
Abstract
Background
Cosmoscartini (Hemiptera: Cercopoidea: Cercopidae) is a large and brightly colored Old World tropical tribe, currently containing over 310 phytophagous species (including some economically important pests of eucalyptus in China) in approximately 17 genera. However, very limited information of Cosmoscartini is available except for some scattered taxonomic studies. Even less is known about its phylogenetic relationship, especially among closely related genera or species. In this study, the detailed comparative genomic and phylogenetic analyses were performed on nine newly sequenced mitochondrial genomes (mitogenomes) of Cosmoscartini, with the purpose of exploring the taxonomic status of the previously defined genus Okiscarta and some closely related species within the genus Cosmoscarta.
Results
Mitogenomes of Cosmoscartini display similar genomic characters in terms of gene arrangement, nucleotide composition, codon usage and overlapping regions. However, there are also many differences in intergenic spacers, mismatches of tRNAs, and the control region. Additionally, the secondary structures of rRNAs within Cercopidae are inferred for the first time.
Based on comparative genomic (especially for the substitution pattern of tRNA secondary structure) and phylogenetic analyses, the representative species of Okiscarta uchidae possesses similar structures with other Cosmoscarta species and is placed consistently in Cosmoscarta. Although Cosmoscarta bimacula is difficult to be distinguished from Cosmoscarta bispecularis by traditional morphological methods, evidence from mitogenomes highly support the relationships of (C. bimacula + Cosmoscarta rubroscutellata) + (C. bispecularis + Cosmoscarta sp.).
Conclusions
This study presents mitogenomes of nine Cosmoscartini species and represents the first detailed comparative genomic and phylogenetic analyses within Cercopidae. It is indicated that knowledge of mitogenomes can be effectively used to resolve phylogenetic relationships at low taxonomic levels. Sequencing more mitogenomes at various taxonomic levels will also improve our understanding of mitogenomic evolution and phylogeny in Cercopidae.
Similar content being viewed by others


Background
The mitochondrial genome (mitogenome) of most metazoan is circular and compact with relatively conserved gene organization, order and direction. It varies from 14 to 20 kb and contains 13 protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), 22 transfer RNA genes (tRNAs), and a large non-coding region (also referred to as the control region) [1,2,3]. Owing to some unique features like small size, high copy numbers, maternal inheritance, strict orthologous genes, low rate of recombination, and accelerated rate of nucleotide substitution [4, 5], the mitogenome has been extensively used in various study areas, including species identification, population genetics, phylogeny and evolution [6,7,8,9]. Genome-level characters, including nucleotide composition, structural genomic features, and gene rearrangement, have also been widely used for comparative and evolutionary genomics, and phylogenetic inference at different taxonomic levels [7, 10, 11]. TRNAs with different patterns in base composition, gene rearrangement, and secondary structure are proposed as useful ways to study the evolution of mitogenomes and can be used as powerful phylogenetic markers [12,13,14,15]. Furthermore, mitogenomes, especially in intergenic spaces, usually present higher mutation rates than nuclear genes and may provide valuable information for phylogenetic analyses among closely related taxa [10, 16, 17].
Cosmoscartini (Hemiptera: Cercopoidea: Cercopidae) is a large and brightly colored Old World tropical tribe, including more than 310 described species in approximately 17 genera [18,19,20]. Just as other spittlebugs, during the nymphal period, they present the habit of producing copious spittle masses to cover themselves inside through continuously sucking the liquid and nutrients contained in xylem tissue, which may cause serious economic damage to host plants [21, 22]. In terms of ecological function, spittle mass has traditionally been seen as an effective barrier against predation, parasitism, and desiccation [21, 23]. In addition, the spittle mass functions as a light attenuator, which could reduce potentially damaging solar radiation [24]. Furthermore, nymphs of many spittlebug species are known to aggregate in one spittle mass and inflict more serious economic damage [25]. Some species of Cosmoscarta are reported as economically important pests of eucalyptus in China [26]. Therefore, it is very important to accurately elucidate the taxonomic status and phylogenetic relationships of Cosmoscartini species. However, only very limited studies about Cosmoscartini are available except for some scattered taxonomic studies [27, 28] and the detailed biology of Cosmoscartini species is likewise scattered. Additionally, some taxa are difficult to identify to species due to a lack of sufficient diagnostic characters to illustrate distinctions [21]. Even less is understood about the phylogenetic relationship within Cosmoscartini, especially among closely related genera or species. There is a controversy related to whether or not Okiscarta should be raised to genus level [18,19,20]. Therefore, detailed molecular data is required for a comprehensive phylogenetic analysis of the tribe Cosmoscartini.
To date, only one complete mitogenome of Cosmoscartini has been sequenced (GenBank accession number KP064511), which is quite limited and restricts our understanding of the phylogeny of Cosmoscartini. In the present study, we sequenced mitogenomes of nine Cosmoscartini species, including one Ectemnonotum, one Okiscarta (this genus has only two species totally), and seven Cosmoscarta species, respectively. The aims of this study were to: 1) provide a detailed comparative analysis of these mitogenomes, including nucleotide composition, codon usage, secondary structures of RNAs, and novel features of the control region; 2) investigate the phylogenetic relationships among three genera of Cosmoscartini, especially for the taxonomic status of the previously defined genus Okiscarta; 3) explore the relationships among some closely related species, which were difficult to distinguish from traditional morphological methods.
