
Abstract
Background
Pekin duck products have become popular in Asia over recent decades and account for an increasing market share. However, the genetic mechanisms affecting carcass growth in Pekin ducks remain unknown. This study aimed to identify quantitative trait loci affecting body size and carcass yields in Pekin ducks.
Results
We measured 18 carcass traits in 639 Pekin ducks and performed genotyping using genotyping-by-sequencing (GBS). Loci-based association analysis detected 37 significant loci for the 17 traits. Thirty-seven identified candidate genes were involved in many biological processes. One single nucleotide polymorphism (SNP) (Chr1_140105435 A > T) located in the intron of the ATPase phospholipid transporting 11A gene (ATP11A) attained genome-wide significance associated with five weight traits. Eight SNPs were significantly associated with three body size traits, including the candidate gene plexin domain containing 2 (PLXDC2) associated with breast width and tensin 3 (TNS3) associated with fossil bone length. Only two SNPs were significantly associated with foot weight and four SNPs were significantly associated with heart weight. In the gene-based analysis, three genes (LOC101791418, TUBGCP3 (encoding tubulin gamma complex-associated protein 3), and ATP11A) were associated with four traits (42-day body weight, eviscerated weight, half-eviscerated weight, and leg muscle weight percentage). However, no loci were significantly associated with leg muscle weight in this study.
Conclusions
The novel results of this study improve our understanding of the genetic mechanisms regulating body growth in ducks and thus provide a genetic basis for breeding programs aimed at maximizing the economic potential of Pekin ducks.
Similar content being viewed by others

Background
China is the world’s largest producer and consumer of domestic ducks, with Chinese duck production accounting for 75% of duck production worldwide (FAO). Furthermore, duck farming has been growing rapidly in China, and in addition to breast meat and eggs, secondary products, such as duck neck and wing are also very popular. However, these secondary products including the head, neck, feet, and wings are more expensive than breast muscle, and it is therefore necessary to pay more attention to the traits affecting these secondary products.
Previous breeding programs have not focused on the composition of these other body parts in ducks. However, molecular-based breeding programs now have been applied in animal breeding, and numerous quantitative trait loci (QTL) affecting carcass traits have been identified in various animals. Several biological pathways related to carcass traits in cattle have been identified, including peroxisome proliferator-activated receptor signaling, while some significant associations were detected in close proximity to genes with known roles in animal growth, such as glucagon and leptin [1,2,3]. In pigs, the number of copies of the vertebrae development associated gene (VRTN) was related to body length [4], while SSC1 and SSC8 include several loci related to carcass traits in pigs. Three genes, TBC1D1 (encoding TBC1 domain family member 1), BAAT (bile acid-CoA: amino acid N-acyltransferase), and PHLPP1 (PH domain and Leucine-rich repeat protein phosphatase 1) were highlighted as functionally plausible candidate genes for pig growth and fatness traits [5, 6]. In addition to pigs and cattle, similar studies have been conducted in poultry, and numerous candidate genes associated with carcass traits have been found in chickens, including TBC1D1, LCORL (ligand-dependent nuclear receptor corepressor-like), LAP3 (leucine aminopeptidase 3), LDB2 (LIM-domain-binding 2), and TAPT1 (transmembrane anterior posterior transformation 1) [7,8,9,10]. However, compared with other domestic animals, study about the genetic basis of growth traits in ducks is lacking. Previous studies suggested that the fat mass and obesity-associated gene (FTO) [11] and mutations in intron 2 of the growth hormone gene (GH) [12] influenced duck carcass and meat quality traits, while Zhang et al. [13] also showed that the perilin gene (PLIN) affected duck carcass and fat traits.
The current chicken QTL database [14], includes more than 1500 QTLs related to body weight traits, but QTLs for traits related to the composition of subordinate body parts, such as neck length, fossil bone length, and foot weight, are still rare. Furthermore, few studies have been conducted in ducks, and no animal QTL database currently includes duck-related QTL. This lack of information needs to be addressed to support the development of duck breeding projects.
In the present study, we conducted a genome-wide association study (GWAS) in 639 Pekin ducks using genotyping-by-sequencing (GBS) [15]. We measured or derived a total of 18 body size and carcass yields traits, and aimed to identify potential loci and candidate genes affecting these traits. To our knowledge, this is the first large-scale GWAS investigation of duck carcass traits. These QTL information will not only facilitate the study of molecular genetic mechanisms, but may also improve the accuracy of genetic selection for carcass traits in ducks.
Results
Descriptive statistics
Mean values (and standard deviations) for body size and carcass yields traits are shown in Table 1 and the phenotypic correlations are shown in Table 2. The highest phenotypic correlation (0.98) was between DW and EW, and the lowest correlation (− 0.24) was between DP and LMWP. The phenotypic correlations among the four body weight traits (DW, EW, HEW, and BW42) were all > 0.90, while the correlations between leg muscle percentage and the other 17 traits were all < 0.2, and were mostly negative.
