
Abstract
Background
White striping (WS) is an emerging muscular defect occurring on breast and thigh muscles of broiler chickens. It is characterized by the presence of white striations parallel to the muscle fibers and has significant consequences for meat quality. The etiology of WS remains poorly understood, even if previous studies demonstrated that the defect prevalence is related to broiler growth and muscle development. Moreover, recent studies showed moderate to high heritability values of WS, which emphasized the role of genetics in the expression of the muscle defect. The aim of this study was to identify the first quantitative trait loci (QTLs) for WS as well as breast muscle yield (BMY) and meat quality traits using a genome-wide association study (GWAS). We took advantage of two divergent lines of chickens selected for meat quality through Pectoralis major ultimate pH (pHu) and which exhibit the muscular defect. An expression QTL (eQTL) detection was further performed for some candidate genes, either suggested by GWAS analysis or based on their biological function.
Results
Forty-two single nucleotide polymorphisms (SNPs) associated with WS and other meat quality traits were identified. They defined 18 QTL regions located on 13 chromosomes. These results supported a polygenic inheritance of the studied traits and highlighted a few pleiotropic regions. A set of 16 positional and/or functional candidate genes was designed for further eQTL detection. A total of 132 SNPs were associated with molecular phenotypes and defined 21 eQTL regions located on 16 chromosomes. Interestingly, several co-localizations between QTL and eQTL regions were observed which could suggest causative genes and gene networks involved in the variability of meat quality traits and BMY.
Conclusions
The QTL mapping carried out in the current study for WS did not support the existence of a major gene, but rather suggested a polygenic inheritance of the defect and of other studied meat quality traits. We identified several candidate genes involved in muscle metabolism and structure and in muscular dystrophies. The eQTL analyses showed that they were part of molecular networks associated with WS and meat quality phenotypes and suggested a few putative causative genes.
Similar content being viewed by others

Background
During the past few decades, a notable increase in the demand for poultry meat has been observed due to its convenience for cooking and processing, health benefits, and low price. To meet market demand and the world population increase, producers have had to increase their production while reducing the costs. Consequently, the production of broiler chickens has become more efficient mainly thanks to genetic selection. By comparing two genetic strains representative of the broilers being grown in 1957 or in 2001, Havenstein et al. [1] showed that changes over these 44 years resulted in a 2001 broiler that required approximately one-third the time and over a threefold decrease in the amount of feed consumed to reach the market weight of 1,8 kg. Genetic selection brought about 85% to 90% of the change that has occurred in broiler growth rate [1]. Genetics was also the major contributor to changes in the yield of carcass parts that continued to increase over time especially for breast meat yield which currently exceeds one fifth of the weight of the bird [2, 3]. Nevertheless, for nearly a decade now, the poultry industry in many countries (i.e. Italy, the United States, Brazil, the United Kingdom, Finland, France, etc.) has witnessed an increasing prevalence of broiler breast muscle abnormalities such as WS [4].
WS is characterized by white striations parallel to muscle fibers, mainly on breast, but sometimes on thigh and tender muscle of broilers. Histological observations of white-striped meat indicate an increase in degenerative fibers associated with regeneration phenomenon (nuclear rowing, multiple internationalized nuclei), variation in muscle fiber size, mononuclear cell infiltration, adiposis, and fibrosis [5]. This myopathic pattern has significant consequences for meat quality. WS fillets have a higher fat content and a lower protein content than normal fillets, resulting in meat of reduced nutritional quality [6,7,8,9,10]. They also exhibit higher cooking loss (CL), as a result of a lower water holding capacity (WHC), and reduced tenderness [10,11,12,13]. In addition to altering the sensory and technological properties of meat, the severity of WS has negative effects on breast fillet appearance and therefore on consumer purchases [14]. Furthermore, meat affected by breast muscle myopathies can be downgraded or in some cases condemned, leading to serious economic losses. This muscular defect could also result in social acceptability issues regarding animal welfare and meat quality [15].
WS etiology remains poorly understood even if previous studies demonstrated that the prevalence of this defect is directly related to broiler growth performances [6, 11, 16, 17]. Bailey et al. provided the first estimates of genetic parameters of chicken breast muscle myopathies (WS, wooden breast, deep pectoral myopathy) recorded in a high-yielding line or a moderate-yielding line [18]. WS was the most common myopathy in both chicken lines. It was also the most heritable one, with heritability values ranging from 0.19 to 0.34. Although WS incidence was significantly higher in the high-yielding line than in the moderate-yielding line (49,6% vs 14,5%), the estimated genetic correlations with body weight (BW) and breast muscle yield (BMY) remained low to moderate (0.06 to 0.23) in the two studied lines [18]. The second study conducted by Alnahhas et al. showed a strong genetic basis of WS (h2 = 0.65) measured in two divergent lines of broilers selected for meat quality through Pectoralis major pHu [19]. A high genetic correlation between WS and BMY was reported (rg = 0.68) while it was moderate with BW [19]. Difference in magnitude of genetic parameters exist between the two studies (related to differences of genetic backgrounds or of methods of estimation), but they both indicate a role of genetic factors in the expression of WS. However, no information is currently available regarding the genomic architecture of this trait.
The aim of this GWAS was to identify QTL for WS as well as BMY and meat quality traits by taking advantage of the two divergent lines of broilers selected for Pectoralis major pHu and which exhibit the muscular defect. A previous study showed that there is a higher incidence of WS in line with high pHu value [19]. One hypothesis is that the depletion of glycogen reserves observed in the high pHu line would be a predisposing metabolic environment for WS development.
After the first step of QTL mapping, an eQTL detection was performed for a few positional and/or functional candidate genes. This made it possible to better understand the associated molecular mechanisms, and provided candidate genes and markers that could be used in breeding programs to reduce the prevalence of WS and, more generally, to improve meat quality in broilers.
