
Abstract
Background
Cigarette smoking has severe adverse health consequences in adults and in the offspring of mothers who smoke during pregnancy. One of the most widely reported effects of smoking during pregnancy is reduced birth weight which is in turn associated with chronic disease in adulthood. Epigenome-wide association studies have revealed that smokers show a characteristic “smoking methylation pattern”, and recent authors have proposed that DNA methylation mediates the impact of maternal smoking on birth weight. The aims of the present study were to replicate previous reports that methylation mediates the effect of maternal smoking on birth weight, and for the first time to investigate whether the observed mediation effects are sex-specific in order to account for known sex-specific differences in methylation levels.
Methods
Methylation levels in the cord blood of 313 newborns were determined using the Illumina HumanMethylation450K Beadchip. A total of 5,527 CpG sites selected on the basis of evidence from the literature were tested. To determine whether the observed association between maternal smoking and birth weight was attributable to methylation, mediation analyses were performed for significant CpG sites. Separate analyses were then performed in males and females.
Results
Following quality control, 282 newborns eventually remained in the analysis. A total of 25 mothers had smoked consistently throughout the pregnancy. The birthweigt of newborns whose mothers had smoked throughout pregnancy was reduced by >200g. After correction for multiple testing, 30 CpGs showed differential methylation in the maternal smoking subgroup including top “smoking methylation pattern” genes AHRR, MYO1G, GFI1, CYP1A1, and CNTNAP2. The effect of maternal smoking on birth weight was partly mediated by the methylation of cg25325512 (PIM1); cg25949550 (CNTNAP2); and cg08699196 (ITGB7). Sex-specific analyses revealed a mediating effect for cg25949550 (CNTNAP2) in male newborns.
Conclusion
The present data replicate previous findings that methylation can mediate the effect of maternal smoking on birth weight. The analysis of sex-dependent mediation effects suggests that the sex of the newborn may have an influence. Larger studies are warranted to investigate the role of both the identified differentially methylated loci and the sex of the newborn in mediating the association between maternal smoking during pregnancy and birth weight.
Similar content being viewed by others


Background
Cigarette smoking has severe adverse health consequences in adults and in the offspring of mothers who smoke during pregnancy [1]. Despite the efforts of advisory boards to inform women of the risk to the developing fetus, around 10% of mothers continue to smoke during pregnancy [https://www.cdc.gov/prams/]. Previous research has identified associations between maternal smoking during pregnancy and a range of health problems in the offspring [2, 3]. One of the most widely reported effects is low birth weight [4,5,6]. Low birth weight has in turn been associated with various long-term health problems in adulthood. These include increased vulnerability to stress [7], cognitive deficits [8], and chronic somatic disorders, such as cardiovascular disease [9, 10], and obesity [11]. Low birth weight has also been associated with psychiatric disorders, such as schizophrenia and depression [12, 13]. On the basis of such research, previous authors have proposed that chronic disease in adulthood may be initiated during pregnancy as a result of exposure to adverse intrauterine conditions. According to the theory of “fetal programming”, alterations in fetal nutrition and endocrine status result in developmental adaptations, which cause permanent changes in cellular structure, physiology, and metabolism, thereby predisposing to disease in adult life [14, 15].
A plausible mechanism through which exposure to adverse intrauterine conditions may negatively impact longterm health is DNA methylation. Candidate CpG investigations and epigenome-wide association studies (EWAS) have found consistent associations between smoking and differential DNA methylation in adults [16,17,18]. A number of studies have also investigated the impact of maternal smoking during pregnancy on methylation patterns in the offspring. These investigations have comprised candidate gene and genome wide approaches [19,20,21,22,23], as well as EWAS [24,25,26,27,28,29,30,31]. In these EWAS, a number of differentially methylated genes have shown repeated and consistent association [32,33,34]. The top five genes are AHRR, GFI1, MYO1G, CYP1A1, and CNTNAP2.
Previous authors have formulated the hypothesis that maternal smoking impacts birth weight via smoking-induced DNA methylation. According to this theory, differential methylation functions as a mediator, i.e., a variable that accounts for part of the relation between the predictor and the criterion [35, 36]. Initial analyses to determine whether DNA methylation mediates between maternal smoking and birth weight have already been performed. In these investigations, a mediation effect was found for eight CpGs in an analysis of infant cord blood [37], and two CpGs in an analysis of the placenta [38].
The aims of the present study were to replicate the previously reported mediating effects of methylation on the association between maternal smoking and birth weight, and for the first time to investigate whether the observed mediation effects are sex-specific. The analyses focused on high confidence smoking-related sites, and were restricted to CpG sites with: (i) significant association with smoking in a large recent meta-analysis [31]; or (ii) a reported mediating effect in the association between maternal smoking and birth weight [37, 38].
