
Abstract
Background
Cryptophytes are an ecologically important group of algae comprised of phototrophic, heterotrophic and osmotrophic species. This lineage is of great interest to evolutionary biologists because their plastids are of red algal secondary endosymbiotic origin. Cryptophytes have a clear phylogenetic affinity to heterotrophic eukaryotes and possess four genomes: host-derived nuclear and mitochondrial genomes, and plastid and nucleomorph genomes of endosymbiotic origin.
Results
To gain insight into cryptophyte mitochondrial genome evolution, we sequenced the mitochondrial DNAs of five species and performed a comparative analysis of seven genomes from the following cryptophyte genera: Chroomonas, Cryptomonas, Hemiselmis, Proteomonas, Rhodomonas, Storeatula and Teleaulax. The mitochondrial genomes were similar in terms of their general architecture, gene content and presence of a large repeat region. However, gene order was poorly conserved. Characteristic features of cryptophyte mtDNAs included large syntenic clusters resembling α-proteobacterial operons that encode bacteria-like rRNAs, tRNAs, and ribosomal protein genes. The cryptophyte mitochondrial genomes retain almost all genes found in many other eukaryotes including the nad, sdh, cox, cob, and atp genes, with the exception of sdh2 and atp3. In addition, gene cluster analysis showed that cryptophytes possess a gene order closely resembling the jakobid flagellates Jakoba and Reclinomonas. Interestingly, the cox1 gene of R. salina, T. amphioxeia, and Storeatula species was found to contain group II introns encoding a reverse transcriptase protein, as did the cob gene of Storeatula species CCMP1868.
Conclusions
These newly sequenced genomes increase the breadth of data available from algae and will aid in the identification of general trends in mitochondrial genome evolution. While most of the genomes were highly conserved, extensive gene arrangements have shuffled gene order, perhaps due to genome rearrangements associated with hairpin-containing mobile genetic elements, tRNAs with palindromic sequences, and tandem repeat sequences. The cox1 and cob gene sequences suggest that introns have recently been acquired during cryptophyte evolution. Comparison of phylogenetic trees based on plastid and mitochondrial genome data sets underscore the different evolutionary histories of the host and endosymbiont components of present-day cryptophytes.
Similar content being viewed by others


Background
The cryptophyte algae are an evolutionarily significant unicellular eukaryotic lineage inhabiting marine, brackish, and freshwater environments [1, 2]. Cryptophytes are comprised of photosyntheticic, heterotrophic and osmotrophic species [3,4,5,6,7,8,9]; phototrophs contain plastids with chlorophyll-a and -c, as well as phycobilins as accessary pigments. Cryptophytes have a clear phylogenetic affinity to heterotrophic eukaryotes, including goniomonads, kathablepharids and Palpitomonas bilix, which collectively have been proposed to comprise a monophyletic assemblage known as Cryptista [10,11,12]. Cryptophyte cells contain four genomes: host-derived nuclear and mitochondrial genomes, and plastid and nucleomorph genomes of endosymbiotic origin. Given this unusual feature, cryptophytes provide direct evidence for secondary endosymbiosis, a process whereby a photoautotrophic eukaryote is engulfed by a phagotrophic eukaryote [13, 14]. Secondary endosymbiosis has given rise to photosynthetic groups in other protist lineages as well (e.g., euglenoids and chlorarachniophytes from green-algal derived secondary endosymbioses, and cryptophytes haptophytes, stramenopiles, and dinoflagellates, whose plastids are of red-algal origin).
Relatively little is known about mitochondrial diversity in cryptophytes and their non-photosynthetic relatives. Considering eukaryotes as a whole, mitochondrial genomes vary significantly in size, gene content, and genome organization [15,16,17]. The least derived mitochondrial genomes known are found in an obscure protist group called jakobids and harbor up to 100 genes (including protein-coding genes and genes for noncoding RNA molecules) [18], whereas the mitochondrial genomes of Myzozoa (a subgroup of alveolates) code for only three proteins [19]. In terms of genome architecture, the most common structure is a contiguous circular-mapping DNA molecule, but a contiguous linear genome or genomes segmented into multiple circular or linear molecules are known from some taxa [15, 19]. Although considerable progress has been made in exploring the diversity and evolution of mitochondrial genomes across the full breadth of eukaryote phylogeny, representative genome sequences are still limited or altogether lacking for a large number of eukaryotic lineages, especial protists [16]. Hence, further sampling is necessary to get a full picture of the evolutionary history of mitochondria and their genomes.
To date, only two cryptophyte mitochondrial genomes have been sequenced, those of Rhodomonas salina and Hemiselmis andersenii [20, 21]. More recently, the mitochondrial genome of Palpitomonas bilix, a member of the Cryptista, was sequenced [22]. Here we present the sequences of five mitochondrial genomes belonging to the following organisms: the blue/green-colored cryptophyte Chroomonas placoidea, the brown-colored Cryptomonas curvata and the red-colored Proteomonas sulcata, Storeatula sp. CCMP1868 and Teleaulax amphioxeia. We performed a comparative analysis of genome structure and gene re-arrangements and investigated the phylogeny of mitochondrial genes relative to nuclear and plastid genes. Our results provide important insights into the broad evolution of organelle genomes and fine-scale dynamics within cryptophytes.
Results
General features of cryptophyte mitochondrial genomes
Mitochondrial genomes (mtDNAs) were sequenced and analyzed from representatives of three different colored cryptophytes: the red-coloured Proteomonas, Rhodomonas, Storeatula and Teleaulax, the green-coloured Chroomonas and Hemiselmis, and the brown-colored Cryptomonas species (Table 1). The mitochondrial genomes of two species, Storeatula sp. CCMP1868 and Teleaulax amphioxeia, could not be completely assembled, due to the presence of complex repeat regions (Fig. 1), as seen previously (e.g., in the haptophyte Phaeocystis; [23]). All of the newly sequenced cryptophyte mitochondrial genomes were found to have a highly repetitive non-coding region (Table 1), as previously described in two other cryptophyte mitochondrial genomes [20, 21]. The mitochondrial genomes ranged in size from ~ 37 Kbp (Cryptomonas curvata) to ~ 54.5 Kbp (Storeatula sp. CCMP 1868), not including repeat regions (Table 1). They share a core set of 2 rRNAs, 25~ 28 tRNAs and 42 protein-coding genes, including 18 components of the respiratory chain, 5 ATP synthase subunits, 16 ribosomal proteins and 2 subunits of the tat translocase (Tables 2, 3). Notably, the mitochondrion-encoded rps1 and tatA genes in cryptophytes were previously found to be present exclusively in jakobids and/or malawimonads, believed to possess the most ‘primitive’ mitochondrial genomes known [18]. Minor variation was found in tRNA gene content (Table 2). For instance, trnK(UUU) is present in three red-colored cryptophytes, R. salina, Storeatula sp. CCMP1868, and T. amphioxeia, but not Proteomonas sulcata, while the blue/green-colored Ch. placoidea and H. andersenii and the brown-colored Cr. curvata have a unique isotype trnG(GCC) (Table 2).