Results and discussion
Descriptive statistics
The descriptive statistics describing the distribution of carcass composition and meat quality traits of the 558 broilers used for the current GWAS are shown in Table 1. A logarithmic transformation was applied to the thiobarbituric acid-reactive substance index (TBA-RS), a marker of lipid peroxidation, in order to normalize its distribution. A chi2 homogeneity test was performed to characterize the distribution of normal fillets, fillets that were moderately affected, and fillets severely affected by WS defect between lines (pHu + and pHu-). The proportion of fillets with each degree of WS severity was not the same in both lines (p = 2.6 × 10− 15). There was a higher proportion of normal fillets in the pHu- line than in the pHu + line (57.3% vs 32.4%, respectively) and incidence of severe WS was higher in the pHu + line than in the pHu- line (27.7% vs 4.3%, respectively) (p = 7.5 × 10− 16).
These results are in agreement with previous studies reporting that meat with myopathy is often characterized by a higher pHu value than normal meat [11, 12, 19,20,21,22].
QTL detection
White striping
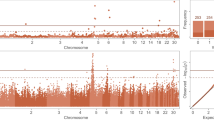
The GWAS performed on the whole population created from the two divergent lines identified three SNPs that were significantly associated with WS at the chromosome level and located on GGA1, GGA17, and GGA18 (Fig. 1, Table 2). This finding was not in favor of a monogenic inheritance of WS since several QTLs seemed to be involved in addition to the polygenic effect included in the model (see Methods section). Moreover, no co-localization with SNPs previously identified for pHu in the same genetic population was observed [23], which is not in favor of common genetic variants between the two traits. In order to explore whether pHu could indirectly influence WS, intra-line QTL detection of WS was performed (Additional file 1: Figure S1). No significant SNPs were detected in the pHu- line, while eight SNPs were significant at the chromosome threshold in the pHu + line (Table 3). They confirmed the two regions previously detected on GGA1 and GGA17 in the entire population and identified a new one on GGA20. The SNP identified with the lowest p-value was also the most significant SNP associated with WS in the entire population (Gga_rs13899127 on GGA1). Five SNPs significant at the GGA1 threshold co-localized with this SNP. Furthermore, we recovered the same SNP on GGA17 as the one associated with WS in the entire population (Gga_rs15804842).

Manhattan plot showing the association of SNPs with WS. Black line represents the 5% genome-wide threshold, red line the 5% GGA1-wide threshold, blue line the 5% GGA17 and GGA18-wide threshold
For this SNP, the minor allele frequency (MAF) was similar in both lines (MAF = 0.446 and MAF = 0.478 in the pHu + and pHu- lines, respectively), but the SNP effect was higher in the pHu + line than in the pHu- line (β = − 3.2 × 10− 1 and β = − 6.5 × 10− 2, respectively). This illustrated that unless no co-localization was observed between WS and pHu QTLs, there might be an interaction between the genetic control of WS and the metabolic and physiological status of the two lines.
Body composition and meat quality traits
Three SNPs were significant at the genome threshold, while 39 SNPs were significant at the chromosome level for body composition and meat quality traits (Table 2). These SNPs were mostly associated with CL and drip loss (DL), color parameters, and Pectoralis major and minor yields (PMY and PmY, respectively). They defined 15 QTL regions distributed over 11 chromosomes. As for WS, these results strongly suggested a polygenic inheritance of the studied carcass and meat quality parameters.
More than one third of the detected SNPs were located on GGA4 and defined two QTL regions (QTL3, QTL4). The first one (QTL3) was defined by a unique SNP (GGaluGA263381 at 65.97 Mb) which controlled both PMY and BMY. Interestingly, this SNP was also close to significance for WS (p = 3.5 × 10− 5, not shown in the table). The second one (QTL4) (90.82 to 91.20 Mb) contained three genome-wide significant SNPs for CL as well as several chromosome-wide significant SNPs for DL, lightness (L*) and yellowness (b*) of the meat. There were two common SNPs between DL, CL, L* and b* (Gga_rs1654745 at 91.09 Mb and Gga_rs14506759 at 91.11 Mb) and one more common SNP between CL and DL at 91.20 Mb (Gga_rs15648179). As illustrated in Fig. 2, the QTL4 region appeared to be a pleiotropic region controlling both the WHC (through DL and CL), and the meat color (through L* and b*). Although strong genetic correlations have already been reported between these parameters and the meat pHu in chickens [24, 25], no QTL for pHu was found nearby [23]. Similarly, we did not observe any co-localization with QTL of WS, which can also impair WHC and meat color [11].

Manhattan plots showing the pleiotropic region on GGA4. Manhattan plots of CL (a), DL (b), L* (c) and b* (d). Black line represents the 5% genome-wide threshold and orange line the 5% GGA4-wide threshold
Two additional pleiotropic regions were evidenced (QTL6, QTL15), one for DL and CL on GGA5 (20.55 to 20.97 Mb), and the other, for DL and b* on GGA24 (4.33 Mb).
A significant positive genetic relationship between WS and intramuscular fat content (IMF) had previously been reported in this population [19], suggesting that IMF could be a useful quantitative predictor of WS. However, no significant SNPs were detected for IMF in the current study, maybe because this trait was controlled by many genes with too small effects. In the same way, no QTLs were found for the measurements of TBA-RS index, a marker of lipid peroxidation.
Identification of candidate genes
WS phenotype
Identification of genomic regions involved in the control of WS is an opportunity to improve our understanding of the molecular mechanisms underlying the appearance of this defect. For that, we looked for candidate genes within several regions of interest. They comprised the three significant WS-QTL regions (on GGA1, GGA17, and GGA18), the GGA4 region for which a pleiotropic effect with BMY was suggested, and two additional suggestive regions for which SNPs (Gga_rs16576992 at 4.51 Mb on GGA7 and GGaluGA071697 at 16.43 Mb on GGA10) close to significance were detected (p = 7.52 × 10− 5 and 6.60 × 10− 5 respectively).
Using an R software package (Gviz) [26], we visualized the gene environment around these different regions of interest (Additional file 2: Figure S2, Additional file 3: Figure S3, Additional file 4: Figure S4, Additional file 5: Figure S5 and Additional file 6: Figure S6). Potential candidate genes were selected based on their proximity with a significant SNP and/or their biological function (Table 4). Most of the selected genes are involved in muscular structure and processes related to muscle fiber regeneration and repair (i.e., MYH15, MYH1E, MYH1B, MYH1F, MYH13, MYOCD), adiposis and fibrosis (PDGFRα), extracellular matrix or sarcolemma composition (i.e., COL6A3, FN1, SGCB), muscle metabolism (PNPLA7), and human neuromuscular disorders (i.e., FN1, COL6A3, SGCB, LRSAM1).