Analyses were also performed to determine whether the observed mediation effects were sex-specific, since: (i) methylation levels show substantial sex differences [39]; (ii) the observed sex differences are present from birth [40], and (iii) differing methylation patterns might have different outcomes.
Methods
Sample description
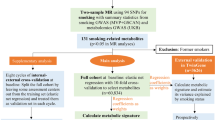
Data for the present study were derived from the POSEIDON study ("Pre-, peri- and postnatal Stress in human and non-human offspring: A translational approach to study Epigenetic Impact on DepressiON"). A detailed description of the POSEIDON study is provided elsewhere [41]. The study protocol was approved by the Ethics Committee of the Medical Faculty Mannheim of the University of Heidelberg, and the study was conducted in accordance with the Declaration of Helsinki. Data from 410 mothers and 405 infants recruited during the third trimester of pregnancy from hospitals in the Rhine-Neckar Region of Germany were analyzed. Briefly, maternal exclusion criteria for the present analyses were: (i) a history of hepatitis B, hepatitis C, or HIV-infection; (ii) any current or previous diagnosis of schizophrenia or any substance dependency other than nicotine; and (iii) any current psychiatric disorder requiring inpatient treatment. Exclusion criteria in the offspring were: birth weight <1.500 g; gestational age at delivery <32 weeks; multiple birth; and the presence of a congenital disease, malformation, deformation, or chromosomal abnormality.
A structured interview and a questionnaire battery were used to collect information concerning environmental-, sociodemographic-, medical-, and psychosocial risk factors for stress. A detailed description of these instruments is provided elsewhere [42]. Sociodemographic data are presented in the supplement (Additional file 1: Table S1). For the purposes of the present analyses, the women were divided into two subgroups: smokers and non-smokers. The smoking group comprised women who had smoked throughout pregnancy. The non-smoking group comprised women who had either not smoked at all during the pregnancy, or who had smoked during early pregnancy only. This approach was taken since previous studies as well as our data have revealed no differences in the methylation patterns of infants whose mothers smoked during early pregnancy compared to infants whose mothers did not smoke at all [43].
Blood collection, DNA extraction, genome-wide methylation assay
Whole cord blood was collected immediately after birth from n=313 newborn singletons. For n=299 newborns, automated genomic DNA extraction was performed using the chemagic Magnetic Separation Module I (Chemagen Biopolymer-Technologie AG; Baesweiler; Germany). For n=14 newborns, a low volume of umbilical cord blood (<2 mL) was obtained. For these 14 samples, DNA was isolated using the QIAamp DNA Blood Midi Kit (Qiagen GmbH; Hilden; Germany). All genomic DNA samples were stored at -20 °C prior to analysis.
DNA methylation was measured using the Illumina Infinium HumanMethylation450K Beadchip.
Statistical analysis
Applied software
Quality control (QC) and all statistical analyses were performed using the R version 3.2.5 statistical analysis software [44], and the R-packages minfi [45], wateRmelon [46], Enmix [47], sva [48], limma [49], and mediation [50].
Data preprocessing, QC, and filtering
Methylation intensity signals were extracted from raw intensity data using the preprocessENmix procedure [47]. This includes background noise correction using out-of-band Infinium1 intensities as well as correction for dye bias based on internal control probes [51]. Data were then quantile normalized followed by Beta Mixture Quantile Dilation [BMIQ] [52]. Samples with insufficient DNA quality (average of medians of methylated and unmethylated signals <10.5; outlier status with regard to either averaged total intensity values or beta value distribution; insufficient bisulfite conversion; or failure in detection (detection P-value > 0.01) at more than 1% of positions) were excluded. Probes were excluded if any of the following criteria were met: beadcount <3; detection failure in more than 1% of samples; located within 10bp of a single nucleotide polymorphism (SNP); cross reactivity; X- or Y- linked status.
Data transformation, batch correction, and cell type adjustment
For all downstream analyses, methylation intensity data were converted to M-Values [53]. To detect batch effects, a principal component analysis was performed, based on the 10,000 sites with highest variance. The first 5 principal components (PC) were extracted. Extracted PCs were tested for association with possible batch effects using MANOVA and visual inspection of scatter plots. Detected batch effects were then removed using the ComBat procedure, as implemented in the sva package [54]. Following the removal of Sentrix ID and position related effects, no further batch effects were evident. Cell type composition was estimated using the Houseman reference based method, as implemented in the minfi package [55]. Adjustment for cell type composition was then performed by including the first five of a total of six cell type estimators as covariates in the regression models used for association testing.