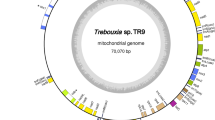
Circular map of the mitochondrial genome of seven cryptophytes. The protein coding genes, rRNA and tRNA genes (single letter) are labeled outside the circle. The genes are color-coded according to the functional categories in the index
Further consideration of mitochondrial genome structure revealed that while all genes were located on the same strand in the Chroomonas placoidea, Hemiselmis andersenii and Proteomonas sulcata genomes, some genes were located on the opposite strand in Cryptomonas curvata (31 genes), Storeatula sp. CCMP1868 (41 genes), Teleaulax amphioxeia (39 genes), and Rhodomonas salina (53 genes) (Figs. 1, 2, Table 1).

Gene content and arrangement of cryptophyte mitochondrial genomes. Twelve synthetic blocks (A–L) and two rRNAs are described relative to a phylogenetic tree of cryptophytes. Putative gene rearrangements are indicated with numbers (1–23). Tandem repeat regions are marked with red triangles. The most conserved syntenic blocks among cryptophytes are marked; * = 100%, and † = broken by one gene. The hairpin structures with palindromic sequences are marked blue (hairpin symbol). The tRNAs are coded with single letters and tRNAs with palindromic sequences are highlighted red
Genome re-arrangements
The most evident feature of the cryptophyte mitochondrial genomes sequenced herein was that, although gene content is very stable, gene order is highly variable. The 42 protein-coding genes were arranged together and analyzed in the context of 12 syntenic blocks including rRNA and tRNA genes, each block consisting of 2–6 genes (Fig. 2). These blocks were as follows: A) cox1-cob-nad11, B) atp8-atp4-rps4-atp9, C) nad3-rps2, D) tatC-nad7, E) cox2-tatA-cox3-rpl12-rpl7-rps19, F) rps3-rpl16-rpl14-rpl5-rps14, G) nad8-nad6-nad1, H) sdh3-sdh4, I) nad4L-nad5-nad4-nad2, J) atp1-rps1-orf166, rpl31, K) rps8-rpl6-rps13-rps11-atp6, L) nad10-nad9, and 2 rRNAs (Fig. 2). Two syntenic blocks were found to be common to all cryptophyte mitochondrial genomes (Fig. 2, J and L).
To assess the extent of mitochondrial genome rearrangements more closely, genomes were aligned using the Mauve genome aligner (Additional file 1: Figure S1). Synteny was broadly conserved within the three differently colored cryptophyte lineages. Overall, we documented 23 instances of mitochondrial genome rearrangements, suggesting that extensive scrambling has occurred since the evolutionary split of these species (Fig. 2 and Additional file 1: Figure S1). Comparing Proteomonas sulcata with the three other red-colored cryptophyte mitochondrial genomes, 14 gene-order rearrangements were detected (Fig. 2 ①–⑭). The genes in the mitochondrial genome of Teleaulax amphioxeia appear to have been rearranged via transposition of synteny block C (①), an inversion of a set of consecutive genes in blocks G to L (②), and a combination of transposition and inversion involving synteny block H (③). Genes in the mitochondrial genome of Rhodomonas salina were rearranged with transposition of nad3 and rps2 from broken block C (in a manner different than that seen for T. amphioxeia) (④, ⑤), transposition of block D–E (⑥), an inversion of a set of consecutive genes in blocks G to J (⑦), and transposition of atp6 within block K (⑧). The mitochondrial genome of Storeatula sp. CCMP1868 was rearranged with transposition of synteny block G–L (⑨), transposition of tatC and nad7 from a broken block D (⑩), an inversion of block E (⑪), an inversion of block F (⑫), inversion of block B with rRNAs and nad11 being split from block A (⑬), and an inversion of block C (⑭).
In terms of mitochondrial gene order, the brown-colored cryptophyte Cryptomonas curvata is more similar to the red-colored lineage than to the blue-greens. Relative to the genome of the red-colored Proteomonas sulcata, four gene-order rearrangements were detected (Fig. 2 ⑮–⑱): a combination of an inversion and break in block E–F (⑮–⑯), an inversion of a string of genes in blocks G to L (⑰), and a combination of inversion and transposition block C (⑱). The gene order of blue/green-colored cryptophytes is very different from the other groups. Between the two blue/green-colored cryptophytes Hemiselmis andersenii and Chroomonas placoidea, five gene-order rearrangements were detected (Fig. 2 ⑲– ): transposition of G and partial I block (⑲), transposition of rps4 (⑳) and atp9 (
): transposition of G and partial I block (⑲), transposition of rps4 (⑳) and atp9 ( ) from broken block B, transposition of sdh4 from broken block H (
) from broken block B, transposition of sdh4 from broken block H ( ), and transposition of cox2 break free from the block E (
), and transposition of cox2 break free from the block E ( ). Clearly there have been extensive mitochondrial genome rearrangements in each of these lineages during cryptophyte evolution.
). Clearly there have been extensive mitochondrial genome rearrangements in each of these lineages during cryptophyte evolution.
Palindromic sequences and large non-coding regions with tandem repeats
We found between 3 and 20 A-T rich palindromic sequences in the intergenic regions of each of the mitochondrial genomes of cryptophytes (Fig. 3, Additional files 2 and 3: Figures S2 and S3), similar to those reported previously in the large non-coding regions of Rhodomonas salina [20] and Hemiselmis andersenii [21]. Interestingly, between 4 and 9 tRNA genes are present at the palindromic endpoints (Additional file 3: Figure S3).

Representative tRNA palindromic sequences in the mitochondrial genome of Proteomonas sulcata. The non-tRNA sequences are marked with blue and palindromic sequences are marked with purple. All hairpin structures or tRNAs with palindromic sequences in cryptophtye mitochondrial genome are illustrated in Additional file 2: Figure S2 and Additional file 3: Figure S3
Cryptophyte mtDNA structure also appears to have been impacted by the presence of tandem repeats in the large non-coding region, with individual repeats ranging from 1.1Kbp (P. sulcata) to 19.7Kbp (H. andersenii), each of which contains multi-copy palindromic sequences [20, 21] (Figs. 1 and 2 marked as triangle). The repeats are inverted in the genomes of R. salina, Storeatula species CCMP1186 and T. amphioxeia, but in the other four completely assembled genomes, large non-coding regions are instead dispersed or arranged in tandem throughout the large non-coding region (Table 1).