The Myosin Heavy chain 15 gene (MYH15) located in the region of interest on GGA1 (Additional file 2: Figure S2) is involved in the contraction, development, and regeneration of avian skeletal muscles [27]. The most significant SNP associated with WS is located in the gene itself.
The Platelet Derived Growth Factor Receptor Alpha gene (PDGFRα), which is located upstream of the SNP of interest for WS and BMY on GGA4 (Additional file 3: Figure S3), is known to be a marker of fibro-adipogenic precursors (FAPs) recently characterized as playing a role in muscle regeneration and repair. Under pathological conditions like muscular dystrophy, FAPs differentiate into fibroblasts and adipocytes, leading to fat and connective tissue infiltration. In addition, FAPs promote myoblast differentiation in co-cultivation experiments [28]. The Sarcoglycan Beta gene (SGCB) is another gene of interest for the GGA4 region (Additional file 3: Figure S3) as it codes for the beta sarcoglycan protein, a member of the sarcoglycan complex located to the sarcolemma. This subcomplex of the dystrophin-associated glycoprotein complex (DAG complex) contributes to muscle tissue structure maintenance and stability and transfer of mechanical strength all along the sarcolemma during muscle contraction. Mutations in this gene were identified in human limb-girdle (LGMD1E) muscular dystrophy [29]. It has been shown that transgenic beta-sarcoglycan-deficient mice exhibited progressive muscular dystrophy with extensive degeneration and regeneration of muscle fibers [30]. These mice also exhibited muscular hypertrophy and whitish stripes within the muscles [30]. This makes SGCB an interesting positional and functional candidate gene for the QTL3 region, for which a pleiotropic effect on WS and BMY was suggested.
The Fibronectin 1 gene (FN1) is the closest gene to the WS QTL region on GGA7 (Additional file 4: Figure S4) and encodes a glycoprotein, which contributes to the creation of fibers and extracellular matrix during tissue repair after degeneration. In addition, an increase in fibronectin levels is a biomarker of Duchenne muscular dystrophy [31]. The Collagen type VI Alpha 3-chain gene (COL6A3) is another interesting gene for this region (Additional file 4: Figure S4). It produces collagen molecules found in the extracellular matrix and surrounding cells that make up the muscles used for movement. Mutations in this gene are associated with Bethlem myopathy and Ullrich congenital myopathy [32].
One candidate gene in the region of interest on GGA17 (Additional file 5: Figure S5) is the Leucine-rich Repeat and Sterile Alpha Motif containing 1 gene (LRSAM1). It encodes an ubiquitin-protein ligase with a role in sorting internalized cell-surface receptor proteins. Mutations in the LRSAM1 gene have been shown to cause an axonal form of Charcot-Marie-Tooth hereditary neuropathy (CMT2P), characterized by progressive muscle weakness and atrophy [33]. A second candidate gene, Patatin-like Phospholipase domain containing 7 gene (PNPLA7), is an insulin-regulated lysophospholipase expressed in muscle and fat. The regulation of its expression by nutritional status and insulin suggests a role in the catabolism of lipid precursors and/or mediators that affect energy metabolism in mammals [34]. Furthermore, another member of the patatin family, PNPLA2, is involved in neutral lipid storage disease associated with myopathy that is characterized by the accumulation of triglycerides in multiple tissues [35]. The last candidate gene of this region, Tubulin Beta-4B chain (TUBB4B), codes for a protein (tubulin) which is the major constituent of microtubules. Microtubules are major constituents of the cytoskeleton and have major roles in maintenance of the cell structure and intracellular transport.
Finally, in addition to MYH15 on GGA1, four fast myosin heavy chain isoforms (i.e. MYH1E, MYH1B, MYH1F, and MYH13) and myocardin (MYOCD) were selected. As shown in Additional file 6: Figure S6, these genes are grouped within a cluster located at the beginning of GGA18 (0.35 to 0.93 Mb), near the QTL region associated with WS (2.77 Mb). Out of all the groups of myosins, we chose to focus on them because they are fairly well annotated. Furthermore, a previous histological study on the same genetic lines highlighted a greater expression of embryonic (MYH1B) and neonatal (MYH1F) myosin heavy chain isoforms and a lower expression of adult myosin heavy chain (MYH1E) in the pHu + than in the pHu- muscle, which is consistent with a stronger muscle fiber regeneration process [36]. In addition, a transcriptomic study on the same model showed differential expression of MYH13 and MYOCD genes between normal and WS fillets [37].
Other meat quality phenotypes
Because of its high level of significance and pleiotropic effect on CL, DL, L* and b*, we looked for candidate genes within the QTL4 region (Table 2). Visualization of the gene environment showed that the dysferlin (DYSF) gene (91.21–91.27 Mb) is close to the QTL region (90.82 to 91.20 Mb) (Additional file 7: Figure S7). The dysferlin gene encodes a protein that is found in the sarcolemma. It is involved in sarcolemma repair and some results suggest that dysferlin may also be involved in the formation of new muscle fibers (regeneration) and inflammation [38]. Furthermore, dysferlin was identified as a gene that is mutated in limb-girdle muscular dystrophy and some congenital myopathies. Some studies demonstrated that dysferlin-null mice developed progressive muscular dystrophy caused by failure of plasma membrane repair in muscle [38, 39]. Two genes encoding for skeletal muscle proteins (i.e. caveolin 3 and calpain 3) were added to the set of genes of interest both for their interaction with the dysferlin gene [40, 41], and their involvement in human limb-girdle muscular dystrophies (LGMD1C and LGMD2A, respectively) [42, 43]. Caveolin 3 (CAV3) may play a key role in the fusion of myoblasts into myotubes during the maturation process of muscle fibers. Calpain 3 (CAPN3) is an intracellular protease specific to the muscle, involved in the calcium-dependent proteolytic system. They are known to catalyze the limited proteolysis of proteins (i.e., desmin and vimentin) involved in cytoskeletal modelling or signal transduction and to play a role in regeneration processes [44, 45].