Methylation association analysis
To avoid introducing additional heterogeneity with regard to birth weight, mothers whose pregnancy was complicated by premature delivery (gestational age <259 days) or treatment for gestational diabetes were excluded prior to analysis. To minimize the risk of spurious associations due to small sample size and thus avoid false positives, the analysis was strictly limited to high confidence smoking-related CpG sites that had shown significant association with smoking in a large recent meta-analysis [31], and CpG sites previously reported to mediate the effect of maternal smoking on birth weight [37, 38]. Association testing for methylation levels was performed using general linear models as implemented in the R-package limma. Gestational age, sex of the newborn, and parental height were included as covariates, since research has shown that these factors impact birth weight [56]. Adjustment was also made for maternal age. Cell type composition was taken into account, as described above. Sex and gestational age (weeks) were reported by the responsible obstetrician. Sex specific analyses were then conducted using the covariates above, with the exception of sex.
To exclude other significant loci in this sample, we also performed an EWAS on smoking (data not shown).
Mediation analysis
Sites showing significant association with smoking (FDR<0.05) were then used to test whether methylation levels partly mediate the effects of maternal smoking on birth weight. This was performed using a quasi-Bayesian approach as proposed in Imai et al. ([57]; for a more detailed explanation please see Additional file 1: Text), as implemented in R-package mediation [57], running 10,000 simulations. The same possible confounders as named above were included as additional covariates in the mediator and outcome regression models (gestational age, sex of the newborn, parental height, maternal age, cell type composition; in contrast to Kupers et al. [37] socioeconomic status was not included in the model as it was not associated with birth weight in our sample). For sex specific analyses sex of newborn was excluded from the models as above.
Gene-based analysis of Early Growth Genetics (EGG) Consortium GWAS results
Data on the trait “birth weight” were obtained from the EGG Consortium via the UK Biobank Resource. These data were downloaded from www.egg-consortium.org [58]. Gene-based analysis was performed using the ´birth weight summary statistic data 2016´ (file: BW3_EUR_summary_stats.txt.gz from http://egg-consortium.org/birth-weight-2016.html) and MAGMA v1.06[59]. The applied linkage disequilibrium (LD) structure was that of the 1000 Genomes project (http://www.internationalgenome.org/data). SNPs were assigned to a gene if the variant was located within 20kb flanking its transcript.
Results
After technical QC, a total of 405,654 sites and 311 individuals were in principle available for analysis. A total of n=5,527 of these CpG sites had previously been reported associated with smoking (see above) and were therefore selected for association testing. After excluding prematurely delivered newborns and individuals receiving anti-diabetic treatment during pregnancy the final sample size was n=282 individuals (138 male and 144 female). Of these, 13 males and 12 females were the offspring of mothers who had smoked throughout pregnancy. The general characteristics of the participants included in the present study are provided in Additional file 1: Table S1.
Impact of maternal smoking on birth weight
-
a)
The average difference in birth weight between the smoking and non-smoking groups was 209g. The newborns of smokers had a mean birth weight of 3,267g. The newborns of non-smokers had a mean weight of 3,476g (see Table 1; see also Additional file 1: Figure S2 for a more fine grained comparison between different groups).
-
b)
In female newborns (n=144), the average birth weight was 189g lower in the smoking subgroup compared to the non-smoking subgroup, i.e., birth weight was 5.6% lower when mothers smoked. This difference however did not reach statistical significance. In male newborns (n=138), the average birth weight was significantly lower by 242g in the smoking subgroup compared to the non-smoking subgroup, i.e., birth weight was 6.7% lower when mothers smoked (see Table 1).
Methylation association analysis
Following correction for multiple testing, a total of 30 CpG sites showed significant differential methylation in the smoking subgroup (see Table 2). These CpGs map to 13 genes (AHRR, CNTNAP2, CYP1A1, FRMD4A, GFI1, ITGB7, MIR548F3, MYO1G, PIM1, RNF157, SAMD3, TFEB, UNC45B).
The EWAS on smoking revealed no further epigenome-wide associated CpG sites (data not shown.
There was no significant association between DNA extraction method and methylation levels using a linear modelling approach as well as including the extraction methods as a covariate (data not shown).
Mediation analysis
Of the 30 CpG sites found differentially methylated after maternal smoking in our sample, the following were found to mediate the effect of maternal smoking on birth weight: cg25325512 (PIM1, p=0.005); cg25949550 (CNTNAP2, p=0.008); and cg08699196 (ITGB7, p=0.045). Sex-specific analyses for these three CpG sites revealed that cg25949550 (CNTNAP2, p=0.022) mediated the effect of maternal smoking on birth weight in male newborns (see Table 3).