Genetic parameters
The genetic parameters of all traits are shown in Table 2. The estimated heritability of these 18 traits ranged from 0.30–0.91. LMWP showed the highest heritability, suggesting a considerable genetic contribution to carcass yields and body size. There was also a high genetic correlation between FBL and body weight traits, with correlation coefficients > 0.75, and a high genetic correlation between body weight and internal organ weight, with a correlation coefficient > 0.90.
Loci-based analysis
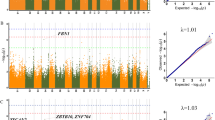
A total of 37 significant QTLs (P < 3.48E− 05) across 14 chromosomes were identified by loci-based analysis (Table 3; Additional file 1: Figure S1). The estimated genomic inflation factor λ ranged from 1.01–1.06, suggesting no population stratification in the studied population (Additional file 1: Figure S2). One result indicated a genome-wide significant QTL (SNP Chr1_140105435 A > T) (Fig. 1).

Manhattan plots showing associations of all SNPs with five traits and quantile-quantile (Q-Q) plots of the GLM (black dots) for five traits (a. DW, b. EW, c. HEW, d. BW42, e. WW). One SNP reached genome-wide significance for the five above traits. In Manhattan plots, SNPs are plotted on the x-axis according to their position on each chromosome, against association with these traits on the y-axis (shown as −log10P-value). Blue solid line indicates suggestive significance association (P = 3.48E− 05), and red solid line shows genome-wide significance with a P-value threshold of 1.74E− 06. In Q-Q plots, expected P-values under the null hypothesis are plotted on the x-axis and observed P-values on the y-axis. The estimated genomic inflation factor λ ranged from 1.03–1.06
Eight non-overlapping QTLs were obtained for body size traits. The most significant of these (Chr2_24791499_A > T; located in the intron between exons 8 and 9 of LOC101799835) was related to BrW (P = 7.67E− 06) and accounted for 10.8% of the genetic variance. A SNP on chromosome 1 (Chr1 130,709,943 A > T; located in the intron between exons 8 and 9 of the Xg blood group gene, XG) was the only locus significantly associated with NL. Three significant QTLs in separate regions of Chr2 were associated with FBL, with the most significant (SNP chr2_52398515_C > T; P = 1.13E− 05) accounting for 6.2% of the genetic variance.
We identified 29 non-overlapping QTLs related to carcass traits. The traits DW, EW, HEW, BW42, and WW shared one genome-wide significant QTL (SNP Chr1_140105435 A > T) (Fig. 2), located in the intron between the last two exons of ATP11A. This QTL was also significantly (P = 5.27E− 08) associated with HEW, accounting for 5.4% of the genetic variance. There were two significant QTL for FW (Chr1_148339868 A > G, P = 7.73E-06; Chr4_59568289 A > G, P = 1.62E-05), one in the region of the ATP-binding cassette subfamily C member 4 gene (ABCC4) and the other near LDB2. We identified two significant QTLs (Chr3_11176648 C > T and Chr10_1656050 A > G) for BMW, located near the transmembrane protein 17 gene (TMEM17) and LOC101804888, respectively. Three significant QTLs for BMWP were identified on Chr1, of which the most significant locus LOC101803092 accounted for 8.1% of the genetic variance for this trait. No QTL was identified for LMW, and the SNP Chr20_2198171 C > G, located near the autism susceptibility candidate 2 gene (AUTS2), was the only potentially significant QTL for LMWP.

Region of significant loci for DW (a), EW (b), HEW (c), BW42 (d), and WW (e). Blue curve represents the minor allele frequency; dot color represents the linkage coefficient between the most significant locus and other loci (red highest)
Gene-set analysis
We further examined the significance of the candidate genes within the QTL using gene set analysis (Table 4). Among the 18 traits, three candidate genes were found to be significant for the four traits BW42, EW, HEW, and LMWP: LOC101791418, tubulin gamma complex-associated protein 3 gene (TUBGCP3), and ATP11A (P < 1E− 05). The start and end positions of the three genes were concentrated within the genomic region between 139 and 140 Mb on Chr1. The highest significance was seen for ATP11A with HEW (P = 3.83E− 07). These results were consistent with the results of the association analysis above.