Of the 16 candidate genes considered in the study, whether for WS or meat quality phenotypes, more than half are components of the extracellular matrix, sarcolemma, or muscles. Interestingly, several of them (i.e., COL6A3, CAV3, CAPN3, DYSF, SGCB) are associated with, or are members of the dystrophin complex. This complex contains dystrophin, which is at the origin of the genetic defect causing Duchenne muscular dystrophy (DMD) in humans, and establishes a direct link between the actin cytoskeleton and the extracellular matrix [46]. Thus, it participates in maintaining the extracellular matrix and sarcolemma. A perturbation within this complex or these associated proteins, such as a loss of the mechanical link between the cytoskeleton, extracellular matrix, and sarcolemma, may result in muscle fragility, contraction-induced damage, and necrosis [47].
eQTL detection
GWAS can be applied in order to map eQTLs that control the transcript level of genes. Co-localization between phenotypic QTLs and eQTLs is particularly useful in order to identify candidate genes, which are potentially causative [48]. In the current study, the expression of all identified candidate genes was quantified using RT-qPCR. After GWAS analysis, 132 SNPs were found to be significantly associated with molecular phenotypes including 24 SNPs significant at the genome threshold (Additional file 8: Table S1). They defined 21 eQTL regions located on 16 chromosomes.
eQTLs are categorized as cis or trans, where cis eQTLs represent a polymorphism physically located near the gene itself, for example, a promoter polymorphism that gives rise to differential expression of the gene [48]. In the current study, we defined as cis eQTLs, SNPs that were within 1 Mb of the annotated start or stop site of the corresponding structural gene. eQTLs that did not fulfill this condition were considered to be trans eQTLs. These regions could, for example, contain a polymorphism in a transcription factor that correspondingly modulates the level of transcripts for target genes [48]. Of the 21 eQTL regions, three were cis eQTLs and 18 were trans eQTLs.
Cis eQTLs
The first cis eQTL relates to LRSAM1 gene. Out of 31 significant SNPs associated with its expression, 28 were located in a region close to the gene (eQTL15) and one was in the gene itself (Gga_rs15034052 at 2.04 Mb). Eleven out of these 29 SNPs were significant at the genome threshold (Fig. 3, Additional file 8: Table S1).

Manhattan plot showing the association of SNPs with LRSAM1 expression. Black line represents the 5% genome-wide threshold and blue line the 5% GGA17-wide threshold
The second cis eQTL relates to MYH1F gene. Of the 16 SNPs significantly associated with its expression, two were significant at the genome threshold on GGA18 (Additional file 9: Figure S8, Additional file 8: Table S1). The expression of MYH1F is associated with 14 SNPs, which are close to the gene (eQTL16) and located within a region containing the cluster of myosin heavy chain isoforms.
The last cis eQTL relates to CAV3 gene. GWAS analysis identified 29 SNPs significantly associated with gene expression and which defined two eQTL regions, one cis eQTL on GGA12 (eQTL13) (Additional file 10: Figure S9) and one trans eQTL on GGA18 (eQTL17) (Additional file 8: Table S1). In contrast to the two other cis eQTLs, the CAV3 cis eQTL was significant only at the chromosome level.
Trans eQTLs
Among all trans eQTLs (Additional file 8: Table S1), four regions seem to control several molecular phenotypes. The first one is on GGA2 (eQTL3), where a single SNP is associated with the expression of PNPLA7 and DYSF genes (Gga_rs14139566 at 12.55 Mb). The second, on GGA6 (eQTL9), includes several SNPs for the MYH1B gene, and one single SNP for MYH1B, DYSF and PDGFRα (Gga_rs14581613 at 2.03 Mb). The third region is located on GGA22 (eQTL18) where two single SNPs are identified (GGaluGA186934 at 2.90 Mb and GGaluGA186952 at 2.94 Mb), for LRSAM1, PNPLA7 and TUBB4B gene expression. Finally, one last region appears to control several molecular phenotypes on GGA4 (eQTL6). This region is associated with the expression of FN1, MYH13, COL6A3, and CAPN3. Of the six SNPs significantly associated with these molecular phenotypes, half are associated with at least two gene expressions: one single SNP between FN1 and MYH13 (Gga_rs16454745 at 91.10 Mb), another between FN1 and COL6A3 (Gga_rs14506759 at 91.11 Mb), and finally a last between the four genes (Gga_rs15648179 and 91.20 Mb) (Fig. 4). This last region seems particularly interesting since it regulates the expression of three genes involved in human muscle diseases, as a biological marker (FN1) or genetic markers (COL6A3 and CAPN3).

Manhattan plot showing the association of SNPs with FN1 (a), COL6A3 (b), MYH13 (c) and CAPN3 (d) expressions. Black line represents the 5% genome-wide threshold and orange line the 5% GGA4-wide threshold
Co-localization between QTLs and eQTLs
Interestingly, several co-localizations between QTL and eQTL regions were observed on GGA4, GGA5, and GGGA17, which could suggest causative genes and gene networks involved in the variability of meat quality traits or breast meat yield.
White striping
The first co-localization concerns the region of GGA17 associated with both the expression of LRSAM1 gene and WS defect (eQTL15 and QTL12) (Additional file 8: Table S1, Additional file 11: Table S2). The level of significance was much higher for the molecular phenotype than for the WS phenotype, which suggested a tighter regulation of the gene expression by this region. Although three genes of interest (LRSAM1, PNPLA7, TUBB4B) were tested for this region, only LRSAM1 was significantly regulated and appeared to be a putative causative gene. In addition to being a positional and expressional candidate gene, LRSAM1 is also a functional candidate gene, since a mutation in this gene results in a form of hereditary motor and sensory neuropathy where slowly progressive distal muscle weakness and atrophy were observed [33].