Gene-based analysis of EGG Consortium GWAS results
In the gene-based-analysis, PIM1, CNTNAP2, and ITGB7 were tested for association with birth weight. ITGB7 showed significant association (p=8.24x10-7). PIM1 and CNTNAP2 failed to achieve nominal significance (p>0.05). Single marker p-values of the SNPs at the ITGB7 locus are listed in Additional file 1: Table S2.
Discussion
The aims of the present study were to replicate the finding that the association between maternal smoking and birth weight is mediated by methylation, and to investigate whether the observed mediation effects are sex-specific. Differentially methylated CpG sites were detected in 13 genes, including AHRR, GFI1, MYO1G, CYP1A1, and CNTNAP2. These represent the top five differentially methylated genes reported in adult smokers [16,17,18, 60, 61], and in previous studies of newborns exposed to maternal smoking [25,26,27, 31, 32, 37].
Mediation analysis of the 30 CpG sites revealed that CpG sites in the genes PIM1, CNTNAP2, and ITGB7 mediated the effect of maternal smoking on birth weight in the complete sample. The serine/threonine-protein kinase PIM1 has a marked anti-apoptotic effect, and its level is increased in lung tissue following exposure to cigarette smoke [62]. Interestingly, it has been shown that an enhanced gene expression of PIM1 - corresponding to a decreased methylation level - protects against cell death induced by cigarette smoke and neutrophilic airway inflammation [62]. In our sample, the methylation level of cg25325512 in PIM1 is lower in smokers than in non-smokers. A lower methylation level can lead to an upregulation of gene expression. Thus, upregulation of PIM1 in smokers could be an adaptive process to protect against negative effects of cigarette smoking. In monkeys, research has shown that PIM1 is associated with body mass indeces after calorie restriction [63]. PIM1 belongs to the PIM serine/threonine kinase family, which is involved in the regulation of cell survival. PIM kinases are constitutively active, and regulate cell growth, differentiation, and apoptosis [64]. The Contactin Associated Protein-Like 2 gene (CNTNAP2) encodes a member of the neurexin family, whose members function as cell adhesion molecules and receptors in the nervous system of vertebrates. Genetic variation in CNTNAP2 has been associated with the regulation of body weight [65]. Interestingly, research has also implicated CNTNAP2 in multiple neurodevelopmental disorders, including Gilles de la Tourette syndrome, schizophrenia, epilepsy, autism, attention deficit and hyperactivity disorder, and mental retardation [66]. CNTNAP2 may play a role in the formation of functionally distinct domains critical for the saltatory conduction of nerve impulses in myelinated nerve fibers. The methylation level of the mediating CpG cg25949550 in CNTNAP2 is lower in infants whose mothers smoked during pregnancy, presumably leading to higher gene expression. As loss of CNTNAP2 has been shown to lead to deficits in axonal excitability [67], lower methylation of this gene might also be an adaptive process to protect against negative effects of cigarette smoking. The Integrin Subunit Beta 7 gene (ITGB7) encodes a protein that is a member of the integrin superfamily. Members of this family are adhesion receptors, which are involved in signaling from the extracellular matrix to the cell. High expression of ITGB7 has been found to be associated with poor survival of cancer cells [68]. In the present study, the methylation level of cg08699196 in ITGB7 is increased, which may reduce its gene expression. A reduction in of ITGB7 expression might result in higher survival of cancer cells and, thus, oncogenesis.
The genes PIM1, CNTNAP2, and ITGB7 were not implicated in previous EWAS of birth weight [69,70,71]. However, in a gene-based analysis of genetic data obtained from a genome-wide association analysis of birth weight by the EGG Consortium [58], the present authors found that ITGB7 was strongly associated with birth weight (p=4, 2x10-7). Furthermore, ITGB7 was found to be associated with the pathophysiology of childhood obesity in a Hispanic population [72].
As methylation levels are known to be sex-specific already at birth, the present study involved sex-specific mediation analyses. Unsurprisingly, these suggest that the mediation effects can also be dependent on sex. The CpG site in CNTNAP2 had a more pronounced effect in male newborns, and failed to reach significance in females. Further sex-specific mediation effects are possible. However, their detection will require larger samples.
In contrast to previous studies, parental height was included as a covariate in the present analyses, as it had a pronounced influence on birth weight in our study. Performance of the analysis without this covariate did indeed obtain 45 rather than 30CpG sites associated with smoking. In the mediation analysis of these 45 CpG sites, three further sites became significant in addition to PIM1 and CNTNAP2 (see Additional file 1: Table S3). Determining whether the respective findings are true or false positives is problematic, and can only be resolved through the performance of larger studies and metaanalyses.