Ribosomal proteins and bacterial-like operons
Ribosomal protein gene clusters have been suggested to represent vestiges of bacterial operons in the cryptophyte mitochondrial genome [20]. Our comparisons of mitochondrial gene data provide further support for this idea. The ribosomal protein clusters rps3-rpl16-rpl14-rpl5-rps14 and rps8- rpl6-rps13-rps11 in Ch. placoidea, H. andersenii, Cr. curvata, R. salina and P. sulcata exhibit the same relative gene order as in the operons of the bacteria Escherichia coli and Rickettsia prowazekii; this arrangement is also seen (with little variation) in the gene-rich mtDNAs of several other protists (Jakoba libera, Acanthamoeba catellanii, Phytophthora infestans, and the green alga Nephroselmis olivacea) as well as the liverwort M. polymorpha (Additional file 4: Figure S4). However, the bacterial-operon cluster rps8- rpl6-rps13-rps11 was inverted in the cryptophytes Storeatula sp. CCMP 1868 and T. amphioxeia (Additional file 4: Figure S4). Furthermore, the other conserved ribosomal protein gene cluster, rps12-rps7-rps19, is translocated in the mitochondrial genome of Storeatula sp. CCMP1868.
Phylogeny
An ML tree was reconstructed using 4257 amino acids from 16 representative genes conserved in the mtDNAs of all chlorophyll-c containing algal groups (Fig. 4), as well as diverse photosynthetic and non-photosynthetic lineages from across the eukaryotic tree. The sequences of dinoflagellates and euglenoids were not included in this analysis due to the limited available data of mitochondrial genes. A monophyletic cryptophyte clade was strongly supported (MLB = 100%) and showed well-resolved internal relationships. The four red-colored taxa (i.e., Storeatula, Rhodomonas, Teleaulax and Guillardia) group together while the brown-colored Cryptomonas curvata forms a sister relationship with the blue/green-colored Chroomonas and Hemiselmis species (Fig. 4). Although our taxon sampling was limited, these results are consistent with earlier studies. In particular, the red-colored cryptophyte species (Rhodomonas, Storeatula, Teleaulax, and Proteomonas) form a monophyletic lineage in both mitochondrial and plastid phylogenomic studies to the exclusion of other cryptophytes ([24], this study), in contrast to single gene analyses of nuclear SSU rDNA [6, 8].

Phylogenetic tree of cryptophyte mitochondrial proteins. The tree was constructed using a dataset of 16 concatenated proteins (4257 amino acids). The numbers on each node represent RAxML bootstrap values. The scale bar indicates the inferred number of substitutions/site
Discussion
Mitochondrial DNA gene content was found to be essentially identical amongst all cryptophytes examined, although gene order rearrangements were detected. Similar mtDNA architectures, including gene-dense regions, a single large repetitive intergenic region and absolute strand polarity, are seen in the stramenopile Thraustochytrium aureum and the green algae Pedinomonas minor and Pycnococcus provasolii [25, 26]. Structurally speaking, the mitochondrial genomes of haptophycean algae are similar to those of cryptophytes, and show variation of strand polarity within the group. For example, absolute strand polarity is seen in the mtDNAs of Chrysochromulina tobin and Emiliania huxleyi but not in Pavlova lutheri (32/49 genes) and Phaeocystis globosa (40/45 genes) [23, 27, 28].
Interestingly, the mitochondrial genomes of various other major algal lineages have also undergone extensive rearrangements. These include the green algae [29, 30], red algae [31], Eustigmatophyceae [32], haptophytes [28], and chlorarachniophytes [33]. The significant structural variation seen in cryptophyte mitochondrial genomes is interesting when compared to the plastid genomes of these same organisms, where gene order was recently shown to be nearly identical [24].
Palindromic sequences and repeat structures relative to recombination sites
The fact that palindromic sequences are found concentrated near the ends of syntenic blocks in cryptophyte mitochondrial genomes suggests that they have played a role in mediating genomic rearrangements. In fungal mtDNAs, double-hairpin elements have been suggested to act as mobile DNA elements and to mediate lateral gene transfer [34, 35]. In any case, a notable consequence of the presence of such repeats is that they facilitate genome rearrangements [36,37,38,39]. A number of possible mechanisms have been proposed. Inversions may occur in a specific location due to the presence of short repeat elements subject to homologous recombination [40, 41]. Even in un-rearranged plastid genomes, small inversions regularly occur in intergenic areas, caused by short inverted repeats forming hairpins that can easily flip the orientation of the intervening sequences [42, 43]. Such a mechanism could explain the highly shuffled genes seen in the cryptophytes mitochondrial genomes described herein (Fig. 2). In addition, tRNA genes associated with gene re-arrangement breakpoints have been reported in plastid genomes [41, 44]. It has been suggested that tRNA genes at inversion endpoints may be due to the presence of short repeats within or near the tRNA genes in the highly rearranged chloroplast genome of the charophyte Chaetospheridium globosum [45], as well as in the flowering plant Trachelium [46].
The direct repeat arrangements we observed are strikingly similar to the larger (35 kb) repeat structure found in the mitochondrial genome of the diatom Phaeodactylum tricornutum [47]. The haptophytes Chrysochromulina tobin, Emiliania hyxleyi, Phaeocystis antarctica, Phaeocystis globosa and Pavlova lutheri, and the chlorophytes Pedinomonas minor and Acutodesmus obliquus also contain large tandem repeat regions (>4 kb) in their genomes [23, 25, 27, 28, 48]. In animal mitochondrial genomes, the tandem repeats are located in the control region and help explain the evolutionary origins of tandem repeats among species, populations and even individuals [49, 50]. However, in most algal mitochondrial genomes the tandem repeat manifests itself in a pattern that is species-specific. The repeat sequences within the mitochondrial genomes of cryptophytes described herein do not retain obvious sequence or structural similarity across species bounds.
Gene content of cryptophyte mitochondrial genomes
The most gene-rich mitochondrial genomes reported to date are those of the excavate group Jakobida, with 61–69 protein-coding genes and 30–34 RNAs [18]. Cryptophyte mitochondrial genomes are also noteworthy in retaining genes that are not found in most other eukaryotes (Fig. 5). The mtDNAs of cryptophytes contain 42 conserved protein genes (excluding non-conserved ORFs in each genome), more than found in glaucophytes (30 CDSs), Bacillariophyceae (34 CDSs), Eustigmatophyceae (36 CDSs), Phaeophyceae (34 CDSs), Rhaphidophyceae (34 CDSs), rhodophytes (22 CDSs), haptophytes (22 CDSs), Synurophyceae (32 CDSs), chlorarachniophytes (26 CDSs) and green algae (11–39 CDSs). Most of the cryptophyte mitochondrial genes are associated with electron transport systems belonging to a set of five complexes, as summarized below.