Functional annotation of significant regions
A total of 36 candidate genes were detected by GWAS analysis. We performed QTL annotation of these 36 chicken ortholog genes using the chicken QTL database [14] (Additional file 1: Figure S3). Twenty genes were annotated with QTL information in chickens. The QTL information for seven genes was consistent with the carcass traits measured in this study. ATP11A, calcium voltage-gated channel subunit alpha1 C (CACNA1C), which were associated with 42-day weight-related QTL in chicken studies, were also related to DW, EW, and HEW in ducks in the current study, respectively. Genes TMEM17 and ATPase plasma membrane Ca2+ transporting 1 (ATP2B1), as candidate genes associated with BMW and BMWP, were associated with breast muscle weight/percentage QTL in chicken studies. We also annotated the candidate genes using the GO and KEGG databases (Additional file 2: Tables S1 and S2). A total of 25 genes were annotated. Clustering analysis showed that these genes are not significantly enriched in any specific function or biological pathway. The gap junction protein alpha 1 gene (GJA1) plays an important role in gap junctions, which are essential for many physiological events, including embryonic development, electrical coupling, metabolic transport, apoptosis, and tissue homeostasis. ATP2B1 is associated with the calcium signaling pathway and adrenergic signaling in cardiomyocytes. CACNA1C was found to be associated with seven pathways, including the calcium signaling pathway, gonadotropin-releasing hormone signaling pathway, and regulation of the actin cytoskeleton.
Discussion
Heritability and correlation coefficients
The results of the current study indicated that body size and carcass traits demonstrated moderate to high heritability in Pekin ducks (0.30–0.91). Xu [16] previously estimated that breast muscle weight (BMW) and breast muscle weight percentage (BMWP) had moderate heritabilities (0.23 and 0.16, respectively), while the heritability of body weight was high (0.48). Furthermore, the heritability of BW42 in broilers in Nunes’ study was 0.31 [17]. Compared with these results, we found similar heritabilities of most traits, other than BMW. In this study, we constructed a kinship matrix using SNPs and estimated the heritability of BMW as 0.63, which was obviously higher than Xu’s results. This apparent discrepancy may have been due to population differences or differences in the estimation methods. However, estimations derived from a constructed pedigree from genomic information are likely to be more accurate than those from traditional pedigree records as the genomic information reflect more real genetic relationship than traditional pedigree.
The largest phenotypic correlations were between DW and EW and between DW and HEW, while the genetic correlation between BW42 and DP was small (0.39). Nunes et al. [17] estimated the genetic parameters related to body weight, chemical carcass composition, and yield in a broiler-layer cross and found a genetic correlation between BW42 and DP of 0.33, which was in accord with the current results. We found highly positive genetic (0.74) and phenotypic correlations (0.63) between BW42 and BMW. Venturini et al. [18] also found genetic and phenotypic correlation coefficients of 0.86 and 0.84 between BW42 and BMW, respectively, which were consistent with our results. However, compared with Venturini et al.’s result (0.7), we found a smaller genetic correlation between BW42 and LMW (0.3), likely due to the use of a different species. Overall, our results indicated that the genetic parameters related to body weight traits in the Pekin duck population were consistent with previous studies.
Candidate genes related to body size, body weight and carcass traits
In this study, ATP11A was significant in both loci-based model GWAS and gene-set GWAS, and was associated with DW, EW, HEW, BW42, WW, and LMWP. Based on chicken QTL information, ATP11A was also associated with body weight and internal organ weight in chickens. Also, Segawa et al. [19] noted that the major flippases at the plasma membrane in most mammal cells are encoded by ATP11A and ATP11C. ATP11A encodes a complete membrane ATPase involved in the transport of phospholipids. Transporting creates membrane phospholipid asymmetry and initiates the biogenesis of transport vesicles. We hypothesize that this gene may be involved in metabolism in duck fat cells, and may be highly significantly related to five traits because body weight is a complex trait that involves the development of multiple organs. This gene may thus be involved in basic growth and development processes, especially in relation to fat deposition; however, more experiments are needed.
The measurement of carcass traits in poultry breeding is both expensive and difficult, and can only be conducted after death. In contrast, BrW is an important live body measurement that is closely related to body weight and breast muscle production [20]. The current results revealed very high genetic and phenotypic correlation coefficients between BrW and EW, HEW, and BW42, respectively. All of them share the same SNP (Chr2_19666206 A > G). It was one of the most significant SNPs for BrW, accounting for 9.8% of the observed genetic variance, with a MAF of 0.479. The candidate gene was PLXDC2, which coordinates the development and differentiation of nerve cells in various animals, including humans, mice, and chickens [21,22,23]. Our results demonstrated correlations between this site and BW42, DW, EW, HEW, and BrW, and chicken QTL annotation showed that the gene was associated with both body weight and leg muscle weight.