A cis eQTL for the neonatal myosin heavy chain form MYH1F (0.30 to 0.83 Mb) was also observed near the QTL associated with WS on the region of interest on GGA18 (2.77 Mb). Embryonic (MYH1B) and neonatal (MYH1F) forms of myosin heavy chain are found in breast muscle during its early development or in muscle fibers regeneration. A characterization of the muscle fibers was previously performed in the pHu + and pHu- lines. The muscle fibers of the pHu + birds were characterized by a higher number of cells expressing the developmental embryonic and neonatal isoforms of the fast myosin heavy chain [36], as well as a higher incidence of WS than was found in pHu- muscle fibers [19]. MYH1F could be an interesting molecular marker of WS in relation to the regeneration process. However, we cannot conclude that there is a single mutation affecting this molecular phenotype and WS, since the positions of the QTL and eQTL were rather distant.
Body composition and meat quality traits
A first co-localization was found on GGA5 between PMY QTL (QTL5) and SGCB-eQTL (eQTL7) (Additional file 8: Table S1, Additional file 11: Table S2). As previously reported, mutations within this gene result in a loss or a large decrease in the sarcoglycan complex [30]. Mice carrying the mutation are hypertrophic and larger than wild-type phenotypes. No significant SNP for WS was found in this region in the current study, which did not support any role of the region in the genetic relationship observed between PMY and WS in this population [19]. Nevertheless, our results indicated that SGCB could be an interesting molecular actor to consider in future studies on the control of muscle development and integrity.
Another co-localization was found on GGA4, between the previously identified pleiotropic region (QTL4) containing the SNPs strongly associated with CL, DL, and color parameters (L* and b*), and the eQTL region controlling FN1, COL6A3, MYH13, and CAPN3 expression (eQTL6). This region seems to regulate meat quality phenotypes possibly related to muscle cell integrity and expression of genes involved in human neuromuscular disorders, as well as in the structure and composition of muscle fibers.
The QTL mapping revealed several regions of importance for the control of meat quality phenotypes. The eQTL analysis suggested some candidate genes and molecular networks associated with WS and meat quality traits. On the other hand, the eQTL analysis for MYH15, SGCB, and DYSF did not evidence any co-localizations with WS nor with meat quality traits. However, we cannot exclude an effect of these genes. They remain interesting candidate genes whose action could be modulated by mutations in the coding region, leading to variations in protein stability or enzymatic activity, or by post-translation modification [48]. This was the case for the RN gene (Rendement Napole) in pigs where a loss of function mutation in the PRKAG3 gene is associated with an excess of glycogen content in the skeletal muscles [49]; or for the MSTN gene (myostatin), for which a loss of function mutation results in muscular hypertrophy in cattle and sheep [50]. Further analyses are needed to elucidate the role of these genes in the initiation of biological processes involved in the appearance of WS defect and more generally in poultry meat quality variations.
Conclusions
This study reported the first QTLs for WS in chicken. It did not support the existence of a major gene, but was in favor of a polygenic inheritance of the defect and of the other studied meat quality traits. We identified several candidate genes involved in muscle metabolism and structure, and also in neuromuscular disorders for some of them. The eQTL analyses confirmed that they were part of molecular networks associated with WS and meat quality phenotypes and suggested a few putative causative genes. This study provides a first set of molecular and genetic markers for WS that will have to be validated and enriched by the study of additional genetic populations and the integration of molecular and genetic information.
Methods
Birds and housing
Meat pHu reflects the level of muscle glycogen content and is a determining criterion of meat quality. In this study we used two broiler lines divergently selected for Pectoralis major pHu according to a breeding scheme described in Alnahhas et al. [25] and which exhibit WS defect.
WS, body composition, and meat quality traits were evaluated in 558 birds (253 males and 305 females) from the 6th generation of divergent selection (G6). Of this total, 278 broilers (135 males and 143 females) were from the pHu + line (selected for high breast pHu value) and 280 broilers (118 males and 162 females) from the pHu- line (selected for low breast pHu value). Birds were reared as a single population (males and females from both lines), following standard rearing practices and had ad libitum access to feed and water. They were produced in two successive batches and slaughtered at 6 weeks of age at the PEAT experimental unit (INRA, Centre Val de Loire, Nouzilly, France). After hanging the birds on the processing chain, they were automatically stunned in a water bath using a constant electric current and slaughtered manually by cutting the carotid artery and the jugular vein at the ventral surface of the neck from inside the oral cavity. Samples of P. major were collected fifteen minutes after slaughter, snap-frozen in liquid nitrogen and stored at − 80 °C until further analysis. Breasts were visually graded a day after slaughter by one trained person. The categories of WS were defined according to a modified version of the scale of Kuttappan et al. [14] to account for the lower degree of severity in our experimental population (0 = normal, absence of WS, 1 = moderate white striping, corresponding to striation thickness ≤ 1 mm, and 2 = severe occurrence of white striping, corresponding to striation thickness ≥ 1 mm). Body composition was characterized for all birds through the measurement of BMY, abdominal fat percentage (AFP), PMY, PmY and thigh yield (TY) expressed in relation to body weight (BW). Breast meat quality was evaluated on Pectoralis major muscle through the measurement of color parameters (L*, a*, b*), CL, DL, Warner-Bratzler shear force (SF) of cooked meat, curing-cooking yield (CCY), IMF, and TBA-RS index. All these measurements were realized as described in Alnahhas et al. [19].
Genotyping
The 558 birds were genotyped at Labogena Laboratory (Jouy en Josas, France) using the Illumina chicken SNP 57 K Beadchip containing 57,636 SNPs. Quality control of the genotype data was performed using PLINK 1.9 and included animal call rate (> 95%), SNP call rate (> 95%), and minor allele frequency (> 5%). Finally, after filters, all animals were kept and 40,195 out of 57,636 SNPs distributed along 28 autosomal chromosomes were validated for further analyses.
Gene expression
Gene expression was determined by RT-qPCR performed on one batch of animals from the 6th generation of divergent selection (145 pHu+, 136 pHu-), as described by Beauclercq et al. [36]. Total RNA was extracted from Pectoralis major muscle sampled for each individual, using RNA NOW (Ozyme, St Quentin en Yvelines, France). Ten μg of RNA from each sample were reverse-transcribed using RNase H−MMLV reverse transcriptase (Superscript II, Invitrogen, Illkirch, France) and random primers (Promega, Charbonnières les Bains, France). Primers targeting the studied genes were designed with Primer3 version 4.0.0 [51, 52]. The list of primer sequences is available in Additional file 11: Table S2. Their products of amplification were analyzed by electrophoresis and further sequenced.