The present study had several limitations in terms of the investigated smoking phenotype. Smoking status was conceptualized as a dichotomized trait, and no distinction was made between light and heavy smokers. Furthermore, smoking was assessed using retrospective self-reports rather than prospective and objective measures such as cotinine levels. This may have led to an underestimation of the number of smoking mothers, and thus to an underestimation of the direct effect, and an overestimation of the mediation effect, of smoking. This phenomenon was reported recently by Valeri et al. in a study on the Norwegian MoBa cohort [73]. However, unreliable self-reporting of smoking status in the present cohort is unlikely for two reasons. First, the subjects had a high relationship of confidence as they received regular obstetric care at the recruitment centers throughout pregnancy. Second, the direct effect of smoking on birth weight in the present cohort (average reduction in birth weight of ~200g) was more pronounced than that reported in the MoBa study of Valeri et al (~90g). The rate of false self-reporting is likely to be in accordance with rates reported in previous studies.
Conclusions
The present study supports reported findings that DNA methylation may represent a biological mechanism through which maternal smoking impacts birth weight. Unsurprisingly, this effect may be sex-dependent, as suggested for the first time in the present analyses. Further studies are warranted to investigate the role of the identified differentially methylated loci in mediating the association between maternal smoking during pregnancy and birth weight, and their role in determining offspring phenotypes in later life.
Abbreviations
- ACME:
-
Average Causal Mediation Effect
- ADE:
-
Average Direct Effect
- CI:
-
Confidence Intervall
- DNA:
-
Desoxyribonucleic Acid
- EWAS:
-
Epigenome-Wide Association Study
- LD:
-
Linkage Disequilibrium
- MANOVA:
-
Multivariate Analysis of Variance
- QC:
-
Quality Control
- SD:
-
Standard Deviation
- SNP:
-
Single Nucleotide Polymorphism
References
WHO: WHO recommendations for the prevention and management of tobacco use and second-hand smoke exposure in pregnancy; 2013.
Power C, Atherton K, Thomas C. Maternal smoking in pregnancy, adult adiposity and other risk factors for cardiovascular disease. Atherosclerosis. 2010;211(2):643–8.
Syme C, Abrahamowicz M, Mahboubi A, Leonard GT, Perron M, Richer L, Veillette S, Gaudet D, Paus T, Pausova Z. Prenatal exposure to maternal cigarette smoking and accumulation of intra-abdominal fat during adolescence. Obesity (Silver Spring, Md). 2010;18(5):1021–5.
Rayfield S, Plugge E. Systematic review and meta-analysis of the association between maternal smoking in pregnancy and childhood overweight and obesity. J. Epidemiol. Community Health. 2017;71(2):162–73.
Knopik VS, Maccani MA, Francazio S, McGeary JE. The epigenetics of maternal cigarette smoking during pregnancy and effects on child development. Dev Psychopathol. 2012;24(4):1377–90.
Alexander BT, Dasinger JH, Intapad S. Fetal programming and cardiovascular pathology. Compr. Physiol. 2015;5(2):997–1025.
Wust S, Entringer S, Federenko IS, Schlotz W, Hellhammer DH. Birth weight is associated with salivary cortisol responses to psychosocial stress in adult life. Psychoneuroendocrinology. 2005;30(6):591–8.
National Center for Chronic Disease P. Health Promotion Office on S, Health: Reports of the Surgeon General. In: The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General. edn. Atlanta. Centers for Disease Control and Prevention (US): GA. p. 2014.
Barker DJP, Osmond C, Forsen TJ, Kajantie E, Eriksson JG. Trajectories of growth among children who have coronary events as adults. N. Engl. J. Med. 2005;353(17):8.
Phipps K, Barker DJP, Hales CN, Fall CHD, Osmond C, Clark PMS. Fetal growth and impaired glucose tolerance in men and women. Diabetologia. 1993;36:4.
Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ. Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation. 1996;94(12):3246–50.
Kunugi H, Nanko S, Murray RM. Obstetric complications and schizophrenia: prenatal underdevelopment and subsequent neurodevelopmental impairment. Br J Psychiatry Suppl. 2001;40:s25–9.
Schlotz W, Phillips DI. Fetal origins of mental health: evidence and mechanisms. Brain Behav. Immun. 2009;23(7):905–16.