Results
Genome structure
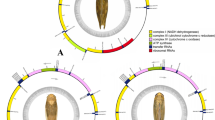
This study presented five complete and four nearly complete mitogenomes with the absence of the control region (Fig. 1). The total length of each complete mitogenome ranged from 15,024 bp in Cosmoscarta bimacula to 15,677 bp in Cosmoscarta rubroscutellata (Additional file 1: Table S1). The sequenced mitogenomes contained the entire set of 37 genes. The J-strand carried most of the genes (9 PCGs and 14 tRNAs), while the remaining genes (4 PCGs, 8 tRNAs and two rRNAs) were located on the N-strand (Fig. 1). Gene arrangement was identical within Cosmoscartini and was consistent with the putative ancestral type of insects [2, 7]. All the sequenced genomes were relatively compact with genes overlapping at 12–15 locations, in which the longest overlap (8 bp) was between trnW and trnC. These overlapping sequences were highly conserved in length among all sequenced Cosmoscartini mitogenomes. Two PCG pairs, including atp8-atp6 and nad4-nad4l, both overlapped 7 bp and shared the similar sequence (ATGNTAA), which had also been reported in many other insect mitogenomes [29, 30]. There were also 5–7 intergenic spacers (in addition to the control region), with the longest intergenic spacer located between trnS2 and nad1. In addition, 14–19 pairs of genes were directly adjacent with one another including the pairs of rrnL-trnV, trnV-rrnS, rrnS-CR, and CR-trnI.

Circular diagram of the nine Cosmoscartini mitogenomes. Different color is performed to show the nucleotide identity of BLAST hits, with the reference genome represented by C. bimacula. The rings are arranged in an order that the most similar genome is placed closest to the outer edge of the circle
To better visualize the sequence identity in Cosmoscartini mitogenomes, the comparable circular diagram was generated (Fig. 1). Pairwise comparisons between C. bimacula and other Cosmoscartini species revealed an overall similarity from 86% (C. bimacula vs. Ectemnonotum fruhstorferi) to 92% (C. bimacula vs. C. rubroscutellata). The rrnL, rrnS, and some tRNA genes (e.g., trnI, trnL1, and trnL2) exhibited much higher levels of sequence conservation. Conversely, control region was the most variable region. Within the PCGs, cytochrome oxidase genes were more conserved with cox1 and cox2 presented the highest conservation, whereas NADH dehydrogenase subunit genes were more variable with nad3 showed maximal variation.
Nucleotide composition and codon usage
The nucleotide composition was observed to be similar among the five complete Cosmoscartini species, with the overall A + T content ranging from 78.1% in Cosmoscarta heros to 79.1% in C. bimacula. These results confirmed an AT-bias, which were also higher than other Cercopidae species (73.8–77.4%) (Fig. 2a; Additional file 1: Table S1). The segment with highest A + T content was found in control region (80.5–84.8%), which was generally higher than other Cercopidae species (74.5–82.5%). The lowest A + T content within Cosmoscartini was found in PCGs (76.5–78.7%), which was also higher than other Cercopidae species (73.1–76.2%). The biased usage of A + T nucleotides was also reflected in codon frequencies. Relative synonymous codon usage (RSCU) of Cosmoscartini revealed that degenerate codons were biased to use more A/T than G/C in the third codon position (Additional file 2: Table S2). Some GC-rich codons were seldom utilized in the Cosmoscartini species, such as GCG and CGC which were absent in six and four of all nine species, respectively. Conversely, the four most prevalent codons in Cosmoscartini, including TTT (trnF), TTA (trnL2), ATT (trnI), and ATA (trnM), were all composed of A and/or T. AT-skews (from 0.119 to 0.169) and GC-skews (from − 0.227 to − 0.141) in Cosmoscartini mitogenomes were similar to patterns typically found in other Cercopidae species, i.e., positive AT-skew and negative GC-skew for the J-strand (Fig. 2b).

Base composition of various datasets among Cercopidae mitogenomes. (a) Hierarchical clustering of Cercopidae species (y-axis) based on their A + T content; (b) Clustering of species in terms of AT-skew and GC-skew
Protein-coding genes
Orthologs from the Cosmoscartini mitogenomes had similar start and stop codons (Additional file 3: Table S3). Most PCGs exhibited the typical start codon ATN, but cox1, nad2, and nad5 in all the species initiated with TTG. While most PCGs ended with the termination codon TAA or TAG, truncated codon T was also detected in cox2, nad4, and nad5 in the tribe. Truncated stop codons were common in insect mitogenomes and might be completed by post-transcriptional polyadenylation [31]. In order to assess the evolutionary patterns of PCGs, the values of Ka, Ks, and Ka/Ks were calculated (Fig. 3), with atp8 and cox1 presented the highest and lowest evolutionary rate, respectively. In addition, the Ka/Ks ratio for each PCG was far lower than 1.

Evolutionary rate of each PCG among Cosmoscartini species. Ks, synonymous nucleotide substitutions per synonymous site; Ka, nonsynonymous nucleotide substitutions per nonsynonymous site
Comparison of tRNA secondary structures
All 22 tRNAs typical of mitogenomes of bilateral animals were found in the nine Cosmoscartini mitogenomes (Fig. 4). Most tRNAs could be folded into the canonical cloverleaf structure except for trnS1, with its dihydrouracil (DHU) arm forming a simple loop, which was considered a typical feature in metazoan mitogenomes [32]. Moreover, we found that trnS1 in all nine Cosmoscartini mitogenomes had an unusual anticodon stem, with an unpaired nucleotide. Similar pattern was also found in the trnR acceptor stem. This was an unusual phenomenon, but had also been reported in the anticodon and acceptor stems of some other hemipterans [33, 34].