The level of mRNA expression of candidate genes was quantified using a Fluidigm Biomark microfluidic device (Fluidigm, South San Francisco, CA, USA) according to the manufacturer’s protocol (ADP37). Because MYH15 and PDGFRα genes were added to the list of candidate genes later, their expression was quantified by RT-qPCR using a Roche LightCycler® 480 II (Roche Applied Science, Penzberg, Upper Bavaria, Germany) and the Takyon® (Eurogentec, Liege, Belgium), according to the manufacturer’s recommendations. Quantitative PCR conditions were set at 95 °C for 5 min, followed by forty-five cycles of 10 s at 95 °C, 20 s at 60 °C and 10 s at 72 °C. Two references were used: PDE3B (phosphodiesterase 3B), which is invariant whatever the sex and degree of severity of WS within and between the lines, was used as the housekeeping gene to normalize the Ct values; and a mix of chicken Pectoralis major muscle cDNA. The calculation of absolute mRNA levels was based on the PCR efficiency and the threshold cycle (CT) deviation of an unknown cDNA versus the control cDNA according to the equation proposed by Pfaffl [53]. The normalized Ct values (ΔΔCt) were used as the molecular phenotype for the GWAS analyses.
QTL and eQTL detections
Genome-wide association studies were performed on classical and molecular phenotypes using an univariate linear mixed model (LMM) implemented in GEMMA (Genome-wide Efficient Mixed Model Association) software [54]. The model used for analyses was:
where y is a n-vector of phenotypes for n individuals, W the n x c matrix of covariates which contains fixed effects of sex and batch (only sex effect for eQTL detection) and including a column of 1 s for the general mean, α the c-vector of the corresponding coefficients including the intercept, x the n-vector of marker genotypes, β a p-vector of marker effects, u the n vector of polygenic effects, and ϵ the n-vector of the residuals. u follows a multivariate normal distribution: u ~ MVNn(0, λτ− 1K) where τ− 1 is the variance of the residual errors, λ is the ratio between the genetic variance (variance explained by the SNPs) and the residual variance, and K is a known n x n relationship matrix estimated from genotypes, using a n x p matrix of genotypes (X):
This expression was used to calculate K where x i is the ith column representing genotypes of ith SNP, \( {\overline{x}}_i \) is the sample mean and 1n is a n × 1 vector of 1’s. ϵ follows a multivariate normal distribution: ϵ ~ MVNn(0,τ−1I n ) where I n is an n x n identity matrix.
To control the family-wise error rate (FWER) in the context of multiple hypothesis testing, a Bonferroni correction at 0.05 significant level was applied for both genome-wide and chromosome-wide thresholds (significant level = α/number of SNPs). Any SNP with a P-value < 1.21 × 10− 6 was considered to be significantly associated at the genome-wide threshold. The chromosome-wide significance thresholds ranged from 7.56 × 10− 6 to 3.36 × 10− 4, depending on the chromosome size.
References
Havenstein GB, Ferket PR, Qureshi MA. Growth, livability, and feed conversion of 1957 versus 2001 broilers when fed representative 1957 and 2001 broiler diets. Poult Sci. 2003;82(10):1500–8.
Havenstein GB, Ferket PR, Qureshi MA. Carcass composition and yield of 1957 versus 2001 broilers when fed representative 1957 and 2001 broiler diets. Poult Sci. 2003;82(10):1509–18.
Petracci M, Mudalal S, Soglia F, Cavani C. Meat quality in fast-growing broiler chickens. World's Poultry Science Journal. 2015;71:363–73.
Kuttappan VA, Hargis BM, Owens CM. White striping and woody breast myopathies in the modern poultry industry: a review. Poult Sci. 2016;95(11):2724–33.
Kuttappan VA, Shivaprasad HL, Shaw DP, Valentine BA, Hargis BM, Clark FD, McKee SR, Owens CM. Pathological changes associated with white striping in broiler breast muscles. Poult Sci. 2013;92(2):331–8.
Kuttappan VA, Brewer VB, Apple JK, Waldroup PW, Owens CM. Influence of growth rate on the occurrence of white striping in broiler breast fillets. Poult Sci. 2012;91(10):2677–85.
Kuttappan VA, Goodgame SD, Bradley CD, Mauromoustakos A, Hargis BM, Waldroup PW, Owens CM. Effect of different levels of dietary vitamin E (DL-α-tocopherol acetate) on the occurrence of various degrees of white striping on broiler breast fillets. Poult Sci. 2012;91(12):3230–5.
Kuttappan VA, Huff GR, Huff WE, Hargis BM, Apple JK, Coon C, Owens CM. Comparison of hematologic and serologic profiles of broiler birds with normal and severe degrees of white striping in breast fillets. Poult Sci. 2013;92(2):339–45.
Petracci M, Mudalal S, Babini E, Cavani C. Effect of white striping on chemical composition and nutritional value of chicken breast meat. Ital J Anim Sci. 2014;13(3138):179–83.
Mudalal S, Babini E, Cavani C, Petracci M. Quantity and functionality of protein fractions in chicken breast fillets affected by white striping. Poult Sci. 2014;93(8):2108–16.
Petracci M, Mudalal S, Bonfiglio A, Cavani C. Occurrence of white striping under commercial conditions and its impact on breast meat quality in broiler chickens. Poult Sci. 2013;92(6):1670–5.
Mudalal S, Lorenzi M, Soglia F, Cavani C, Petracci M. Implications of white striping and wooden breast abnormalities on quality traits of raw and marinated chicken meat. Animal. 2015;9(4):728–34.
Tijare VV, Yang FL, Kuttappan VA, Alvarado CZ, Coon CN, Owens CM. Meat quality of broiler breast fillets with white striping and woody breast muscle myopathies. Poult Sci. 2016;95(9):2167–73.
Kuttappan VA, Lee YS, Erf GF, Meullenet JF, McKee SR, Owens CM. Consumer acceptance of visual appearance of broiler breast meat with varying degrees of white striping. Poult Sci. 2012;91(5):1240–7.
Lorenzi M, Mudalal S, Cavani C, Petracci M. Incidence of white striping under commercial conditions in medium and heavy broiler chickens in Italy. J Appl Poult Res. 2014;23:754–8.