Like BrW, FBL can also be measured easily and is significantly correlated with weight traits, and can thus be used for trait selection indirectly. The results of the present study showed that all the genetic and phenotypic correlation coefficients between FBL and BW42, DW, EW, HEW, and WW, respectively, were high. We also found that the SNP Chr2_52398515_C > T (located in intron of TNS3), which was significantly associated with FBL, accounted for 6.2% of the genetic variance. The TNS gene family plays an important role in the development and formation of bones. Dedicator of cytokinesis 5 (Dock5) and TNS3 genes have been shown to have a synergistic effect on the maintenance of osteoclast activity to ensure the correct organization of podosomes [24]. QTL information indicated that the QTL related to body weight and breast muscle weight/percentage localized to TNS3. However, the trait of FBL was not included in the chicken QTL information and we were unable to find any previous reports of FBL in poultry. We therefore provide the first report of a QTL associated with FBL in ducks, as an important reference for future research.
Secondary duck products after slaughter represent a large market share in Asia, and a better understanding of the genetic bases of the relevant economic traits will help to increase yield. In our study, the candidate gene for FW was LDB2, which encodes a LIM-domain-binding family protein that binds to a variety of transcription factors and plays a crucial role in brain development and angiogenesis [25, 26]. Wang et al. [27] conducted a GWAS in 400 chickens from a conservation population of a local Chinese breed (Jinghai Yellow chickens). They identified five SNPs with genome-wide significance for FW, including one for which the candidate gene was LDB2, located on Chr4, as in the current study. Gu et al. [8] also found that the SNP within LDB2 had the strongest association with late growth (body weight from 7 to 12 weeks old and average daily weight gain from 6 to 12 weeks old).
The current study identified four suggestive sites for HW, distributed on four different chromosomes. The candidate gene for one of these, GJA1, encodes connexin43 and connexin45, which comprise part of the gap junction consisting of an array of intercellular channels that provides a pathway for the diffusion of low-molecular-weight substances between cells [28]. Proteins are thought to play a key role in the synchronous contraction of the heart and in embryonic development [29]. Recent studies have also demonstrated that GJA1 is an important functional gene in chicken growth and development, especially of chicken breast muscle [7, 30]. Other functions of this gene have yet to be explored; however, the high correlation between BMW and HW suggests that GJA1 might be an important gene in terms of increasing breast muscle yield.
We also identified genes such as that encoding Kruppel-like factor 5 (KLF5) in relation to DW, EW, HEW, and HW, and ATP2B1 for BMWP. KLFs are zinc-finger transcription factors that act as key regulators of cellular differentiation and growth in adipocytes [31]. KLF5 is involved in the biological process that regulates lipid storage, and thus affects body weight-related traits. However, further studies are needed to clarify the role of ATP2B1 in poultry.
The use of molecular markers, revealing polymorphism at the DNA level, has been playing an increasing part in animal genetics studies. If we obtained more QTLs for duck, it would be possible to integrate these SNPs information in the genomic selection scheme to improve the selection accuracy in ducks.
Conclusions
In this study, we conducted a GWAS of 18 carcass traits in Pekin ducks. We detected 37 QTLs distributed across 26 chromosomes, and identified 36 candidate genes related to body size and carcass traits. These findings further our understanding of poultry genetics and provide a genetic basis for breeding programs aimed at maximizing the economic potential of Pekin ducks.
Methods
Ducks and phenotypes
A total of 639 (males: 314; females: 325) 21-day-old Pekin ducks from the same flock were randomly selected at Beijing Golden Duck Co., Ltd. (Beijing, China). They were divided randomly into five batches (120, 114, 117, 134, and 154 ducks each). The interval between each batch was 5 days. All ducks were fed with the same diet and maintained under the same lighting condition, as described previously [32]. Ducks were weighed at 21 and 42 days old. After fasting for 6 h, all the ducks were slaughtered at d 42 according to standard commercial procedures. Euthanasia was performed by cervical dislocation. A total of 18 traits were measured according to the performance terms and measurement of poultry guidelines issued by the Ministry of Agriculture of China (NY/T 823–2004), including neck length (NL), fossil bone length (FBL), breast width (BrW), dressed weight (DW), dressed percentage (DP), eviscerated weight (EW), eviscerated weight percentage (EWP), half-eviscerated weight (HEW), percentage of half-eviscerated yield (HEWP), 42-day body weight (BW42), heart weight (HW), liver weight (LW), foot weight (FW), wing weight (WW), breast muscle weight (BMW), breast muscle weight percentage (BMWP), leg muscle weight (LMW), and leg muscle weight percentage (LMWP) [32]. The following traits were measured using a caliper and weighing scale and calculated as follows:
Neck length: the distance between the first cervical vertebra and the end of neck;
Fossil bone length: The distance between the anterior and the posterior border of the breast-bone crest;
Breast width: the distance vertically between the backbone and the beginning of the breast-bone crest;
Dressed weight: Weight after bloodletting and removal of feathers, foot cuticles, toes, and clam shells;
Dressed percentage (%) = Dressed weight/slaughter weight × 100;
Half-eviscerated weight: Weight of carcass after removal of trachea, esophagus, crop, intestines, spleen, pancreas, gallbladder, reproductive organs, stomach contents, and keratinocytes;
Percentage of half-eviscerated yield (%) = Half-eviscerated weight/slaughter weight × 100;
Eviscerated weight: Half-eviscerated weight minus the weight of the heart, liver, stomach, lungs, and abdominal fat;
Percentage of eviscerated yield (%) = Eviscerated weight/slaughter weight × 100;
Slaughter weight/42-day body weight: Weight after fasting for 6 h;
Percentage of breast muscle (%) = Breast muscle weight/eviscerated weight × 100;
Leg muscle: Total leg muscle weight after removing leg bones, skin, and subcutaneous fat;
Leg muscle weight percentage of (%) = Leg muscle weight/eviscerated weight × 100.