Secondary structures of tRNA families in Cosmoscartini mitogenomes. The nucleotide substitution pattern for each tRNA is modeled using as reference the structure predicted for C. bimacula. Watson-Crick base pairings, GU bonds, and mismatches are illustrated by dashes, solid dots, and hollowed dots, respectively
Except for the most conserved region of anticodon arm, the conservation of each stem was always higher than the corresponding loop. Conversely, the TΨC loop was the most variable structure, followed by the DHU loop and “extra” arm as the second and third most variable, respectively. According to the substitution patterns proposed by Negrisolo et al. [35], two patterns were summarized: (1) fully compensatory base changes (cbcs) (e.g., G-C vs. A-U); (2) hemi-cbcs (e.g., G-U vs. A-U). All the stem-base changes presented in Cosmoscartini tRNAs could be explained by these two patterns, except for several potential mismatches (e.g. A-A vs. C-A in the acceptor stem of trnW). However, the nucleotide substitutions on loops could not be modeled clearly because of a high level of variation. In some conserved tRNAs (e.g., trnI and trnD), nucleotide substitutions were generally restricted to TΨC and DHU loops and extra arms (Fig. 4), with changes on stems reduced to only 0–1 fully cbcs (e.g., G-C vs. A-T in the TΨC stem of trnI).
Comparison of rRNA secondary structures
The secondary structure of rrnL consisted of 44 helixes in five domains (I-II, IV-VI) of insect rrnL that do not have domain III [36]. The multiple alignment of Cosmoscartini rrnL extended over 1258 positions and contained 969 conserved (77.03%) and 289 variable sites (22.97%), respectively. Conserved nucleotides were distributed unevenly, with domains IV and V more conserved than domains I, II, and VI (Fig. 5).

Predicted secondary structure of the rrnL in the mitogenome of C. bimacula. Filled circle, nucleotide conserved in nine Cosmoscartini mitogenomes; hollowed circle, nucleotide not conserved. Roman numerals denote the conserved domain structure. Watson-Crick pairs are connected by dashes, whereas GT pairs are joined by dots
The rrnS contained three domains and 27 helixes, with a limited number of non-canonical pairings (e.g., G-A on helix H944). The multiple alignments of Cosmoscartini rrnS spanned 776 positions and contained 633 conserved (81.57%) and 143 variable (18.43%) positions, respectively. Nucleotide conservation among domains and helixes was distributed unevenly (Fig. 6). Compared to domain I and II, domain III was structurally more conserved within Cosmoscartini.

Predicted secondary structure of the rrnS in the mitogenome of C. bimacula. Filled circle, nucleotide conserved in nine Cosmoscartini mitogenomes; hollowed circle, nucleotide not conserved. Roman numerals denote the conserved domain structure. Watson-Crick pairs are connected by dashes, whereas GT pairs are joined by dots
Non-coding regions
Except for the control region, Cosmoscartini mitogenomes were highly economized in size with only 5–7 intergenic spacers, most of which were shorter than 5 bp. However, the longest intergenic spacer was found between trnS2 and nad1 (17–26 bp) in all Cosmoscartini mitogenomes (Additional file 4: Figure S1), sharing a conserved sequence of AAC(C/T)A(A/C)AAA(T/C)AATGAA.
Overall, Cosmoscartini control regions presented distinct sequence and structure features, such as size variation and different tandem repetitions (Additional file 5: Figure S2). The repeat units ranged from 15 to 355 bp, and the copy number ranged from 1.8 to 4.8. Most of the length variation of control regions among species was due to the variable lengths of tandem repeats, such as the control region (1135 bp in total) of Cosmoscarta sp., the majority of which was made up of a 721 bp tandem-repeat region, containing 2 large tandem duplications with the high similarity of 98%.
Phylogenetic analysis
The tree topologies obtained from all BI and ML analyses were identical (Fig. 7). The monophyly of Cosmoscartini was highly supported, with genus Ectemnonotum (represented by the species E. fruhstorferi) inferred as sister to other Cosmoscartini species (BI = 1.0 and ML = 100). However, Cosmoscarta was recovered as a non-monophyletic group in all analyses, with respect to the previously mentioned Okiscarta uchidae,

Phylogenetic tree inferred from mitogenomes of Cosmoscartini. Squares at the nodes are Bayesian posterior probabilities (PP) for 1, 3, 5, and 7, Bootstrap values (BS) for 2, 4, 6, and 8. Dataset of P123, 1 and 2; P12, 3 and 4; P123R, 5 and 6; P12R, 7 and 8. * indicates PP = 1.00 and BS = 100 in all inferences
To further investigate the phylogenetic results described above, the AU, KH, and SH tests for constrained topologies were performed (Table 1). The genus Cosmoscarta was paraphyletic in the topology of Fig. 7. The alternative phylogeny implying its monophyly is rejected in all topology tests (P < 0.05).
Within Cosmoscarta, although C. bimacula is difficult to be distinguished from Cosmoscarta bispecularis due to their highly similar morphology, phylogenetic analyses based on mitogenome data robustly supported the relationships of (C. bimacula + C. rubroscutellata) + (C. bispecularis + Cosmoscarta sp.). From the aspect of comparative mitogenomics analysis, C. bimacula also had the highest nucleotide similarity with C. rubroscutellata. C. bispecularis and Cosmoscarta sp. not only shared the identical length in each PCG, but also had similar substitution patterns in the secondary structures of tRNAs.