Venn diagram comparing gene content among the mitochondrial genomes of chlorophyll-c containg algae. Only genes/proteins with predicted functions are included. Total protein gene numbers are indicated for each group (parentheses). The few genes found in specific subgroups are colored: light-blue in Cyanidiophyceae, green in Chryso/Bacillario/Raphiophyceae, light-green in Eustigmatophyceae, purple in the genus Pavlova (Haptophyta). The 16 genes conserved among all groups used to construct phylogenetic trees are highlighted red
The genes encoding certain subunits of NADH dehydrogenase (complex I; nad7, nad8, nad9, nad10, and nad11) are only rarely found in mtDNA. The nad7, nad9 and nad11 genes are missing from all haptophyte and rhodophyte mitochondrial genomes. On the other hand, these three genes are present in all cryptophytes, Palpitomonas, stramenopiles and jakobid mitochondrial genomes (Fig. 5). Comparison of the mtDNA gene order in red-/brown-colored cryptophytes reveals the presence of three NADH dehydrogenase gene clusters (cluster G: nad8-nad6-nad1, cluster I: nad4L-nad5-nad4-nad2, and cluster L: nad10-nad9). However, two clusters (G and I) were separated into one or two genes, respectively (G: nad8-nad1 and nad6, cluster I: nad4L-nad5 and nad4-nad2), in blue/green-colored cryptophytes (Fig. 2).
The succinate dehydrogenase (complex II) is made up of four protein subunits; two are hydrophilic and belong to the catalytic portion of the complex (sdh1 and sdh2 genes) and the other two, encoded by the sdh3 and sdh4 genes, are hydrophobic and act as anchors to the entire complex [51, 52]. The sdh2, sdh3, and sdh4 genes are sometimes found in mitochondrial genomes, whereas sdh1 is transferred to the nuclear genome and its protein product is imported from the cytosol. The sdh2 gene is present in Rhodophyta except in Galdieria sulphuraria [31]. Many other algae have also lost the sdh2 gene from their mitochondrial genomes. Mitochondrial genes for two subunits of succinate dehydrogenase (sdh3 and sdh4) are present in only a few green algae, the liverwort Marchantia polymorpha, several red algae [31, 53], all jakobid flagellates including Reclinomonas americana [54, 55] and all known cryptophytes ([20, 21], this study).
The genes for subunits of the cytochrome bc 1 complex (complex III) and cytochrome c oxidase (complex IV) are present in the mtDNAs of all cryptophytes and most algae. The cob and cox1 genes are always together in all cryptophyte genomes sequenced thus far (Fig. 2A). The cox2 and cox3 genes are found together in almost all cryptophytes, the exception being the cox2 gene in Hemiselmis andersenii, which is separated from cluster E (Fig. 2E). Interestingly, the cox1 gene of Storeatula sp. CCMP1868, Teleaulax amphioxeia, and Rhodomonas salina was found to contain a putative group II intron with a coding region for an intron encoded protein (IEP). In Chroomonas placoidea, the cox1 gene was split into two parts with 13 genes in between. The first exon encodes a maturase sequence and is located between the nad8-trnD and atp8 genes. The cob gene in Storeatula sp. CCMP1868 also has an intron with an IEP (Additional file 5: Figure S5). These introns belong to group II, which often harbor three distinct protein-coding regions corresponding to reverse transcriptase, maturase, and C-terminal DNA binding domains [56]. The Storeatula sp. intron coding regions show similarity to an IEP in the atpA gene in the mitochondrial genome of the liverwort Marchantia polymorpha [57].
The mtDNAs of jakobid flagellates contain the biggest complement of ATP synthase genes (atp1, 3, 4, 6, 8, 9). Three atp genes (atp6, 8, 9) are present in the mitochondrial genomes of green algae, glaucophytes, haptophytes and stramenopiles (Fig. 5). The atp1 and atp4 genes are present in the mitochondrial genome of green algae, jakobids and cryptophytes. The heterolobosean Naegleria fowleri and Tsukubamonas globose (a member of the Tsukubamonadidae) have atp3 in their mtDNA but lack atp4. All cryptophytes and Palpitomonas bilix possess most of the atp gene set (atp1, 4, 6, 8, 9) with the exception of atp3 ([22], this study).
The two genes for subunits of the twin-arginine protein translocation system transporters (encoded by the tatA and tatC genes) are generally conserved in diverse prokaryotes as well as in plastids [58]. The tatC homologs are present in some mitochondrial genomes [59], and the mitochondrial tatA gene was thought to be limited to the jakobids [60]. However, a recent review of algal mitochondrial genomes [61] noted the presence of tatA in some eukaryotic groups, including diatoms, raphidophytes, chrysophytes and cryptophytes; these genes are found in our newly sequenced cryptophyte genome as well (Fig. 5).
Phylogenetic relationships
Curiously, our 16-protein mitochondrial genome phylogeny shows a strong monophyletic relationship between cryptophytes and rhodophytes. This is unexpected given that the cryptophyte mitochondrion is derived from the host component of the original secondary endosymbiotic partnership that gave rise to modern-day cryptophytes. Based on the monophyletic relationship between chlorophyll-c containing groups and red algae seen in many (but not all) plastid genome phylogenies, the hypothesis that a single secondary endosymbiotic uptake of a red alga in a common ancestor of all ‘chromist’ algae has been explored [24, 62,63,64,65,66,67,68,69,70]. However, recent large-scale studies using nuclear genome data show topologies that are incongruent with this hypothesis. The occurrence of multiple secondary and serial endosymbioses has been proposed to reconcile the apparent incongruence between host and endosymbiont-associated phylogenies (e.g., [12, 69, 71,72,73,74]). We examined phylogenies inferred from each of the single genes/proteins used in our multi-gene tree, and could see no consistent signal for a relationship between cryptophytes and rhodophytes to the exclusion of other eukaryotic groups (Additional file 6: Figure S6). In only one of our protein trees (cob) did the cryptophytes branch with Palpitomonas bilix, to which it is clearly related on the basis of nuclear multi-gene trees (e.g., [10, 12]). Clearly more mitochondrial genome data are needed from plastid-lacking lineages that are related to cryptophytes, such as goniomonads and kathablepharids. Nevertheless, our mitochondrial genome-based phylogeny is consistent with previously published phylogenies, including a 250 nuclear gene dataset [12], in suggesting that the cryptophytes and haptophytes are not specifically related to one other. The monophyly of haptophytes + Opisthokonta/Amoebozoa + Stramenopiles/Rizaria + other relatives is supported by nuclear and mitochondrial analyses ([12], this study).