GBS
Fresh blood was collected from the ducks before slaughtering and genomic DNA was extracted from the blood using a phenol–chloroform-based method. Genotyping was performed using GBS, as described previously [15]. A total of 544 million clean reads (63.25 GB) were generated, of which 96.12% (523 million reads) were mapped to the duck genome, with an average mapping rate of 96.25%. The data were deposited in the NCBI sequence read archive (SRP068685). GBS procedures are available on Zhu Feng’s article [15]. Restriction enzyme (MSe1) for the PCR-RFLP assay was selected using information from REBASE [33]. A set of variable barcode adapters that recognize Mse1-compatible sequences were ligated to the digested DNA fragments. The restriction fragments were enriched by PCR amplification with adapter-specifc primers [34]. Te data of 2 × 125 bp pair-end reads were generated by the Illumina HiSeq2500.
Single nucleotide polymorphism (SNP) identification
Clean data were mapped to a reference genome using BWA (version 1.73) [35]. The reference genome was a chromosome-assembly version from BGI duck 1.0 reference (GCA_000355885.1) based on the RH map [36] with ALLMAP [37]. VCFtools [38] and PLINK (1.90) [39] were used for quality control of the data. SNP detection was performed using the GATK HaploCaller (3.7) [40]. All parameters were kept at default settings, except for -stand_call_conf 30. The data were imputed using Beagle (4.1) [41], using R2 > 0.3 for low-quality filtering. A total of 62,067 SNPs met one or more of the following conditions: minor allele frequency (MAF) of > 1%, sample call rate ≥ 95%, and the Hardy–Weinberg equilibrium test (P > 10− 6). All phenotype measurements were normalized using the rank transformation method, and the effects of batch and gender on the phenotype were examined using the variance test for subsequent covariates of the mixed model. Population substructure analysis was performed using EIGENSOFT (2.04), and the top ten PCA [42] components were used as covariates for further analysis. GCTA-LDMC [43] was used to estimate the genetic parameters. SNPs used in this study listed in Additional file 2: Table S3.
Statistical analysis
Loci-based analysis was performed using the generalized linear mixed model implemented in GEMMA [44], where the kinship matrix was calculated using the center method. The mixed model was mainly based on the additive effect of sites:
where y is the vector phenotypes of per duck; μ is the overall mean; X is the covariance matrix (mainly containing the gender effect, batch effect, and first ten PCA principal components obtained from the analysis of population substructure); b is the estimator vector of fixed effects; u ~ (0, G\( {\upsigma}_{\mathrm{u}}^2 \)) is the additive polygenic effect, with G the genomic kinship matrix, and \( {\upsigma}_{\mathrm{u}}^2 \) is the additive effect variance; S is the design matrix containing the corresponding SNP sites; α is the substitution effect size corresponding to each site; and e~N (0, I\( {\upsigma}_{\mathrm{e}}^2 \)) is the vector of random residual effects, with I the identity matrix and \( {\upsigma}_{\mathrm{e}}^2 \) the residual variance.
Gene-set analysis used the MAGMA Top model [45]. Gene analysis in MAGMA is based on a multiple linear principal components regression model, using an F-test to compute the gene P-value. The association level for each gene was the weighted sum of the associated statistics for the SNP sites in the region. Using this model can improve the statistical power for the identification of a candidate gene. To perform the gene-set analysis, for each gene g the gene p-value pg computed with the gene analysis is converted to a Z-value zg = Φ− 1(1–pg), whereΦ− 1 is the probit function. This yields a roughly normally distributed variable Z with elements z g that reflects the strength of the association each gene has with the phenotype, with higher values corresponding to stronger associations.
The genomic inflation factor (λ) was calculated using R package qqman [46]. Multiple test thresholds were calculated using the simpleM method [47]. A total of 28,707 valid inspections were obtained. The genome-wide significance level was 1.741735E− 06 (0.05/28,707), and the suggestive significance level was 3.483471E− 05 (1/28,707). LD statistic were performed using PLINK (−-r2) to calculates inter-variant allele count correlations [39]. Multiple consecutive significant sites were defined as separate QTL regions, and site-effect values were calculated using the equation 2pqβ2/σ2, with allele frequencies p and q, β is corresponding effect size of SNP identified in association study, σ2 is phenotypic variance.
