
Li XY, Zhang QY, Zhang J, Zhou L, Li Z, Zhang XJ, et al. Extra microchromosomes play male determination role in polyploid gibel carp. Genetics. 2016;203:1415–24.
Sun S, Li W, Xiao S, Lin A, Han Z, Cai M, et al. Genetic sex identification and the potential sex determination system in the yellow drum (Nibea albiflora). Aquaculture. 2018;492:253–8.
Martínez P, Viñas AM, Sánchez L, Díaz N, Ribas L, Piferrer F. Genetic architecture of sex determination in fish: applications to sex ratio control in aquaculture. Front Genet. 2014;5:340.
Centeno-Cuadros A, Tella JL, Delibes M, Edelaar P, Carrete M. Validation of loop-mediated isothermal amplification for fast and portable sex determination across the phylogeny of birds. Mol Ecol Resour. 2017;18:251–63.
Gray CM, Hamer KC. Food-provisioning behaviour of male and female Manx shearwaters, Puffinus puffinus. Anim Behav. 2001;62:117–21.
Malison JA, Held JA, Kaatz SE. Sex determination of yellow perch by external morphology. N Am J Aquacult. 2011;73:285–7.
Chiotti JA, Boase JC, Hondorp DW, Briggs AS. Assigning sex and reproductive stage to adult lake sturgeon using ultrasonography and common morphological measurements. N Am J Fish Manage. 2016;36:21–9.
Nazeri S, Amiri BM, Nazeri MR, Mirvaghefi AR. Sexing of farmed immature beluga (Huso huso) using steroid hormone levels as indicators. Comp Clin Path. 2014;23:631–5.
Wang X, Jiang J, Gao J, Liu J, Qi J, Wang Z, et al. Identification of two novel female-specific DNA sequences in half-smooth tongue sole, Cynoglossus semilaevis. Aquaculture. 2013;388:49–53.
Wang N, Zhang ZW. The novel primers for sex identification in the brown eared-pheasant and their application to other species. Mol Ecol Resour. 2009;9:186–8.
Koyama T, Ozaki A, Yoshida K, Suzuki J, Fuji K, Aoki JY, et al. Identification of sex-linked SNPs and sex-determining regions in the yellowtail genome. Mar Biotechnol. 2015;17:502–10.
Keenan CP, Davie PJ. Mann DL. A revision of the genus Scylla de Haan, 1833 (Crustacea: Decapoda: Brachyura: Portunidae). Raffles Bull Zool. 1998;46:217–45.
Sarower MG, Shahriar SIM, Nakamura H, Rouf MA, Okada S. Taxonomic confirmation of mud crab species (genus Scylla) in Bangladesh by nuclear and mitochondrial DNA markers. Mitochondrial DNA A. 2016;28:935–40.
Keenan CP. The fourth species of Scylla. In: Mud crab aquaculture and biology, ACIAR proceedings; 1999. p. 48–58.
Ma H, Ma C, Ma L. Molecular identification of genus Scylla (Decapoda: Portunidae) based on DNA barcoding and polymerase chain reaction. Biochem Syst Eco. 2012;41:41–7.
Ikhwanuddin M, Azmie G, Juariah HM, Zakaria MZ, Ambak MA. Biological information and population features of mud crab, genus Scylla from mangrove areas of Sarawak, Malaysia. Fish Res. 2011;108:299–306.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376.
Etter PD, Bassham S, Hohenlohe PA, Johnson EA. Cresko WA. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol Biol. 2012;772:157–78.
Houston RD, Davey JW, Bishop SC, Lowe NR, Mota-Velasco JC, Hamilton A, et al. Characterisation of QTL-linked and genome-wide restriction site-associated DNA (RAD) markers in farmed Atlantic salmon. BMC Genomics. 2012;13:244.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Acknowledgements
We would like to thank Hanafiah Fazhan, Qingyang Wu, Yin Zhang, Shaobin Fang, Zhuofang Xie and Mengyun Guan for sample collection and laboratory technical assistance.
Funding
This study was supported by the National Key Research & Development Program of China (No. 2018YFD0900201), the National Natural Science Foundation of China (No. 31772837), the “Sail Plan” Program for the Introduction of Outstanding Talents of Guangdong Province, China, the Program of Ocean and Fishery Department of Guangdong Province (Yueyu2018–11), the Science and Technology Project of Shantou City (2016–44), the National Program for Support of Top-Notch Young Professionals, the STU Scientific Research Foundation for Talents (No. NTF17006), and the Programme for Innovation and Enhancement of School of Department of Education of Guangdong Province (No. 2017KCXTD014). The funding bodies did not play any roles in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Sequencing data were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (accession number SRP135178) and GenBank (accession number MH133208).
Author information
Authors and Affiliations
Contributions
H.M. conceived and designed the research. H.M., X.S. and G.M. performed the research. H.M. and X.S. analyzed the data. H.M., X.S., K.W., G.A., J.C.B. and E.T.Q. contributed reagents and materials. X.S. wrote the manuscript. H.M., K.W., F.L. and J.J.A. revised the manuscript. X.L., M.I., Y.Z., S.L., H.Z. and W.L. provided substantial comments toward improving the content of the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The sampling location is not privately owned or protected, and no specific permission is required. The collection of cultured crabs was permitted by the animal owners who agreed to be involved in the project through their respective associations with Marine Biology Institute of Shantou University. All animal experimental procedures used in this study were approved by the committee for Animal Experiments of the Marine Biology Institute of Shantou University. The methods applied in this study were carried out in strict accordance with the Laboratory Animal Management Principles of China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
The cluster analysis and RAD assembly of sample “F2A”. (DOCX 14 kb)
Additional file 2:
The RAD-seq alignments between twenty samples and sample “F2A”. (DOCX 16 kb)
Additional file 3:
Agarose gel separation of PCR amplification products with female-specific and control primers in 24 females and 24 males from a full-sib family cultured in a pond of Raoping, China. Female-specific band (320 bp): PCR products amplified with SPFS primers; Control band (282 bp): PCR products amplified with SPC primers; M: marker; A: the results for 12 females and 12 males. B: the results for another 12 females and 12 males. (DOCX 680 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shi, X., Waiho, K., Li, X. et al. Female-specific SNP markers provide insights into a WZ/ZZ sex determination system for mud crabs Scylla paramamosain, S. tranquebarica and S. serrata with a rapid method for genetic sex identification. BMC Genomics 19, 981 (2018). https://doi.org/10.1186/s12864-018-5380-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5380-8