Conclusions
We have sequenced mitochondrial genomes from five cryptophyte algae with a wide range of color pigmentation: the red-colored Proteomonas sulcata, Storeatula sp. CCMP1868 and Teleaulax amphioxeia, the blue/green-colored Chroomonas placoidea, and the brown Cryptomonas curvata. These newly sequenced genomes will aid in the identification of general trends in mitochondrial genome evolution, not just within cryptophytes but between cryptophytes and their closest plastid-lacking relatives such as goniomonads and kathablepharids once these sequence data become available. While most of the cryptophyte genomes we have sequenced are highly conserved with respect to coding capacity, extensive gene arrangements have shuffled gene order. Such rearrangements appear to have been mediated by palindromic sequences, tRNAs, and/or repeat regions. This pattern lies in stark contrast to the high degree of synteny seen in the plastid genomes of these very same organisms, underscoring the dramatic differences in the tempo and mode of organellar genomes of different evolutionary history, even when they reside within the same cell.
Methods
DNA isolation and sequencing
Cultures of Chroomonas placoidea CCAP 978/8 and Proteomonas sulcata CCMP 705 were obtained from the Culture Collection of Algae and Protozoa (CCAP), whereas Storeatula species CCMP 1868 came from the National Center for Marine Algae and Microbiota (NCMA). Teleaulax amphioxeia collected from Gomso Bay, Korea (35° 40′ N, 126° 40′ E) was established as clonal cultures from single-cell isolates and the strain is available from the Culture collection at the Chungnam National University, Korea. Cryptomonas curvata collected from Cheongyang pond, Korea (36° 30′ N, 126° 47′ E) was established as clonal cultures from single-cell isolates and the strain Cryptomonas curvata FBCC 300012D is available from the Freshwater Bioresources Culture Collection at the Nakdong-gang National Institute of Biological Resources Korea. All cultures were grown in AF-6 medium [75] with distilled water for the freshwater strain (Cr. curvata) or distilled seawater for marine strains, and were maintained at 20 °C under conditions of a 14:10 light:dark cycle with 30 μmol photons·m− 2·s− 1 from cool white fluorescent tubes. DNA was extracted using the QIAGEN DNEasy Blood Mini Kit (QIAGEN, Valencia, CA, USA) as per manufacturer’s instructions. Next-generation sequencing (NGS) was carried out using the Ion Torrent PGM platform (Thermo Fisher Scientific, San Francisco, California, USA). Sequencing libraries were prepared using the Ion Xpress™ Plus gDNA Fragment Library Preparation Kit for 200 bp or 400 bp-sized sequencing library prearation and the Ion OneTouch™ 200 or 400 Template Kit (Thermo Fisher Scientific, San Francisco, CA, USA) according to the manufacturer’s protocol. Genomes were sequenced on an Ion Torrent Personal Genome Machine (PGM) using the Ion PGM sequencing 200 or 400 Kit (Thermo Fisher Scientific, San Francisco, CA, USA). On the MiSeq (Illumina, San Diego CA), the amplified DNA was fragmented and tagged using the NexteraXT protocol (Illumina), indexed, size selected, and pooled for sequencing using the small amplicon targeted resequencing run, which performs paired end 2 × 300 bp sequencing reads using the MiSeq Reagent Kit v3 (Illumina), according to the manufacturer’s recommendations.
Genome assembly and annotation
The raw reads obtained from both NGS platforms (i.e., Ion Torrent and Illumina MiSeq) were trimmed separately using the following settings: base = 80 bp, error threshold = 0.05, n ambiguities = 2. Assemblies were also carried out separately. For the Ion Torrent data, the assembly was carried out using MIRA4 (http://mira-assembler.sourceforge.net/docs/DefinitiveGuideToMIRA.html), whereas SPAdes 3.10 (http://bioinf.spbau.ru/spades) was used for Illumina data. For each genome, the two assemblies were compared and the most ‘complete’ assembly was chosen. The mitochondrial origin of the assembled contigs was verified according to the following criteria and Jung et al. [76]: (i) reads were mapped onto the final consensus contig for each cryptophyte mitochondrial genome using Bowtie2 (similarity = 95%, length fraction = 75%; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) with preset options (sensitive-local: -D 15 -R 2 -N 0 -L 20 -i s,1,0.75); (ii) BLAST searches using commonly known mitochondrial genes against the entire assembly produced hits to these contigs; and (iii) genome sizes consistent with those of other photosynthetic cryptophyte mitochondrial genomes were obtained.
Protein coding genes as well as rRNA and tRNA genes were compiled from all previously sequenced cryptophyte mitochondrial genomes. Preliminary annotation of protein coding genes was performed using GeneMarkS (http://opal.biology.gatech.edu/GeneMark/). The final annotation file was checked in Geneious Pro 10.2.2 (http://www.geneious.com/) using the ORFfinder with genetic code 4 (Protozoan Mitochondrial Code). Predicted ORFs were checked manually and annotated accordingly.
Transfer RNA genes were identified using the tRNAscan-SE version 2.0 server (http://lowelab.ucsc.edu/tRNAscan-SE/) with default settings and the “Mold/Protozoan Mito” model. The rRNA genes were identified by BLASTn searches against a set of known, previously published mitochondrial rRNA sequences of cryptophytes. To determine intron types, mitochondrial genomes were submitted to the RNAweasel server (http://megasun.bch.umontreal.ca/cgi-bin/RNAweasel/RNAweaselInterface.pl). Physical maps were designed with the OrganellarGenomeDRAW program (http://ogdraw.mpimp-golm.mpg.de/).
Palindromic sequence elements were detected using the EMBOSS explorer (http://emboss.bioinformatics.nl/cgi-bin/emboss/palindrome) with the following parameters: 8 for minimum length of palindromes, 100 for maximum length of palindromes, 10 for maximum gap between elements, and no mismatches within the palindrome. To find palindromic sequences in tRNAs, the maximum gap between elements was set at 100 bp (due to the normal tRNA gene length of less 90 bp). Some detected sequences were manually excluded if a loop region was longer than a stem region. Genome sequences were deposited into the NCBI GenBank database under the following accession numbers (Table 1).
Gene arrangement comparisons
Two previously published cryptophyte mitochondrial genome sequences were downloaded from GenBank [20, 21]. For structural and synteny comparisons, the genomes were aligned using the Mauve Genome Alignment tool version 2.2.0 [77] with default settings. For the purposes of visualization, we arbitrarily designated the beginning of the cox1 gene as position 1 in each genome (pointing in the direction of cob).