Godfrey KM, Barker DJP. Fetal programming and adult health. Public Health Nutr. 2001;4(2B)
Reichetzeder C, Dwi Putra SE, Li J, Hocher B. Developmental Origins of Disease - Crisis Precipitates Change. Cell. Physiol. Biochem. 2016;39(3):919–38.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S, et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016;9(5):436–47.
Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Le Calvez-Kelm F, Kaaks R, Barrdahl M, Boeing H, Aleksandrova K, Trichopoulou A, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8(5):599–618.
Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812.
Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009;180(5):462–7.
Guerrero-Preston R, Goldman LR, Brebi-Mieville P, Ili-Gangas C, LeBron C, Witter FR, Apelberg BJ, Hernández-Roystacher M, Jaffe A, Halden RU, et al. Global DNA hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics. 2014;5(6):539–46.
Murphy SK, Adigun A, Huang Z, Overcash F, Wang F, Jirtle RL, Schildkraut JM, Murtha AP, Iversen ES, Hoyo C. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene. 2012;494(1):36–43.
Drake AJ, O'Shaughnessy PJ, Bhattacharya S, Monteiro A, Kerrigan D, Goetz S, Raab A, Rhind SM, Sinclair KD, Meharg AA, et al. In utero exposure to cigarette chemicals induces sex-specific disruption of one-carbon metabolism and DNA methylation in the human fetal liver. BMC Med. 2015;13:18.
Bouwland-Both MI, van Mil NH, Tolhoek CP, Stolk L, Eilers PH, Verbiest MM, Heijmans BT, Uitterlinden AG, Hofman A, van Ijzendoorn MH et al.: Prenatal parental tobacco smoking, gene specific DNA methylation, and newborns size: the Generation R study. Clin Epigenetics 2015, 7(1):83.
Breton CV, Siegmund KD, Joubert BR, Wang X, Qui W, Carey V, Nystad W, Haberg SE, Ober C, Nicolae D, et al. Prenatal tobacco smoke exposure is associated with childhood DNA CpG methylation. PLoS One. 2014;9(6):e99716.
Markunas CA, Xu Z, Harlid S, Wade PA, Lie RT, Taylor JA, Wilcox AJ. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2014;122(10):1147–53.
Richmond RC, Simpkin AJ, Woodward G, Gaunt TR, Lyttleton O, McArdle WL, Ring SM, Smith AD, Timpson NJ, Tilling K, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet. 2015;24(8):2201–17.
Lee KW, Richmond R, Hu P, French L, Shin J, Bourdon C, Reischl E, Waldenberger M, Zeilinger S, Gaunt T, et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ Health Perspect. 2015;123(2):193–9.
Rzehak P, Saffery R, Reischl E, Covic M, Wahl S, Grote V, Xhonneux A, Langhendries JP, Ferre N, Closa-Monasterolo R, et al. Maternal Smoking during Pregnancy and DNA-Methylation in Children at Age 5.5 Years: Epigenome-Wide-Analysis in the European Childhood Obesity Project (CHOP)-Study. PLoS One. 2016;11(5):e0155554.
Ivorra C, Fraga MF, Bayon GF, Fernandez AF, Garcia-Vicent C, Chaves FJ, Redon J, Lurbe E. DNA methylation patterns in newborns exposed to tobacco in utero. J Transl Med. 2015;13:25.
Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120(10):1425–31.
Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu CJ, et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am J Hum Genet. 2016;98(4):680–96.
Andersen AM, Dogan MV, Beach SR, Philibert RA. Current and Future Prospects for Epigenetic Biomarkers of Substance Use Disorders. Genes (Basel). 2015;6(4):991–1022.
Reese SE, Zhao S, Wu MC, Joubert BR, Parr CL, Haberg SE, Ueland PM, Nilsen RM, Midttun O, Vollset SE, et al. DNA Methylation Score as a Biomarker in Newborns for Sustained Maternal Smoking during Pregnancy. Environ Health Perspect. 2016;
Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, Monick M, Brody GH, Tan K, Beach SR, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151.
Baron RM, Kenny DA. The Moderator-Mediator Variable Distinction in Social Psychological Research: Conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 1986;51(6):10.
Richiardi L, Bellocco R, Zugna D. Mediation analysis in epidemiology: methods, interpretation and bias. Int. J. Epidemiol. 2013;42(5):1511–9.
Kupers LK, Xu X, Jankipersadsing SA, Vaez A, la Bastide-van Gemert S, Scholtens S, Nolte IM, Richmond RC, Relton CL, Felix JF, et al. DNA methylation mediates the effect of maternal smoking during pregnancy on birthweight of the offspring. Int. J. Epidemiol. 2015;44(4):1224–37.