Functional annotation
The gene position information was annotated using BEDTools [48]. The functions of the genes were annotated using the KEGG and GO databases, and enrichment analysis was performed using the R package GOSeq [49]. Due to the absence of a duck QTL database, we used the orthologues of chicken genes and their QTL information for candidate genes in the Animal QTL Database [14].
Abbreviations
- BMW:
-
Breast muscle weight
- BMWP:
-
Breast muscle weight percentage
- BrW:
-
Breast width
- BW42:
-
42-day body weight
- DP:
-
Dressed percentage
- DW:
-
Dressed weight
- EW:
-
Eviscerated weight
- EWP:
-
Eviscerated weight percentage
- FAO:
-
Food and Agriculture Organization
- FBL:
-
Fossil bone length
- FW:
-
Foot weight
- GBS:
-
Genotyping-by-Sequencing
- GO:
-
Gene ontology
- GWAS:
-
Genome-wide association study
- HEW:
-
Half-eviscerated weight
- HEWP:
-
Percentage of half-eviscerated yield
- HW:
-
Heart weight
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LD:
-
Linkage disequilibrium
- LMW:
-
Leg muscle weight
- LMWP:
-
Leg muscle weight percentage
- LW:
-
Liver weight
- NL:
-
Neck length
- PCA:
-
Principal component analysis
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
- WW:
-
Wing weight
References
Sanchez M, Govignon-Gion A, Croiseau P, Fritz S, Hozé C, Miranda G, Martin P, Barbat-Leterrier A, Letaïef R, Rocha D, et al. Within-breed and multi-breed GWAS on imputed whole-genome sequence variants reveal candidate mutations affecting milk protein composition in dairy cattle. Genet Sel Evol. 2017;49(1):68.
Inoue K, Honda T, Oyama K. Genetic relationships between internal diseases diagnosed at slaughter and carcass traits in Japanese black cattle. J Anim Sci. 2015;93:2714–21.
Doran AG, Berry DP, Creevey CJ. Whole genome association study identifies regions of the bovine genome and biological pathways involved in carcass trait performance in Holstein-Friesian cattle. BMC Genomics. 2014;15:837.
Nakano H, Sato S, Uemoto Y, Kikuchi T, Shibata T, Kadowaki H, Kobayashi E, Suzuki K. Effect of VRTN gene polymorphisms on Duroc pig production and carcass traits, and their genetic relationships. Anim Sci J. 2015;86:125–31.
Guo Y, Qiu H, Xiao S, Wu Z, Yang M, Yang J, Ren J, Huang L. A genome-wide association study identifies genomic loci associated with backfat thickness, carcass weight, and body weight in two commercial pig populations. J Appl Genet. 2017;58:499–508.
Sanchez M, Tribout T, Iannuccelli N, Bouffaud M, Servin B, Tenghe A, Dehais P, Muller N, Del Schneider M, Mercat M, et al. A genome-wide association study of production traits in a commercial population of large white pigs: evidence of haplotypes affecting meat quality. Genet Sel Evol. 2014;46:12.
Shahjahan M, Liu RR, Zhao GP, Zhang JJ, Zheng MQ, Li QH, Wen J. Polymorphisms in GJA1 and their association with growth traits in chicken. Genet Mol Res. 2015;14:18839–50.
Gu X, Feng C, Ma L, Song C, Wang Y, Da Y, Li H, Chen K, Ye S, Ge C, et al. Genome-wide association study of body weight in chicken F2 resource population. PLoS One. 2011;6:e21872.
Wang Y, Xu HY, Gilbert ER, Peng X, Zhao XL, Liu YP, Zhu Q. Detection of SNPs in the TBC1D1 gene and their association with carcass traits in chicken. Gene. 2014;547:288–94.
Venturini GC, Stafuzza NB, Cardoso DF, Baldi F, Ledur MC, Peixoto JO, El FL, Munari DP. Association between ACTA1 candidate gene and performance, organs and carcass traits in broilers. Poult Sci. 2015;94:2863–9.
Gan W, Song Q, Zhang NN, Xiong XP, Wang DM, Li L. Association between FTO polymorphism in exon 3 with carcass and meat quality traits in crossbred ducks. Genet Mol Res. 2015;14:6699–714.
Wu Y, Pan AL, Pi JS, Pu YJ, Du JP, Liang ZH, Shen J. One novel SNP of growth hormone gene and its associations with growth and carcass traits in ducks. Mol Biol Rep. 2012;39:8027–33.