Discussion
This study presents nine mitogenomes of Cosmoscartini,in which five species were completely sequenced. However, due to the high A + T content and complicated secondary structure, we were unable to amplify control regions in the four other species. Among the five complete mitogenomes, the control region also exhibited more variation in length (varying from 484 bp in C. bimacula to 1152 bp in C. rubroscutellata) in comparison to the other regions. Furthermore, although stem-loop structures could be found at the 3′-end of the control regions in all the complete Cosmoscartini species, no obvious conserved functional motifs could be detected. Additionally, two microsatellite-like repeat regions were found in C. bispecularis, but not in the other species. These results showed that the control region was a fast-evolving sequence with some taxon-specific characteristics, which might be valuable genetic markers for evolutionary and population genetic studies within species groups. Among all nine newly sequenced mitogenomes, each PCG was found to be the same in length, except for cob, where C. bispecularis and Cosmoscarta sp. sharing the same length (1137 bp) but having 3 nucleotides more than that of other species (1134 bp). Because of stable secondary structures, there was also limited length variation in all tRNAs among different species, ranging from 1467 bp in C. bispecularis to 1478 bp in O. uchidae. The rRNAs also shared the generally consistent lengths, ranging from 2015 bp in Cosmoscarta sp. to 2034 bp in C. bimacula.
It was indicated that the evolutionary patterns of PCGs were highly consistent among Cosmoscartini and presented some common characters: (1) atp8 appeared to have the highest evolutionary rate and might be suitable for analyzing intraspecies relationships [37]; (2) cox1, which was used as the DNA barcoding marker [38], had the lowest evolutionary rate. Similarly, two other cytochrome oxidase genes (cox2 and cox3) and the cytochrome b gene (cob) also had the relatively lower evolutionary rates than the remaining genes, suggesting that they might be potential barcoding markers. (3) The Ka/Ks ratio for each PCG was far lower than 1, indicating that all PCGs were evolving under purifying selection and could be used to investigate phylogenetic relationships within Cosmoscartini.
Based on the comparative secondary structures of tRNAs, nucleotide substitutions on both stems (especially cbcs and hemi-cbcs) and loops characterizing taxa at different taxonomic levels might provide evidence for the phylogenetic analyses. For example, the variable tRNAs of E. fruhstorferi were quite different from those of other species (Fig. 4), e.g., A-T presented in the trnG anticodon stem of E. fruhstorferi, while the Cosmoscarta and Okiscarta species exhibited the G-C pair. Secondary structures of tRNAs between C. bispecularis and Cosmoscarta sp. appeared to be generally invariable. These substitution patterns supported the results of phylogenetic analyses that E. fruhstorferi was distantly related to other Cosmoscartini species and that C. bispecularis was closely related to Cosmoscarta sp.
Interestingly, even mismatches in tRNAs, including U-U, A-C, A-A, and C-U pairs, were associated with the Cosmoscartini phylogenetic relationships. For example, U-U in the acceptor stem of trnL1 was highly consistent among Cosmoscartini; U-U in the acceptor stem of trnT was conserved among Cosmoscarta, but presented U-A in E. fruhstorferi; A-A in the acceptor stem of trnW was conserved among seven of nine Cosmoscartini species, but presented as C-A in C. bimacula and C. rubroscutellata. These mismatches in tRNA stems were common in arthropod mitogenomes and could be restored by post-transcriptional editing processes [39] or represented unusual pairings [36].
The double-helix stems, which play a key role in the formation of secondary structures of tRNAs, might experience strong evolutionary constraints [40]. Different substitution patterns in the stems of tRNAs were associated with different evolutionary history, and thus could provide valuable signals in phylogeny. As shown in Fig. 4, strong heterogeneity was observed between E. fruhstorferi (30 individual changes in the stems of 15 tRNAs) and other Cosmoscartini species (0–16 individual changes among different species). However, compared with some of the Cosmoscarta species (e.g., C. heros and Cosmoscarta mandarina exhibited 16 and 9 substitutions, respectively), O. uchidae even presented fewer individual nucleotide substitutions (4 individual changes) in the stems of tRNAs. This similar substitution pattern between O. uchidae and Cosmoscarta indicated that O. uchidae shared similar evolutionary history with Cosmoscarta species, and might be a member of Cosmoscarta.
Conserved nucleotides were distributed unevenly among rRNA genes. For rrnL, domains IV and V were more conserved than domains I, II, and VI (Fig. 5). Within domain IV, six helixes (H1775, H1792, H1830, H1906, H1925, and H1935) were highly conserved, with only 0–2 nucleotide substitutions. In domain V, most helixes were conserved, except for helixes H2077 and H2347 which were also highly divergent among other insect mitogenomes [34, 41]. In the variable domains I, II, and VI, except for the helixes of H183, H461, H563, H589, and H822, there were no obvious conserved helixes within Cosmoscartini. For rrnS, domain III was structurally more conserved than domain I and II (Fig. 6). The primary exceptions were H1074 and H1113, which had more variable sites in secondary structures. Mfold Web Server predicted several possible helix structures for this complex region because of their high A + T content and several non-canonical base pairs. Nevertheless, according to previously published rRNA structures of hemipterans [34, 42], we chose the one with more conserved nucleotides on the paired stems. Other predicted secondary structures, which presented more variable sites on stems, might be taxon-specific and were not accepted in our analyses. Additionally, although the helix H1068 was absent in some hemipterans [43, 44], it was found in the rrnS of Cosmoscartini, as also presented in other insects [45, 46]. In domain I, the helix H47 was highly variable in terms of its sequence and secondary structure. Actually, no consistent structure had been reported for this region in insects [47]. In domain II, although H673 was proposed to form a long stem and a very small loop in some insects [33], this helix formed a relatively short paired stem with a large loop in Cosmoscartini, as reported in other species of Hemiptera [34, 48].