Phylogenetic analysis
Phylogenetic trees were constructed from datasets created by combining amino acid sequences corresponding to 16 protein coding genes from 131 mitochondrial genomes, including those of seven cryptophytes, five haptophytes, 38 stramenopiles, and 36 red algae (Additional file 7: Table S1). The 16 genes are as follows: atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, rps12, and rpl16. The dataset was concatenated (4257 amino acid positions) and final refinements were performed by eyes in the MacGDE2.5 program [78].
Maximum likelihood (ML) phylogenetic analyses were performed using RAxML version 8.0.0 [79] with the Le and Gascuel gamma (LG + GAMMA) model [80] for amino acid data chosen by ProtTest 3 [81]. We used 1000 independent tree inferences using the -# option to identify the best tree. The model parameters with gamma correction values and the proportion of invariable sites in the combined dataset were obtained automatically by the RAxML program. Bootstrap support values (MLBS) were calculated using 1000 replicates with the same substitution model.
Abbreviations
- CDS:
-
Coding sequences
- IEP:
-
Intron encoded protein
- ML:
-
Maximum likelihood
- MLB:
-
Maximum likelihood bootstrap
- mtDNAs:
-
mitochondrial genomes
- NGS:
-
Next-generation sequencing
- ORF:
-
Open reading frame
- PGM:
-
Personal Genome Machine
References
Graham LK, Wilcox LW. The origin of alteration of generations in land plants: a focus on matrotrophy and hexose transport. Philo Trans R Soc Lond B Biol Sci. 2000;255:757–66.
Shalchian-Tabrizi K, Minge MA, Espelund M, Orr R, Ruden T, et al. Multigene phylogeny of choanozoa and the origin of animals. PLoS One. 2008;3:e2098.
Hill DRA, Rowan KS. The biliproteins of the Cryptophyceae. Phycologia. 1989;28:455–63.
McFadden GI, Gilson PR, Hill DRA. Goniomonas: rRNA sequences indicate that that this phagotrophic flagellate is a close relative to the host component of cryptomonads. Eur J Phycol. 1994;29:29–32.
Hoef-Emden K, Marin B, Melkonian M. Nuclear and nucleomorph SSU rDNA phylogeny in the Cryptophyta and the evolution of cryptophyte diversity. J Mol Evol. 2002;55:161–79.
Deane JA, Hill DRA, Brett SJ, McFadden GI. Cryptomonad evolution: nuclear 18S rDNA phylogeny versus cell morphology and pigmentation. J Phycol. 2002;38:1236–44.
von der Heyden S, Chao E, Cavalier-Smith T. Genetic diversity of goniomonads: an ancient divergence between marine and freshwater species. Eur J Phycol. 2004;39:343–50.
Hoef-Emden K. Molecular phylogeny of phycocyanin-containing cryptophytes: evolution of biliproteins and geographical distribution. J Phycol. 2008;44:985–93.
Donaher N, Tanifuji G, Onodera NT, Malfatti SA, Chain PS, et al. The complete plastid genome sequence of the secondarily nonphotosynthetic alga Cryptomonas paramecium: reduction, compaction, and accelerated evolutionary rate. Genome Biol Evol. 2009;1:439–48.
Yabuki A, et al. Palpitomonas bilix represents a basal cryptist lineage: insight into the character evolution in Cryptista. Sci Rep. 2014;4:4641.
Cavalier-Smith T, Chao EE, Lewis R. Multiple origins of Heliozoa from flagellate ancestors: new cryptist subphylum Corbihelia, superclass Corbistoma, and monophyly of Haptista, Cryptista, Hacrobia and Chromista. Mol Phylogenet Evol. 2015;93:331–62.
Burki F, Kaplan M, Tikhonenkov DV, Zlatogursky V, Minh BQ, Radaykina LV, Smirnov A, Mylnikov AP, Keeling PJ. Untangling the early diversification of eukaryotes: a phylogenomic study of the evolutionary origins of Centrohelida, Haptophyta and Cryptista. Proc R Soc B. 2016;283:20152802.
Douglas SE, Murphy CA, Spencer DF, Gray MW. Cryptomonad algae are evolutionary chimaeras of two phylogenetically distinct unicellular eukaryotes. Nature. 1991;350:148–51.
McFadden GI. Second-hand chloroplasts: evolution of cryptomonad algae. In: Callow JA, editor. Advances in botanical research. London: Academic Press Limited; 1993. p. 189–230.
Gray MW, Lang BF, Burger G. Mitochondria of protists. Ann Rev Genet. 2004;38:477–524.
Smith DR. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct Genomics. 2016;15:47–54.
Chen Z, Zhao N, Li S, Grover CE, Nie H, Wendel JF, Hua J. Plant mitochondrial genome evolution and cytoplasmic male sterility. Crit Rev Plant Sci. 2017;36:55–69.
Burger G, Gray MW, Forget L, Lang BF. Strikingly bacteria-like and gene-rich mitochondrial genomes throughout Jakobid protists. Genome Biol Evol. 2013;5:418–38.
Burger G, Jackson CJ, Waller RF. Unusual mitochondrial genomes and genes. In: Bullerwell C, editor. Organelle genetics. New York: Springer; 2012. p. 41–77.
Hauth AM, Maier UG, Lang BF, Burger G. The Rhodomonas salina mitochondrial genome: bacteria-like operons, compact gene arrangement and complex repeat region. Nucleic Acids Res. 2005;33:4433–42.
Kim E, Lane CE, Curtis BA, et al. Complete sequence and analysis of the mitochondrial genome of Hemiselmis andersenii CCMP644 (Cryptophyceae). BMC Genomics. 2008;9:215–27.
Nishimura Y, Tanifuji G, Kamikawa R, Yabuki A, Hashimoto T, Inagaki Y. Mitochondrial genome of Palpitomonas bilix: derived genome structure and ancestral system for cytochrome c maturation. Genome Biol Evol. 2016;13:3090–8.
Smith DR, Arrigo KR, Alderkamp AC, Allen AE. Massive difference in synonymous substitution rates among mitochondrial, plastid, and nuclear genes of Phaeocystis algae. Mol Phylogenet Evol. 2014;71:30–40.
Kim JI, Moore CE, Archibald JM, Bhattacharya D, Yi G, Yoon HS, Shin W. Evolutionary dynamics of cryptophyte plastid genomes. Genome Biol Evol. 2017;9:1859–72.
Turmel M, Lemieux C, Burger G, Lang BF, Otis C, Plante I, Gray MW. The complete mitochondrial DNA sequences of Nephroselmis olivacea and Pedinomonas minor: two radically different evolutionary patterns within green algae. Plant Cell. 1999;11:1717–29.