Zhang HL, Fan HJ, Liu XL, Wu Y, Hou SS. Molecular cloning of the perilipin gene and its association with carcass and fat traits in Chinese ducks. Genet Mol Res. 2013;12:1582–92.
Hu ZL, Park CA, Reecy JM. Developmental progress and current status of the animal QTLdb. Nucleic Acids Res. 2016;44:D827–33.
Zhu F, Cui QQ, Hou ZC. SNP discovery and genotyping using genotyping-by-sequencing in Pekin ducks. Sci Rep. 2016;6:36223.
Xu Y, Hu J, Zhang Y, Guo Z, Huang W, Xie M, Liu H, Hou S, Zhou Z, Lei C, Liu X. Selection response and estimation of the genetic parameters for multidimensional measured breast meat yield related traits in a long-term breeding Pekin duck line. Asian-Australas J Anim Sci. 2018;31(10):1575–80.
Nunes BN, Ramos SB, Savegnago RP, Ledur MC, Nones K, Klein CH, Munari DP. Genetic parameters for body weight, carcass chemical composition and yield in a broiler-layer cross developed for QTL mapping. Genet Mol Biol. 2011;34:429–34.
Venturini GC, Cruz VA, Rosa JO, Baldi F, El FL, Ledur MC, Peixoto JO, Munari DP. Genetic and phenotypic parameters of carcass and organ traits of broiler chickens. Genet Mol Res. 2014;13:10294–300.
Segawa K, Kurata S, Nagata S. Human type IV P-type ATPases that work as plasma membrane phospholipid Flippases and their regulation by caspase and calcium. J Biol Chem. 2016;291:762–72.
Malomane DK, Norris D, Banga CB, Ngambi JW. Use of factor scores for predicting body weight from linear body measurements in three south African indigenous chicken breeds. Trop Anim Health Prod. 2014;46:331–5.
Cheng G, Zhong M, Kawaguchi R, Kassai M, Al-Ubaidi M, Deng J, Ter-Stepanian M, Sun H. Identification of PLXDC1 and PLXDC2 as the transmembrane receptors for the multifunctional factor PEDF. Elife. 2014;3:e5401.
Miller-Delaney SF, Lieberam I, Murphy P, Mitchell KJ. Plxdc2 is a mitogen for neural progenitors. PLoS One. 2011;6:e14565.
Miller SF, Summerhurst K, Runker AE, Kerjan G, Friedel RH, Chedotal A, Murphy P, Mitchell KJ. Expression of Plxdc2/TEM7R in the developing nervous system of the mouse. Gene Expr Patterns. 2007;7:635–44.
Touaitahuata H, Morel A, Urbach S, Mateos-Langerak J, de Rossi S, Blangy A. Tensin 3 is a new partner of Dock5 that controls osteoclast podosome organization and activity. J Cell Sci. 2016;129:3449–61.
Ostendorff HP, Tursun B, Cornils K, Schluter A, Drung A, Gungor C, Bach I. Dynamic expression of LIM cofactors in the developing mouse neural tube. Dev Dyn. 2006;235:786–91.
Javerzat S, Franco M, Herbert J, Platonova N, Peille AL, Pantesco V, De Vos J, Assou S, Bicknell R, Bikfalvi A, Hagedorn M. Correlating global gene regulation to angiogenesis in the developing chick extra-embryonic vascular system. PLoS One. 2009;4:e7856.
Wang W, Zhang T, Wang J, Zhang G, Wang Y, Zhang Y, Zhang J, Li G, Xue Q, Han K, et al. Genome-wide association study of 8 carcass traits in Jinghai yellow chickens using specific-locus amplified fragment sequencing technology. Poult Sci. 2016;95:500–6.
Ishihara Y, Kamioka H, Honjo T, Ueda H, Takano-Yamamoto T, Yamashiro T: Hormonal, pH, and Calcium regulation of connexin 43-mediated dye transfer in osteocytes in chick calvaria. J Bone Miner Res 2008, 23:350–360.
Seki A, Ishikawa T, Daumy X, Mishima H, Barc J, Sasaki R, Nishii K, Saito K, Urano M, Ohno S, et al. Progressive atrial conduction defects associated with bone malformation caused by a Connexin-45 mutation. J Am Coll Cardiol. 2017;70:358–70.
Liu R, Sun Y, Zhao G, Wang F, Wu D, Zheng M, Chen J, Zhang L, Hu Y, Wen J. Genome-wide association study identifies loci and candidate genes for body composition and meat quality traits in Beijing-you chickens. PLoS One. 2013;8:e61172.
Matsubara Y, Aoki M, Endo T, Sato K. Characterization of the expression profiles of adipogenesis-related factors, ZNF423, KLFs and FGF10, during preadipocyte differentiation and abdominal adipose tissue development in chickens. Comp Biochem Physiol B Biochem Mol Biol. 2013;165:189–95.