Morales E, Vilahur N, Salas LA, Motta V, Fernandez MF, Murcia M, Llop S, Tardon A, Fernandez-Tardon G, Santa-Marina L, et al. Genome-wide DNA methylation study in human placenta identifies novel loci associated with maternal smoking during pregnancy. Int. J. Epidemiol. 2016;45(5):1644–55.
Singmann P, Shem-Tov D, Wahl S, Grallert H, Fiorito G, Shin SY, Schramm K, Wolf P, Kunze S, Baran Y, et al. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenetics chromatin. 2015;8:43.
Dukal H, Frank J, Lang M, Treutlein J, Gilles M, Wolf IA, Krumm B, Massart R, Szyf M, Laucht M, et al. New-born females show higher stress- and genotype-independent methylation of SLC6A4 than males. Borderline personality disorder and emotion dysregulation. 2015;2:8.
Nieratschker V. POSEIDON: Pre-, peri- and pOstnatal Stress in human and non-human off-spring: a translational approach to study Epigenetic Impact on DepressiON. In: World Congress of Psychiatric Genetics 2012. Germany: Hamburg; 2012. p. 2012.
Nieratschker V, Massart R, Gilles M, Luoni A, Suderman MJ, Krumm B, Meier S, Witt SH, Nothen MM, Suomi SJ, et al. MORC1 exhibits cross-species differential methylation in association with early life stress as well as genome-wide association with MDD. Transl Psychiatry. 2014;4:e429.
Joubert BR, Haberg SE, Bell DA, Nilsen RM, Vollset SE, Midttun O, Ueland PM, Wu MC, Nystad W, Peddada SD, et al. Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol Biomarkers Prev. 2014;23(6):1007–17.
Team RC. R: A language and environment for statistical computing. Vienna URL http://www.r-project.org/: R Foundation for Statistical Computing; 2014.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
Pidsley R, Wong CCY, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293.
Xu Z, Niu L, Li L, Taylor JA. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic acids research. 2016;44(3):e20.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015;43(7):e47.
Tingley D, Yamamoto T, Hirose K, Keele L, Imai K. mediation: R Package for Causal Mediation Analysis. J. Stat. Softw. 2014;59(5):1–38.
Xu Z, Langie SA, De Boever P, Taylor JA, Niu L. RELIC: a novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genomics. 2017;18(1):4.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–96.
Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics (Oxford, England). 2007;8(1):118–27.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Wills AK, Chinchwadkar MC, Joglekar CV, Natekar AS, Yajnik CS, Fall CH, Kinare AS. Maternal and paternal height and BMI and patterns of fetal growth: the Pune Maternal Nutrition Study. Early Hum. Dev. 2010;86(9):535–40.
Imai K, Keele L, Tingley D. A general approach to causal mediation analysis. Psychol. Methods. 2010;15(4):309–34.
Horikoshi M, Beaumont RN, Day FR, Warrington NM, Kooijman MN, Fernandez-Tajes J, Feenstra B, van Zuydam NR, Gaulton KJ, Grarup N, et al. Genome-wide associations for birth weight and correlations with adult disease. Nature. 2016;538(7624):248–52.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11(4):e1004219.
Philibert RA, Beach SR, Lei MK, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5(1):19.
Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015;7:113.
de Vries M, Heijink IH, Gras R, den Boef LE, Reinders-Luinge M, Pouwels SD, Hylkema MN, van der Toorn M, Brouwer U, van Oosterhout AJ, et al. Pim1 kinase protects airway epithelial cells from cigarette smoke-induced damage and airway inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;307(3):L240–51.
Mitchell AC, Leak RK, Zigmond MJ, Cameron JL, Mirnics K. Gene transcripts associated with BMI in the motor cortex and caudate nucleus of calorie restricted rhesus monkeys. Genomics. 2012;99(3):144–51.
Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer. 2011;11(1):23–34.
Buchner DA, Geisinger JM, Glazebrook PA, Morgan MG, Spiezio SH, Kaiyala KJ, Schwartz MW, Sakurai T, Furley AJ, Kunze DL, et al. The juxtaparanodal proteins CNTNAP2 and TAG1 regulate diet-induced obesity. Mammalian genome : official journal of the International Mammalian Genome Society. 2012;23(7-8):431–42.
Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. European journal of human genetics : EJHG. 2014;22(2):171–8.
Scott R, Sanchez-Aguilera A, van Elst K, Lim L, Dehorter N, Bae SE, Bartolini G, Peles E, Kas MJH, Bruining H, et al. Loss of Cntnap2 Causes Axonal Excitability Deficits, Developmental Delay in Cortical Myelination, and Abnormal Stereotyped Motor Behavior. Cerebral cortex. New York: NY; 1991. p. 2017.
Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, Lin C, Duggan P, Tassone P, Mansoor A, et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. 2011;117(23):6202–13.
Bohlin J, Andreassen BK, Joubert BR, Magnus MC, Wu MC, Parr CL, Haberg SE, Magnus P, Reese SE, Stoltenberg C, et al. Effect of maternal gestational weight gain on offspring DNA methylation: a follow-up to the ALSPAC cohort study. BMC research notes. 2015;8:321.
Adkins RM, Tylavsky FA, Krushkal J. Newborn umbilical cord blood DNA methylation and gene expression levels exhibit limited association with birth weight. Chem Biodivers. 2012;9(5):888–99.
Chen M, Baumbach J, Vandin F, Rottger R, Barbosa E, Dong M, Frost M, Christiansen L, Tan Q. Differentially Methylated Genomic Regions in Birth-Weight Discordant Twin Pairs. Ann. Hum. Genet. 2016;80(2):81–7.
Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA, Butte NF. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One. 2012;7(12):e51954.
Valeri L, Reese SL, Zhao S, Page CM, Nystad W, Coull BA, London SJ. Misclassified exposure in epigenetic mediation analyses. Does DNA methylation mediate effects of smoking on birthweight. Epigenomics. 2017;9(3):253–65.
Acknowledgements
We are very grateful for the essential contributions of the midwives, and would like to thank all families for their participation.
Funding
This work was supported by the a Era-Net Neuron grant to M.R., M.D., and M.Lt., the German Research Foundation [DFG; grant FOR2107; RI908/11-1 to M.R.; WI3429/3-1 to S.H.W., and NO246/10-1 to M.M.N.], the German Federal Ministry of Education and Research (BMBF) through the Integrated Network IntegraMent, under the auspices of the e:Med Programme [01ZX1314G to M.R., and 01ZX1314A to M.M.N.] and by a grant of the Dietmar-Hopp Foundation. M.D. served as PI in phase II and III studies of Johnson & Johnson, Lilly and Roche. He participated in advisory boards of Bristol-Myers Squibb, Lundbeck and Otsuka and received speaker fees of Bristol-Myers Squibb, Janssen, Lundbeck, Mundipharma and Otsuka. The remaining authors declare no conflicts of interest.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due legal restrictions but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
SHW conceived the study and was responsible for its design, interpreted the data, and drafted the manuscript. JF performed epigenetic and statistical analyses, interpreted the data, and drafted the manuscript. MG was responsible for recruitment and data acquisition. MLg performed epigenetic and statistical analyses. JT performed epigenetic and statistical analyses. FS performed epigenetic and statistical analyses. IACW was responsible for recruitment and data acquisition. VP was responsible for recruitment and data acquisition. BS was responsible for recruitment and data acquisition. TSS was responsible for recruitment and data acquisition. SHH performed epigenetic and statistical analyses. SS performed epigenetic and statistical analyses. HD was responsible for biobanking and sample management. JS was responsible for recruitment and data acquisition. MS was responsible for recruitment and data acquisition. JA performed epigenetic and statistical analyses. MLt conceived the study and was responsible for its design. MMN was responsible for analysis and interpretation of data. MD conceived the study and was responsible for its design and data interpretation. MR conceived the study and are responsible for its design, and data interpretation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All data were managed according to the World Medical Association’s Declaration of Helsinki. The study was approved by the Medical Ethics Committee II, Heidelberg University (Medical Faculty Mannheim) and all participating mothers provided written informed consent.
Competing interests
MD served as PI in phase II and III studies of Johnson & Johnson, Lilly and Roche. He participated in advisory boards of Bristol-Myers Squibb, Lundbeck and Otsuka and received speaker fees of Bristol-Myers Squibb, Janssen, Lundbeck, Mundipharma and Otsuka. The remaining authors declare no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Text. The Supplementary Text includes supplementary information with respect to methods (mediation analysis), a description of the differentially methylated genes, Figure S1. (Mediation Modell according to Baron & Kenny, 1986), Figure S2. (Birth weight dependent on maternal smoking status), Table S1. (General characteristics of study sample), Table S2. (Single marker p-values of the SNPs at the ITGB7 locus), and Table S3. (Top differentially methylated CpGs (FDR<0.05; adjusted BMI instead of parental height)). (DOCX 76 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Witt, S.H., Frank, J., Gilles, M. et al. Impact on birth weight of maternal smoking throughout pregnancy mediated by DNA methylation. BMC Genomics 19, 290 (2018). https://doi.org/10.1186/s12864-018-4652-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4652-7