Among the non-coding regions, the spacer of trnS2-nad1 was supposed to contain the binding site for the bidirectional transcription termination factor (DmTTF) [49]. Furthermore, sequence alignments of this region could also be reflective of the relationships of closely related species. It was indicated that C. bimacula, C. rubroscutellata, C. bispecularis, and Cosmoscarta sp. shared the similar length (22–26 bp) and the identical segment of AACCAAAAATAATGAA. Cosmoscarta exultans and O. uchidae also contained the identical sequence of AACTAAAAATAATGAA. It had been proposed that intergenic region was under relaxed selection and had a high nucleotide substitution rate [50]. However, O. uchidae shared the highly conserved nucleotide pattern with other Cosmoscarta species and as a result had a close relationship with them or might be a member of Cosmoscarta.
The species of O. uchidae was classified within the genus Okiscarta [19], but in our studies was placed consistently in Cosmoscarta and grouped with C. exultans. Morphologically, O. uchidae was firstly placed in Cosmoscarta [18]. However, Matsumura [19] proposed that Okiscarta was closely allied to Cosmoscarta, but could be easily distinguished from the latter especially in the following characters: thorax posteriorly produced so as to conceal scutellum except for the apex; tegmina at the apices blunt, approximately twice as long as the pronotum; ovipositor on each side depressed deeply, much shorter than the pygophore. However, after comprehensive review of the type material of the Oriental cercopid spittlebug species, Okiscarta and Cosmoscarta were proposed as generic synonymies [20]. Furthermore, one Cercopoidea phylogenetic analysis based on DNA nucleotide sequence data from six loci demonstrated similar result that O. uchidae was placed within Cosmoscarta, despite having a low support value [21], providing molecular evidence for our placement of Okiscarta species within Cosmoscarta. Here we initially added support for this conclusion with the phylogenetic analyses of mitogenome data. Both the ML bootstrap and BI posterior probability presented strong signals and therefore highly supported the inclusion of O. uchidae within Cosmoscarta, which otherwise would be paraphyletic. This result was also supported by nucleotide similarity among species, the patterns of intergenic spacer trnS2-nad1, the models of substitution models observed in the secondary structures of tRNAs, and the tree topologies tests. These results support the necessity to revise the boundaries of this genus to generate a classification consistent with the evolutionary history of this group of spittlebugs. Given that mitogenomes are powerful molecular markers for inferring phylogenetic relationships at different taxonomic levels within Hemiptera [8, 33, 51,52,53], we concluded that the previously defined Okiscarta species was derived from Cosmoscarta and, therefore, here we transfer O. uchidae to Cosmoscarta.
Although our study provided the first mitochondrial phylogeny of Cosmoscartini, only two genera of Ectemnonotum and Cosmoscarta (if redefined to include Okiscarta) were available. Considering the limited representatives of mitogenomes in each Cosmoscartini genus, a denser taxon sampling from Cosmoscartini is still needed for further clarification of the placement of Okiscarta within Cosmoscarta and the placements of Cosmoscarta and Ectemnonotum within Cosmoscartini.
Conclusions
Our study presents the mitogenomes of nine Cosmoscartini species and is the first representation of the detailed comparative genomic and phylogenetic analyses within Cercopidae. Newly sequenced Cosmoscartini mitogenomes share similar genomic features, but present high variation in control regions among the five complete mitogenomes. The evolutionary patterns of PCGs indicate that their evolution is under purifying selection, in which atp8 and cox1 exhibit the highest and lowest evolutionary rate, respectively. The tRNA secondary structures have diverse substitution patterns in Cosmoscartini, with each stem generally more conserved than the corresponding loop. The secondary structures of rRNAs are predicted for the first time within Cercopidae, with conserved regions distributed unevenly both in rrnL (domain IV and V) and rrnS (domain III). Cosmoscartini mitogenomes are punctuated by non-coding portions highly conserved in size, except for the spacers of trnS2-nad1 and the control region. Among them, the longest spacer is the control region, which appears to be a fast-evolving sequence characterized by variable length of (15–355 bp) repeated motifs. However, the spacer of trnS2-nad1 possesses a conserved motif, which may be the bidirectional transcription termination factor.
Although the status of Okiscarta is still under debate with regards to morphological characters, the previously defined genus Okiscarta is suggested to be a synonym of Cosmoscarta according to the mitogenome data. Within Cosmoscarta, the four closely related species sharing high similarity in morphology are also clearly resolved. Therefore, full mitogenomes provide a better understanding of phylogenetic relationships at a low taxonomic level (genus or species). This study has provided an initial taxonomic recommendation and might assist in future mapping of mitogenomic evolution and phylogenetic relationships in Cercopidae.
Methods
Samples and DNA extraction
Nine adults of Cosmoscartini species were collected from seven locations (Additional file 6: Table S4). All specimens were preserved in absolute ethyl alcohol and stored in − 20 °C freezer in Institute of Zoology, Chinese Academy of Sciences until use. Total genomic DNA was extracted from legs of a single sample using the DNeasy Blood & Tissue kit (Qiagen Hilden, Germany) following the manufacturer’s instructions.