Lin FB, Zhu F, Hao JP, Yang FX, Hou ZC. In vivo prediction of the carcass fatness using live body measurements in Pekin ducks. Poult Sci. 2018;97:2365–71.
Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43(Database issue):D298–9. https://doi.org/10.1093/nar/gku1046.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. https://doi.org/10.1186/1471-2105-13-134.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Rao M, Morisson M, Faraut T, Bardes S, Feve K, Labarthe E, Fillon V, Huang Y, Li N, Vignal A. A duck RH panel and its potential for assisting NGS genome assembly. BMC Genomics. 2012;13:513.
Tang H, Zhang X, Miao C, Zhang J, Ming R, Schnable JC, Schnable PS, Lyons E, Lu J. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 2015;16:3.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Browning BL, Browning SR. Genotype imputation with millions of reference samples. Am J Hum Genet. 2016;98:116–26.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9.
Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH, Robinson MR, Perry JR, Nolte IM, van Vliet-Ostaptchouk JV, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet. 2015;47:1114–20.
Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11:407–9.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
Dessau RB, Pipper CB. “R”--project for statistical computing. Ugeskr Laeger. 2008;170:328–30.
Gao X, Becker LC, Becker DM, Starmer JD, Province MA. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet Epidemiol. 2010;34:100–5.
Quinlan AR. BEDTools: the Swiss-Army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47:11–2.
McCabe M, Waters S, Morris D, Kenny D, Lynn D, Creevey C. RNA-seq analysis of differential gene expression in liver from lactating dairy cows divergent in negative energy balance. BMC Genomics. 2012;13:193.
Acknowledgements
We acknowledged Mr. Guo-Cheng Li, Mr.Guo-Cheng Li helped us to collect samples.
Funding
The work was supported by the National Scientific Supporting Projects of China (2015BAD03B06), the National Waterfowl-Industry Technology Research System (CARS-42-09), Primary Research & Development Plan of Jiangsu Province (BE2017349), and the Leading Technology of Modern Agricultural Science and Technology City (Z181100002418008). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this article and the additional files. The raw duck GBS data was deposited in Sequence Read Archive (SRA) database under the accession numbers PJNA506902 (https://www.ncbi.nlm.nih.gov/sra/PRJNA506902).
Author information
Authors and Affiliations
Contributions
ZCH conceived and designed the experimental plan. MTD, FZ, YZY, FXY, SRC and JPH participated in collecting tissues, sampling maintaining and recording the phenotypic data. FZ, MTD, FXY and YZY analyzed the data and interpreted the results. MTD, FZ, SRC, JPH and ZCH drafted and revised this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal experiments were approved and performed according to the Animal Care and Use Committee of China Agricultural University (permit number: SYXK 2007–0023).
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1 Manhattan plots showing associations of all SNPs with all traits (a. NL, b. FBL, c. BrW, d. DW, e. DP, f. EW, g. EWP, h. HEW, i. HEWP, j. BW42, k. HW, l. LW, m. FW, n. WW, o. BMW, p. BMWP, q. LMW, r. LMWP). SNPs are plotted on the x-axis according to their position on each chromosome, against association with these traits on the y-axis (shown as −log10p-value). Solid blue line indicates suggestive significance association (P = 3.48E-05) and red solid line shows genome-wide significance with a P-value threshold of 1.74E-06. Figure S2 Quantile-quantile (Q-Q) plots of the GLM (black dots) for carcass traits (a. NL, b. FBL, c. BrW, d. DW, e. DP, f. EW, g. EWP, h. HEW, i. HEWP, j. BW42, k. HW, l. LW, m. FW, n. WW, o. BMW, p. BMWP, q. LMW, r. LMWP). Expected P-values under the null hypothesis are plotted on the x-axis and observed P-values on the y-axis. Figure S3 QTL information for ortholog chicken candidate genes. There is no duck QTL database and we therefore used chicken QTL information for the candidate genes in the Animal QTL Database. This figure was based on the shared QTL between our study in ducks and the chicken QTL database. The color intensity represents the degree of enrichment of similar QTL. Liver weight in ducks and carcass proportion in chickens shared the most QTL (9). Foot weight, wing weight, and breast muscle weight in ducks shared no QTL with body size in chickens. (DOCX 855 kb)
Additional file 2:
Table S1 GO analysis of the candidate genes. Table S2 Biological pathways of the candidate genes. Table S3 All SNP information. (XLSX 1050 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Deng, MT., Zhu, F., Yang, YZ. et al. Genome-wide association study reveals novel loci associated with body size and carcass yields in Pekin ducks. BMC Genomics 20, 1 (2019). https://doi.org/10.1186/s12864-018-5379-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5379-1