Kuttappan VA, Brewer VB, Mauromoustakos A, McKee SR, Emmert JL, Meullenet JF, Owens CM. Estimation of factors associated with the occurrence of white striping in broiler breast fillets. Poult Sci. 2013;92(3):811–9.
Russo E, Drigo M, Longoni C, Pezzotti R, Fasoli P, Recordati C. Evaluation of white striping prevalence and predisposing factors in broilers at slaughter. Poult Sci. 2015;94(8):1843–8.
Bailey RA, Watson KA, Bilgili SF, Avendano S. The genetic basis of pectoralis major myopathies in modern broiler chicken lines. Poult Sci. 2015;94(12):2870–9.
Alnahhas N, Berri C, Chabault M, Chartrin P, Boulay M, Bourin MC, Le Bihan-Duval E. Genetic parameters of white striping in relation to body weight, carcass composition, and meat quality traits in two broiler lines divergently selected for the ultimate pH of the pectoralis major muscle. BMC Genet. 2016;17:61.
Dalle Zotte A, Cecchinato M, Quartesan A, Bradanovic J, Tasoniero G, Puolanne E. How does "wooden breast" myodegeneration affect poultry meat quality ? 60th International Congress of Meat. Sci Technol. 2014:476–9.
Bowker B, Zhuang H. Impact of white striping on functionality attributes of broiler breast meat1. Poult Sci. 2016;95(8):1957–65.
Tasoniero G, Cullere M, Cecchinato M, Puolanne E, Dalle Zotte A. Technological quality, mineral profile, and sensory attributes of broiler chicken breasts affected by white striping and wooden breast myopathies. Poult Sci. 2016;95(11):2707–14.
Le Bihan-Duval E, Hennequet-Antier C, Berri C, Beauclercq S, Bourin M, Boulay M, Demeure O, Boitard S: Identification of genomic regions and candidate genes for chicken meat ultimate pH by combined detection of selective sweeps and QTL. BMC Genomics 2017.
Le Bihan-Duval E, Debut M, Berri CM, Sellier N, Sante-Lhoutellier V, Jego Y, Beaumont C. Chicken meat quality: genetic variability and relationship with growth and muscle characteristics. BMC Genet. 2008;9:53.
Alnahhas N, Berri C, Boulay M, Baéza E, Jégo Y, Baumard Y, Chabault M, Le Bihan-Duval E. selecting broiler chickens for ultimate pH of breast muscle: analysis of divergent selection experiment and phenotypic consequences on meat quality, growth, and body composition traits. J Anim Sci. 2014;92(9):3816–24.
Hahne F: The Gviz User Guide. https://bioconductororg/packages/devel/bioc/vignettes/Gviz/inst/doc/Gvizpdf 2014.
Camoretti-Mercado B, Dizon E, Jakovcic S, Zak R: Differential expression of ventricular-like myosin heavy chain mRNA in developing and regenerating avian skeletal muscles. Cell Mol Biol Res 1993, 39(5):425-437.
Joe AW, Yi L, Natarajan A, Grand FL, So L, Wang J, Rudnicki MA, Rossi FM. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12:153–63.
Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, et al. Beta-sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11(3):257–65.
Araishi K, Sasaoka T, Imamura M, Noguchi S, Hama H, Wakabayashi E, Yoshida M, Hori T, Ozawa E. Loss of the sarcoglycan complex and sarcospan leads to muscular dystrophy in beta-sarcoglycan-deficient mice. Hum Mol Genet. 1999;8(9):1589–98.
Cynthia Martin F, Hiller M, Spitali P, Oonk S, Dalebout H, Palmblad M, Chaouch A, Guglieri M, Straub V, Lochmuller H, et al. Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteomics Clin Appl. 2014;8(3–4):269–78.
Bertini E, Pepe G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol. 2002;6(4):193–8.
Guernsey DL, Jiang H, Bedard K, Evans SC, Ferguson M, Matsuoka M, Macgillivray C, Nightingale M, Perry S, Rideout AL, et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-tooth disease. PLoS Genet. 2010;6(8)
Kienesberger PC, Lass A, Preiss-Landl K, Wolinski H, Kohlwein SD, Zimmermann R, Zechner R. Identification of an insulin-regulated lysophospholipase with homology to neuropathy target esterase. J Biol Chem. 2008;283(9):5908–17.
Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297(2):E289–96.
Beauclercq S, Hennequet-Antier C, Praud C, Godet E, Collin A, Tesseraud S, Metayer-Coustard S, Bourin M, Moroldo M, Martins F, et al. Muscle transcriptome analysis reveals molecular pathways and biomarkers involved in extreme ultimate pH and meat defect occurrence in chicken. Sci Rep. 2017;7(1):6447.
Berri C, Petracci M. Poultry quality evaluation, 1st. EDN. 2017;
Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423(6936):168–72.
Bittner RE, Anderson LV, Burkhardt E, Bashir R, Vafiadaki E, Ivanova S, Raffelsberger T, Maerk I, Hoger H, Jung M, et al. Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat Genet. 1999;23(2):141–2.
Matsuda C, Hayashi YK, Ogawa M, Aoki M, Murayama K, Nishino I, Nonaka I, Arahata K, Brown RH Jr. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet. 2001;10(17):1761–6.
Huang Y, Verheesen P, Roussis A, Frankhuizen W, Ginjaar I, Haldane F, Laval S, Anderson LV, Verrips T, Frants RR, et al. Protein studies in dysferlinopathy patients using llama-derived antibody fragments selected by phage display. Eur J Hum Genet. 2005;13(6):721–30.
Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81(1):27–40.
Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, Masetti E, Mazzocco M, Egeo A, Donati M, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18(4):365–8.
Dargelos E, Poussard S, Brule C, Daury L, Cottin P. Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie. 2008;90(2):359–68.
Sorimachi H, Ono Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc Res. 2012;96(1):11–22.
Arvanitidis A, Henriksen K, Karsdal MA, Nedergaard A. Neo-epitope peptides as biomarkers of disease progression for muscular dystrophies and other myopathies. Journal of Neuromuscular Diseases. 2016;3:333–46.