PCR amplification and sequencing
The mitogenomes of all species were generated by amplified overlapping fragments, using a set of newly designed primers (Additional file 7: Table S5). PCR amplification was performed using TaKaRa LA Taq (Takara Bio Inc., Dalian, China), with the following conditions: 2 min at 92 °C; 40 cycles of 30 s at 92 °C, 30 s at 48–55 °C, 12 min at 60 °C; and a final extension step at 60 °C for 20 min. PCR products were electrophoresed in a 1% agarose gel, purified, and sequenced directly or cloned into the pMD™19-T vector (Takara Bio Inc., Dalian, China). All fragments were sequenced on an ABI 3730XL DNA sequencer by Majorbio Biotechnology Company (Beijing, China) with primers walking on both strands.
Bioinformatic analysis
Sequences were assembled using SeqMan program included in the Lasergene software package (DNAStar Inc., Madison, Wisc.). The tRNAs were identified by Mitos WebServer [54], with the Mito genetic code of invertebrate. PCGs, rRNAs and control regions were confirmed by the boundaries of tRNAs, and by alignment with other Cercopidae gene sequences. To ensure the accuracy of gene boundaries, PCGs were also translated into amino acids according to the invertebrate mitochondrial genetic code. The comparable sequence identity map was depicted by the CGView Comparison Tool [55], with query size of 50 bp in each BLAST search. Base composition, codon distribution, and relative synonymous codon usage (RSCU) were calculated by MEGA 6.05 [56]. Composition skew analysis was calculated according to the formulas: AT skew = (A-T)/(A + T) and GC skew = (G-C)/(G + C) [57]. The rate of synonymous substitutions (Ks), the rate of nonsynonymous substitutions (Ka), and the ratio of Ka/Ks were determined with DnaSP 5.0 [58]. Tandem repeats and stem-loop structures of control regions were identified by the tandem repeats finder online server [59] and the Mfold Web Server [60], respectively.
Estimates of rRNA secondary structure
Secondary structures of rRNAs were inferred following the models proposed for some other hemipterans [33, 34, 42, 44]. Helix numbering was named according to the convention established at the Comparative RNA Web (CRW) Site [36]. Sequences lacking significant homology were also folded by the Mfold Web Server.
Phylogeny
Phylogenetic analyses were performed on the nine newly sequenced mitogenomes of Cosmoscartini, with three other Cercopidae species used as outgroups (Additional file 6: Table S4). Nucleotide sequences for all the 13 PCGs were translated into amino acids, aligned using Muscle implemented within MEGA 6.05, and then toggled back into nucleotide alignments. The tRNAs and rRNAs were aligned with MAFFT 7.310 using the Q-INS-i algorithm [61]. Additionally, to eliminate poorly aligned positions and divergent regions, Gblocks 0.91b [62] was employed with default settings. Phylogenetic analyses were conducted with four datasets: (1) P123: all codon positions of 13 PCGs; (2) P12: first and second codon positions of PCGs; (3) P123R: P123, 22 tRNAs, and two rRNAs; and (4) P12R. Each combined dataset was concatenated by BioEdit 7.0.9.0 [63].
Maximum Likelihood (ML) analyses were conducted using IQ-TREE 1.6.5 [64] with 1000 replicates of ultrafast likelihood bootstrap [65]. Partitioned analyses [66] for multi-gene alignments were defined by both gene types (each of 13 PCGs, 22 tRNAs, and two rRNAs) and codon positions (first, second, and third codon positions for each PCG). The best-fit model for each partition was determined by ModelFinder [67] implemented in the IQ-TREE program.
Bayesian inference (BI) was performed with the site-heterogeneous model CAT + GTR implemented in PhyloBayes MPI 1.5a [68] through the online CIPRES Science gateway [69]. Two independent searches were run until the likelihood of the sampled trees stabilized and the two runs converged satisfactorily (maxdiff < 0.1). The initial 25% trees of each run were discarded as burn-in, and the consensus tree was computed from the remaining trees. The phylogenetic trees were visualized in FigTree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Topology tests
To compare alternative phylogenetic hypotheses, ML trees constrained for the monophyly of Cosmoscarta were calculated with IQ-TREE 1.6.5 using the same search strategies described above. P-values were obtained for the Approximately Unbiased (AU) [70], Kishino-Hasegawa (KH) [71], and Shimodaira-Hasegawa (SH) [72] tests with IQ-TREE 1.6.5.
Abbreviations
- atp6 and atp8 :
-
ATPase subunits 6 and 8
- BI:
-
Bayesian Inference
- cob :
-
Cytochrome b
- cox1-cox3 :
-
Cytochrome c oxidase subunits I-III
- mitogenome:
-
Mitochondrial genome
- ML:
-
Maximum Likelihood
- nad1-nad6 and nad4L :
-
NADH dehydrogenase subunits 1–6 and 4 L
- PCG:
-
Protein-coding gene
- rrnL and rrnS :
-
Large and small subunit ribosomal RNA (rRNA)
- RSCU:
-
Relative synonymous codon usage
- tRNA:
-
Transfer RNA
References
Zhang DX, Hewitt GM. Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol. 1997;25(2):99–120.
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27(8):1767–80.
Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410(2):103–23.
Curole JP, Kocher TD. Mitogenomics: digging deeper with complete mitochondrial genomes. Trends Ecol Evol. 1999;14(10):394–8.
Lin CP, Danforth BN. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol Phylogenet Evol. 2004;30(3):686–702.