Turmel M, Otis C, Lemieux C. A deviant genetic code in the reduced mitochondrial genome of the picoplaktonic green alga Pycnococcus provasolii. J Mol Evol. 2010;70:203–14.
Sánchez Puetra MV, Bachvaroff TR, Delwiche CF. The complete mitochondrial genome sequence of the haptophyte Emiliania huxleyi and its relation to heterokonts. DNA Res. 2004;11:1–10.
Hovde BT, Starkenburg SR, Hunsperger HM, Mercer LD, Deodato CR, Jha RK, et al. The mitochondrial and chloroplast genomes of the haptophyte Chrysochromulina tobin contain unique repeat structures and gene profiles. BMC Genomics. 2014;15:604. PMID:25034814
Liu Y, Xue JY, Wang B, Li L, Qiu YL. The mitochondrial genomes of the early land plants Treubia lacunose and Anomodon rugelii: dynamic and conservative evolution. PLoS One. 2011;6:e25836.
Farwagi A, Fučíková K, McManus HA. Phylogenetic patterns of gene rearrangements in four mitochondrial genomes from the green algal family Hydrodictyaceae (Sphaeropleales, Chlorophyceae). BMC Genomics. 2015;16:826.
Yang E, et al. Highly conserved mitochondrial genomes among multicellular red algae of the Florideophyceae. Genome Biol Evol. 2015;7:2394–406.
Ševčíková T, Zbránková Z, Klimeš V, Strnad H, Hroudová M, Vlček Č, Eliáš M. A comparative analysis of mitochondrial genomes in eustigmatophyte algae. Genome Biol Evol. 2016;8:705–22.
Tanifuji G, Archibald JM, Hashimoto T. Comparative genomics of mitochondria in chlorarachniophyte algae: endosymbiotic gene transfer and organellar genome dynamics. Sci Rep. 2016;6:21016.
Paquin B, Laforest MJ, Lang BF. Double-hairpin elements in the mitochondrial DNA of Allomyces: evidence for mobility. Mol Biol Evol. 2000;17:1760–8.
Bullerwell CE, Leigh J, Forget L, Lang BF. A comparison of three fission yeast mitochondrial genomes. Nucleic Acids Res. 2003;31:759–68.
Nedelcu AM, Lee RW. Short repetitive sequences in green algal mitochondrial genomes: potential roles in mitochondrial genome evolution. Mol Biol Evol. 1998;15:690–701.
Bartoszewski G, Katzir N, Harvey MJ. Organization of repetitive DNAs and the genomic regions carrying ribosomal RNA, cob, and atp9 genes in the cucurbit mitochondrial genomes. Theor Appl Genet. 2004;108:982–92.
Beaudet D, et al. Mitochondrial genome rearrangements in Glomus species triggered by homologous recombination between distinct mtDNA haplotypes. Genome Biol Evol. 2013;5:1628–43.
Kamikawa R, Shiratori T, Ishida K, Miyashita H, Roger AJ. Group II intron-mediated trans-splicing in the gene-rich mitochondrial genome of an enigmatic eukaryote, Diphylleia rotans. Genome Biol Evol. 2016;8:458–66.
Palmer JD. Plastid chromosomes: structure and evolution. In: Bogorad L, Vasil IK, editors. Cell culture and somatic genetics of plant, vol. 7A. San Diego: Molecular Biology of Plastids, Academic Press; 1991. p. 5–53.
Knox EB, Downie SR, Palmer JD. Chloroplast genome rearrangements and the evolution of giant lobelias from herbaceous ancestors. Mol Biol Evol. 1993;10:414–30.
Kelchner SA, Wendel JF. Hairpins create minute inversions in non-coding regions of chloroplast DNA. Curr Genet. 1996;30:259–62.
Kelchner SA. The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann Mo Bot Gard. 2000;87:482–98.
Hoot SB, Palmer JD. Structural rearrangements, including parallel inversions, within the chloroplast genome of Anemone and related genera. J Mol Evol. 1994;38:274–81.
Turmel M, Otis C, Lemieux C. The complete mitochondrial DNA sequence of Mesostigma viride identifies this green alga as the earliest green plant divergence and predicts a highly compact mitochondrial genome in the ancestor of all green plants. Mol Biol Evol. 2002;19:24–38.
Haberle RC, Fourcade HM, Boore JL, Jansen RK. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J Mol Evol. 2008;66:350–61.
Oudot-Le Secq M-P, Green BR. Complex repeat structures and novel features in the mitochondrial genomes of the diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana. Gene. 2011;476:20–6.
Nedelcu AM, Lee RW, Lemieux C, Gray MW, Burger G. The complete mitochondrial DNA sequence of Scenedesmus obliquus reflects an intermediate stage in the evolution of the green algal mitochondrial genome. Genome Res. 2000;10:819–31.
Lunt DH, Whipple LE, Hyman BC. Mitochondrial DNA variable number tandem repeats (VNTRs): utility and problems in molecular ecology. Mol Ecol. 1998;7:1441–55.
Casane D, Dennebouy N, de Rochambeau H, Mounolou JC, Monnerot M. Nonneutral evolution of tandem repeats in the mitochondrial DNA control region of lagomorphs. Mol Biol Evol. 1997;14:779–89.
Elorza A, et al. Nuclear SDH2-1 and SDH2-2 genes, encoding the iron-sulfur subunit of mitochondrial complex II in Arabidopsis, have distinct cell-specific expression patterns and promoter activities. Plant Physiol. 2004;136:4072–87.
Bayley JP, Devilee P, Taschner PE. The SDH mutation database: an online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med Genet. 2005;6:39.
Salomaki ED, Lane CE. Red algal mitochondrial genomes are more complete than previously reported. Genome Biol Evol. 2017;9:48–63.
Lang BF, Burger G, Okelly CJ, Cedergren R, Golding GB, et al. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. 1997;387:493–7.
Burger G, Gray MW, Lang BF. Mitochondrial genomes: anything goes. Trends Genet. 2003;19:709–16.
Zimmerly S, Hausner G, Wu X. Phylogenetic relationships among group II intron ORFs. Nucleic Acids Res. 2001;29:1238–50.
Oda K, Yamato K, Ohta E, Nakamura Y, et al. Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA. A primitive form of plant mitochondrial genome. J Mol Biol. 1992;223:1–7.
Berks BC. The twin-arginine protein translocation pathway. Annu Rev Biochem. 2015;84:843–64.
Bogsch EG, et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem. 1998;273:18003–6.
Jacob Y, Seif E, Paquet P-O, Lang BF. Loss of the mRNA-like region in mitochondrial tmRNAs of jakobids. RNA. 2004;10:605–14.