Spence HJ, Chen YJ, Winder SJ. Muscular dystrophies, the cytoskeleton and cell adhesion. BioEssays. 2002;24(6):542–52.
Hansen BG, Halkier BA, Kliebenstein DJ. Identifying the molecular basis of QTLs: eQTLs add a new dimension. Trends Plant Sci. 2008;13(2):72–7.
Milan D, Jeon JT, Looft C, Amarger V, Robic A, Thelander M, Rogel-Gaillard C, Paul S, Iannuccelli N, Rask L, et al. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science. 2000;288(5469):1248–51.
Bellinge RH, Liberles DA, Iaschi SP, O'Brien PA, Tay GK. Myostatin and its implications on animal breeding: a review. Anim Genet. 2005;36(1):1–6.
Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23(10):1289–91.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4.
Acknowledgements
We thank LABOGENA laboratory for the genotyping analyses. We thank the staff of the poultry breeding facilities (PEAT, INRA, 37380, Nouzilly, France) and of the Avian Research Unit (URA, INRA, 37380, Nouzilly, France) for their technical assistance.
Funding
The selection program was funded by INRA. Phenotyping and genotyping of the birds used for QTL detection was funded by the RFI CASDAR program no. 1309 OPTIVIANDE supported by the French Ministry of Agriculture. This funding body had no role in the design of the study and collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
All the data that support the conclusions of the study are included in the paper and additional files.
Author information
Authors and Affiliations
Contributions
ELBD, CB, and YJ are at the origin of the conception of the program and defined the experimental design. ELBD and CB supervised the phenotyping. EP was in charge of QTL and eQTL detection analyses, under the supervision of ELBD, CHA, and SB. CHA and SB contributed to the choice of tools and methodologies used for GWAS analysis. SAB, EG, and EP designed and tested the oligonucleotide primers used for quantitative RT-qPCR analysis. EG and SAB performed RNA extraction. CP and CB supervised the identification of functional candidate genes within QTL regions. EP drafted the first version of the manuscript under the supervision of ELBD. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All the procedures needed for the reproduction and phenotyping of the two lines (described under program 00880.02) were approved by the Ethics Committee for Animal Experimentation in Val de Loire (registered under N°19 by the National Committee). No permissions were required for the collection of the two studied chicken lines owned by INRA.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Manhattan plot showing the association of SNPs with WS in the pHu + (a) and the pHu- (b) line. Black line represents the 5% genome-wide threshold, red line the 5% GGA1-wide threshold, blue line the 5% GGA17-wide threshold and purple line the 5% GGA20-wide threshold. (DOCX 1204 kb)
Additional file 2:
Figure S2. Gene environment of the region of interest for WS on GGA1. The first track corresponds to the chromosome 1 where the region of interest is indicated by a red line. The second track corresponds to the genomic axis for the region of interest. The third track corresponds to the gene model based on GalGal5 assembly. The last track corresponds to the Manhattan plot of the region of interest where the orange line represents the chromosome threshold and the most significant SNP for WS is highlighted in red. (PDF 8 kb)
Additional file 3:
Figure S3. Gene environment of the region of interest for WS and BMY on GGA4. The first track corresponds to the chromosome 4 where the region of interest is indicated by a red box. The second track corresponds to the genomic axis for the region of interest. The third track corresponds to the gene model based on GalGal5 assembly. The two last tracks correspond to the Manhattan plot of BMY and WS respectively, on the region of interest, where the orange line represents the chromosome threshold and the most significant SNP for WS and BMY is highlighted in red. (PDF 10 kb)
Additional file 4:
Figure S4. Gene environment of the region of interest for WS on GGA7. The first track corresponds to the chromosome 7 where the region of interest is indicated by a red box. The second track corresponds to the genomic axis for the region of interest. The third track corresponds to the gene model based on GalGal5 assembly. The last track corresponds to the Manhattan plot of the region of interest where the orange line represents the chromosome threshold and the most significant SNP is highlighted in red. (PDF 8 kb)
Additional file 5:
Figure S5. Gene environment of the region of interest for WS on GGA17. The first track corresponds to the chromosome 17 where the region of interest is indicated by a red box. The second track corresponds to the genomic axis for the region of interest. The third track corresponds to the gene model based on GalGal5 assembly. The last track corresponds to the Manhattan plot of the region of interest where the orange line represents the chromosome threshold and the most significant SNP is highlighted in red. (PDF 9 kb)
Additional file 6:
Figure S6. Gene environment of the region of interest for WS on GGA18. The first track corresponds to the chromosome 18 where the region of interest is indicated by a red box. The second track corresponds to the genomic axis for the region of interest. The third track corresponds to the gene model based on GalGal5 assembly. The last track corresponds to the Manhattan plot of the region of interest where the orange line represents the chromosome threshold and the most significant SNP is highlighted in red. (PDF 10 kb)
Additional file 7:
Figure S7. Gene environment of the pleiotropic region on GGA4. The first track corresponds to the chromosome 4 where the region of interest is indicated by a red box. The second track corresponds to the genomic axis for the region of interest. The third track corresponds to the gene model based on GalGal5 assembly. The last track corresponds to the Manhattan plot of CL, which has the most significant SNP in the pleiotropic region. Orange line represents the chromosome threshold and the most significant SNP is highlighted in red. (PDF 9 kb)
Additional file 8:
Table S1. Significant SNPs for the level of expression of the 16 candidate genes. (DOCX 36 kb)
Additional file 9:
Figure S8. Manhattan plot showing the association of SNPs with MYH1F expression. Black line represents the 5% genome-wide threshold and blue line the 5% GGA18-wide threshold. (DOCX 560 kb)
Additional file 10:
Figure S9. Manhattan plot showing the association of SNPs with CAV3 expression. Black line represents the 5% genome-wide threshold and green line the 5% GGA12-wide threshold. (DOCX 585 kb)
Additional file 11:
Table S2. Primer sequences of the 16 candidate genes and the housekeeping gene (PDE3B). (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pampouille, E., Berri, C., Boitard, S. et al. Mapping QTL for white striping in relation to breast muscle yield and meat quality traits in broiler chickens. BMC Genomics 19, 202 (2018). https://doi.org/10.1186/s12864-018-4598-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4598-9