Avise JC. Mitochondrial DNA polymorphism and a connection between genetics and demography of relevance to conservation. Conserv Biol. 1995;9(3):686–90.
Cameron SL. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 2014;59:95–117.
Li H, Shao RG, Song N, Song F, Jiang P, Li ZH, et al. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci Rep. 2015;5:8527.
Lv L, Peng XX, Jing SL, Liu BF, Zhu LL, He GC. Intraspecific and interspecific variations in the mitochondrial genomes of Nilaparvata (Hemiptera: Delphacidae). J Econ Entomol. 2015;108(4):2021–9.
Qin J, Zhang YZ, Zhou X, Kong XB, Wei SJ, Ward RD, et al. Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes. BMC Genomics. 2015;16:428.
Kim J, Kern E, Kim T, Sim M, Kim J, Kim Y, et al. Phylogenetic analysis of two Plectus mitochondrial genomes (Nematoda: Plectida) supports a sister group relationship between Plectida and Rhabditida within Chromadorea. Mol Phylogenet Evol. 2017;107:90–102.
Littlewood DTJ, Lockyer AE, Webster BL, Johnston DA, Le TH. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic atworms. Mol Phylogenet Evol. 2006;39(2):452–67.
Jühling F, Pütz J, Bernt M, Donath A, Middendorf M, Florentz C, et al. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012;40(7):2833–45.
Wu XY, Xiao S, Li XL, Li L, Shi W, Yu ZN. Evolution of the tRNA gene family in mitochondrial genomes of five Meretrix clams (Bivalvia, Veneridae). Gene. 2014;533(1):439–46.
Xia Y, Zheng YC, Miura I, Wong PBY, Murphy RW, Zeng XM. The evolution of mitochondrial genomes in modern frogs (Neobatrachia): nonadaptive evolution of mitochondrial genome reorganization. BMC Genomics. 2014;15:691.
Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101(4):301–20.
Torriani SFF, Penselin D, Knogge W, Felder M, Taudien S, Platzer M, et al. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet Biol. 2014;62:34–42.
Matsumura S. Die Hemipteren Fauna von Riukiu (Okinawa). Trans Sapporo Nat Hist Soc. 1906;1(1):15–38.
Matsumura S. New species and genera of Cercopidae in Japan, Korea and Formosa, with a list of the known species. J Fac Agric Hokkaido Imperial Univ. 1940;45(2):35–82.
Liang AP. Nomenclatural changes in the oriental Cercopidae (Homoptera). J Entomol Sci. 2001;36(3):318–24.
Cryan JR, Svenson GJ. Family-level relationships of the spittlebugs and froghoppers (Hemiptera: Cicadomorpha: Cercopoidea). Syst Entomol. 2010;35(3):393–415.
Paladini A, Takiyac DM, Urband JM, Cryane JR. New World spittlebugs (Hemiptera: Cercopidae: Ischnorhininae): dated molecular phylogeny, classification, and evolution of aposematic coloration. Mol Phylogenet Evol. 2018;120:321–34.
Henderson G, Hoffman GD, Jeanne RL. Predation on cercopids and material use of the spittle in aphid-tent construction by prairie ants. Psyche. 1990;97:43–53.
Chen X, Meyer-Rochow VB, Fereres A, Morente M, Liang AP. The role of biofoam in shielding spittlebug nymphs (Insecta, Hemiptera, Cercopidae) against bright light. Ecol Entomol. 2018;43:273–81.
Chen X, Liang AP. Identification of a self-regulatory pheromone system that controls nymph aggregation behavior of rice spittlebug Callitettix versicolor. Front Zool. 2015;12:10.
Pang ZH. Current situation and development in trend of eucalytus pest in China. J Guangxi Acad Sci. 2013;29(3):192–206.
Liang AP, Jiang GM, Webb MD. Revision of the oriental spittlebug genera Baibarana Matsumura and Telogmometopius Jacobi (Hemiptera: Cercopidae: Rhinaulacini). J Nat Hist. 2006;40(35–37):2071–99.
Liang AP, Jiang GM. Generic reassignment of Cosmoscarta rutilans Butler (Hemiptera: Cercopoidea: Cercopidae). J Kansas Entomol Soc. 2007;80(1):80–1.
Beckenbach AT, Stewart JB. Insect mitochondrial genomics 3: the complete mitochondrial genome sequences of representatives from two neuropteroid orders: a dobsonfly (order Megaloptera) and a giant lacewing and an owlfly (order Neuroptera). Genome. 2009;52:31–8.
Jiang YL, Garzón-Orduña IJ, Winterton SL, Yang F, Liu XY. Phylogenetic relationships among tribes of the green lacewing subfamily Chrysopinae recovered based on mitochondrial phylogenomics. Sci Rep. 2017;7:7218.
Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290(5806):470–4.
Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992;141:173–216.
Yuan ML, Zhang QL, Guo ZL, Wang J, Shen YY. Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genomics. 2015;16:460.
Li T, Yang J, Li YW, Cui Y, Xie Q, Bu WJ, et al. A mitochondrial genome of Rhyparochromidae (Hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci Rep. 2016;6:35175.
Negrisolo E, Babbucci M, Patarnello T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genomics. 2011;12:221.
Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3(1):2.
Wang Y, Chen J, Jiang LY, Qiao GX. Hemipteran mitochondrial genomes: features, structures and implications for phylogeny. Int J Mol Sci. 2015;16(6):12382–404.
Hebert PDN, Ratnasingham S, de Waard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B. 2003;270:S96–9.