Burger G, Nedelcu AM. Mitochondrial genomes of algae. In: Bock R, Knoop V, editors. Genomics of chloroplasts and mitochondria (advances in photosynthesis and respiration). Dordrecht: Springer; 2012. p. 127–57.
Cavalier-Smith T. Principles of protein and lipid targeting in secondary symbiogenesis: euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryotic family tree. J Eukaryot Microbiol. 1999;46:347–66.
Yoon HS, Hackett JD, Bhattacharya D. A single origin of the peridinin—and fucoxanthin-containing plastids in dinoflagellates through tertiary endosymbiosis. Proc Natl Acad Sci U S A. 2002a;99:11724–9.
Yoon HS, Hackett JD, Pinto G, Bhattacharya D. The single, ancient origin of chromist plastids. Proc Natl Acad Sci U S A. 2002b;99:15507–12.
Keeling PJ. Diversity and evolutionary history of plastids and their hosts. Am J Bot. 2004;91:1481–93.
Archibald JM, Keeling PJ. In: Saap J, editor. On the origin and evolution of plastids. Microbial phylogeny and evolution. New York: Oxford University Press; 2005. p. 238–60.
Bhattacharya D, Yoon HS, Hackett JD. Photosynthetic eukaryotes unite: endosymbiosis connects the dots. BioEssays. 2004;26:50–60.
Bhattacharya D, Archibald JM, Weber APM, Reyes-Prieto A. How do endosymbionts become organelles? Understanding early events in plastid evolution. BioEssays. 2007;29:1239–46.
Archibald JM. The puzzle of plastid evolution. Curr Biol. 2009;19:R81–8.
Kim JI, Yoon HS, Yi G, Kim HS, Yih W, Shin W. The plastid genome of the cryptomonad Teleaulax amphioxeia. PLoS One. 2015;10:e0129284.
Baurain D, Brinkmann H, Petersen J, Rodríguez-Ezpeleta N, Stechmann A, et al. Phylogenomic evidence for separate acquisition of plastids in cryptophytes, haptophytes, and stramenopiles. Mol Biol Evol. 2010;27:1698–709.
Burki F, Okamoto N, Pombert J-F, Keeling PJ. The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc Biol Sci. 2012;279:2246–54.
Stiller JW, Schreiber J, Yue J, Guo H, Ding Q, et al. The evolution of photosynthesis in chromist algae through serial endosymbioses. Nat Commun. 2014;5:5764.
Archibald JM. Genomic perspectives on the birth and spread of plastids. Proc Natl Acad Sci U S A. 2015;112:10147–53.
Watanabe MM, Hiroki M. NIES-collection list of strains. 5th ed. Tsukuba: National Institute for Environmental Studies; 1997. p. 127.
Jung J, Kim JI, Jeong Y-S, Yi G. A robust method for finding the automated best matched genes based on grouping similar fragments of large-scale references for genome assembly. Symmetry. 2017;9:192.
Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–403.
Smith SW, Overbeek R, Woese CR, Gilbert W, Gillevet PM. The genetic data environment: an expandable GUI for multiple sequence analysis. Comput Appl Biosci. 1994;10:671–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–20.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5.
Acknowledgements
The authors thank Jaehee Jung for bioinformatic assistance and two anonymous reviewers for their helpful comments.
Funding
This research was supported by the National Research Foundation (NRF) of Korea funded by the Ministry of Science, ICT & Future Planning, Basic Science Research Program (MSIP; NRF-2013R1A1A3012539) and the Ministry of Education (2015R1D1A1A01057899) to JIK; NRF (2017R1A2B3001923) and the Collaborative Genome Program (20140428) funded by the Ministry of Oceans and Fisheries, Korea to HSY; NRF (2016R1D1A1A09919318), and the Dongguk University Research Fund of 2016 to GY; NRF (MSIP; 2015R1A2A2A01003192 and 2015M1A5A1041808) to WS; and an operating grant from the Natural Sciences and Engineering Research Council of Canada to JMA.
Availability of data and materials
The mitochondrial genome datasets in this project has been deposited in the NCBI GenBank database under the accession numbers MG680941, MG680942, MG680943, MG680944, MG680945. The sequence alignments used for phylogenetic analyses have been deposited in the Treebase repository (accession number 22274). Permission to sample from Gomso Bay and Cheongyang pond was obtained from the Korean government. The new strains described in this study are available from the corresponding author or Culture Collections.
Author information
Authors and Affiliations
Contributions
JIK, HSY, WS, JMA conceived and designed the experiments: JIK, GY performed the experiments and analyzed the data: JIK, HSY, WS, JMA wrote the manuscript: All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Overview of cryptophyte mitochondrial genomes. Linearized maps of the five novel complete mitochondrial genomes are compared to those from previous studies. The color coded syntenic blocks are shown above each genome, and the gene maps are shown below. The syntenic blocks above the horizontal line are on the same strand, and those below the line are on the opposite strand. The horizontal bars inside the syntenic blocks show sequence conservation. The block boundaries correspond to sites where inversion events have occurred. In the gene maps, the genes above the horizontal line are transcribed from left to right, and those below the horizontal line are transcribed from right to left. The rRNA genes are shown in red. (PDF 1860 kb)
Additional file 2:
Figure S2. Hairpin structures wih palindromic sequence in cryptophyte mitochondrial genomes. The non-tRNA sequences are marked with blue and palindromic sequences are marked with purple. (PDF 897 kb)
Additional file 3:
Figure S3. All tRNAs wih palindromic sequences in cryptophyte mitochondrial genomes. The non-tRNA sequences are marked with blue and palindromic sequences are marked with purple. (PDF 1644 kb)
Additional file 4:
Figure S4. Conservation of ribosomal protein gene organization. Gene order found in cryptophyte mitochondrial genomes compared with that of the contiguous bacterial str, S10 spec and alpha operons of Escherichia coli and Rickettsia prowazekii. (PDF 548 kb)
Additional file 5:
Figure S5. Group II introns in cryptophyte cox1 and cob mitochondrial genes. (PDF 352 kb)
Additional file 6:
Figure S6. Phylogenetic trees inferred from single genes. The trees were constructed using amino acid sequences of 16 genes: atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, rps12, and rpl16. The numbers on each node represent ultrafast bootstrap approximation (UFBoot) using IQ-Tree. The scale bars indicate the number of substitutions/site. (PDF 2672 kb)
Additional file 7:
Table S1. The concatenated data set of 16 protein coding genes for phylogenetic reconstruction. (XLSX 22 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, J.I., Yoon, H.S., Yi, G. et al. Comparative mitochondrial genomics of cryptophyte algae: gene shuffling and dynamic mobile genetic elements. BMC Genomics 19, 275 (2018). https://doi.org/10.1186/s12864-018-4626-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4626-9