Lavrov DV, Brown WM, Boore JL. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc Natl Acad Sci U S A. 2000;97(25):13738–42.
Montelli S, Peruffo A, Patarnello T, Cozzi B, Negrisolo E. Back to water: signature of adaptive evolution in Cetacean mitochondrial tRNAs. PLoS One. 2016;11(6):e0158129.
Buckley TR, Simon C, Flook PK, Misof B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol Biol. 2000;9(6):565–80.
Chen ZT, Mu LX, Wang JR, Du YZ. Complete mitochondrial genome of the Citrus spiny whitefly Aleurocanthus spiniferus (Quaintance)(Hemiptera: Aleyrodidae): implications for the phylogeny of whiteflies. PLoS One. 2016;11(8):e0161385.
Li H, Liu H, Shi AM, Štys P, Zhou XG, Cai WZ. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS One. 2012;7(1):e29419.
Wang Y, Huang XL, Qiao GX. Comparative analysis of mitochondrial genomes of five aphid species (Hemiptera: aphididae) and phylogenetic implications. PLoS One. 2013;8(10):e77511.
Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: hymenoptera): structure, organization, and retrotransposable elements. Insect Mol Biol. 2006;15(5):657–86.
Li T, Gao CQ, Cui Y, Xie Q, Bu WJ. The complete mitochondrial genome of the stalk-eyed bug Chauliops fallax Scott, and the monophyly of Malcidae (Hemiptera: Heteroptera). PLoS One. 2013;8(2):e55381.
Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408(1):112–23.
Gao JY, Li H, Truong XL, Dai X, Chang J, Cai WZ. Complete nucleotide sequence and organization of the mitochondrial genome of Sirthenea flavipes (Hemiptera: Reduviidae: Peiratinae) and comparison with other assassin bugs. Zootaxa. 2013;3669:1–16.
Roberti M, Polosa PL, Bruni F, Musicco C, Gadaleta MN, Cantatore P. DmTTF, a novel mitochondrial transcription termination factor that recognises two sequences of Drosophila melanogaster mitochondrial DNA. Nucleic Acids Res. 2003;31(6):1597–604.
Ghikas DV, Kouvelis VN, Typas MA. Phylogenetic and biogeographic implications inferred by mitochondrial intergenic region analyses and ITS1–5.8S-ITS2 of the entomopathogenic fungi Beauveria bassiana and B. brongniartii. BMC Microbiolgy. 2010;10:174.
Song N, Cai WZ, Li H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci Rep. 2017;7:10429.
Liu J, Bu CP, Wipfler B, Liang AP. Comparative analysis of the mitochondrial genomes of Callitettixini spittlebugs (Hemiptera: Cercopidae) confirms the overall high evolutionary speed of the AT-rich region but reveals the presence of short conservative elements at the tribal level. PLoS One. 2014;9:e109140.
Li H, Leavengood JM Jr, Chapman EG, Burkhardt D, Song F, Jiang P, et al. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc R Soc B. 2017;284:20171223.
Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69(2):313–9.
Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView comparison tool. BMC Genomics. 2012;13:202.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA 6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41(3):353–8.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80.
Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–15.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–52.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–8.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–22.
Chernomor O, von Haeseler A, Minh BQ. Terrace aware data structure for phylogenomic inference from supermatrices. Syst Biol. 2016;65:997–1008.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Lartillot N, Rodrigue N, Stubbs D, Richer J. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 2013;62(4):611–5.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees, vol. 14. New Orleans: Gateway Computing Environments Workshop (GCE); 2010. p. 1–8.
Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51(3):492–508.
Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol. 1989;29(2):170–9.
Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–6.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 31572298, 31561163003), awarded to LAP. The funding bodies had no role in the design of the study, collection and analysis of data, interpretation of results, or in writing the manuscript.
Availability of data and materials
The mitogenomes generated and analyzed during the current study are available in the [NCBI] repository, [https://www.ncbi.nlm.nih.gov/], with the GenBank accession numbers of MF621236, MG488215-MG488220, and MG599489-MG599490.
Author information
Authors and Affiliations
Contributions
TS designed the research, performed the molecular experiments, and wrote the paper. TS, BH and KL collected the samples and analyzed the data. AL identified the specimens and coordinated the manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Gene content of our nine sequenced specimens and other published Cercopidae mitogenomes. (XLSX 24 kb)
Additional file 2:
Table S2. Codons usage for the 13 protein-coding genes of nine Cosmoscatini species. (XLSX 24 kb)
Additional file 3:
Table S3. Start and stop codons of 13 protein-coding genes of nine Cosmoscatini species. (XLSX 10 kb)
Additional file 4:
Figure S1. Sequence alignments of the intergenic spacer between trnS2 and nad1 in the nine Cosmoscartini mitogenomes, with the conserved nucleotides marked with *. (TIF 162 kb)
Additional file 5:
Figure S2. Organization of the control regions in the five complete Cosmoscartini mitogenomes. The tandem repeats are presented by colored oval with Arabic numerals inside. Non-repeat regions are shown by gray bars with sequence length inside. (TIF 3876 kb)
Additional file 6:
Table S4. Samples used in this study, collecting data, and GenBank accession numbers. (XLSX 19 kb)
Additional file 7:
Table S5. Primer used in this study. (XLSX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Su, T., He, B., Li, K. et al. Comparative analysis of the mitochondrial genomes of oriental spittlebug trible Cosmoscartini: insights into the relationships among closely related taxa. BMC Genomics 19, 961 (2018). https://doi.org/10.1186/s12864-018-5365-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5365-7