
Abstract
Background
Gastropoda, with approximately 80,000 living species, is the largest class of Mollusca. Among gastropods, apple snails (family Ampullariidae) are globally distributed in tropical and subtropical freshwater ecosystems and many species are ecologically and economically important. Ampullariids exhibit various morphological and physiological adaptations to their respective habitats, which make them ideal candidates for studying adaptation, population divergence, speciation, and larger-scale patterns of diversity, including the biogeography of native and invasive populations. The limited availability of genomic data, however, hinders in-depth ecological and evolutionary studies of these non-model organisms.
Results
Using Illumina Hiseq platforms, we sequenced 1220 million reads for seven species of apple snails. Together with the previously published RNA-Seq data of two apple snails, we conducted de novo transcriptome assembly of eight species that belong to five genera of Ampullariidae, two of which represent Old World lineages and the other three New World lineages. There were 20,730 to 35,828 unigenes with predicted open reading frames for the eight species, with N50 (shortest sequence length at 50% of the unigenes) ranging from 1320 to 1803 bp. 69.7% to 80.2% of these unigenes were functionally annotated by searching against NCBI’s non-redundant, Gene Ontology database and the Kyoto Encyclopaedia of Genes and Genomes. With these data we developed AmpuBase, a relational database that features online BLAST functionality for DNA/protein sequences, keyword searching for unigenes/functional terms, and download functions for sequences and whole transcriptomes.
Conclusions
In summary, we have generated comprehensive transcriptome data for multiple ampullariid genera and species, and created a publicly accessible database with a user-friendly interface to facilitate future basic and applied studies on ampullariids, and comparative molecular studies with other invertebrates.
Similar content being viewed by others


Background
Apple snails are a family (Ampullariidae) of snails belonging to Caenogastropoda, the largest and most diverse clade within the class Gastropoda [1,2,3]. Apple snails seem to have originated on Gondwana [4], with the oldest fossils coming from Early Cretaceous deposits in Africa [5]. After the breakup of Gondwana roughly 100 million years ago, apple snails have undergone diversification in the New World and Old World respectively [4, 6]. Currently, around 120 species of apple snails are recognised in nine genera, including the Old World genera Afropomus, Forbesopomus, Lanistes, Pila and Saulea, and the New World genera Asolene, Felipponea, Marisa and Pomacea [7]. In what follows we abbreviate Pomacea, but not Pila to avoid confusion of these two genera. Ampullariids are distributed in a wide variety of freshwater habitats, including swamps, wetlands, lakes and rivers [7,8,9]. Members of the family exhibit a wide range of morphological, behavioural and physiological adaptations to their inhabited environments [10, 11]. For example, the evolutionary radiation of Lanistes in Lake Malawi contains species with contrasting morphological and behavioural features that have been interpreted as differential adaption to habitats which differ in wave action, food resources, and predators [9, 12]. Due to their long evolutionary history, wide geographic distribution and high diversity, Hayes et al. [4] suggested that ampullariids altogether provide an interesting system to study speciation and phylogeography in freshwater gastropods. Furthermore, several species of apple snails, especially P. canaliculata and P. maculata, are notorious invasive species in Asia and Hawaii, where they cause dramatic agricultural losses [10, 13], and other conservation concerns such as reductions in aquatic plant diversity and shift in wetland ecosystem functions [14, 15]. Therefore, there is substantial interest in the mechanisms of adaptation that have enabled these species to become invasive pests [16, 17], and in their biological control [18, 19].
Ampullariids are well-known for their diverse reproductive behaviours. While they are all dioecious and most genera of apple snails deposit their eggs in a jelly mass underwater, two genera (i.e., Pomacea and Pila) produce calcareous egg clutches that are deposited above the waterline. The shift from aquatic to aerial oviposition thus has occurred at least twice in the evolution of ampullariids, indicating parallel evolution in the genera Pomacea and Pila with respect to the changes in egg deposition behaviour and morphology (e.g., larger lung size and longer siphons [10]). Such behavioural and morphological adaptations in Pomacea are known to be accompanied by biochemical adaptations to predation [20]. In this regard, studies of several Pomacea species have shown that the major proteins of the egg perivitelline fluid (PVF), the fluid that surrounds and nourishes the embryo, possess multiple protective functions against predators including several anti-predation proteins (perivitellins) displaying anti-digestive, anti-nutritive, neurotoxic and aposematic properties [20,21,22,23]. Comparison between the protein-coding genes of P. canaliculata and P. maculata has revealed the involvement of gene duplication and positive selection in the formation and evolution of some PVF proteins [24, 25]. Further comparison with more distantly related genera/species would yield novel insights into the origin and evolution of PVF proteins that may underlie the diversity of reproductive behaviour and morphology in apple snails.
Apart from their use in ecological and evolutionary studies, some ampullariids, including P. canaliculata and M. cornuarietis, have been used in toxicological studies due to their high fecundity and the high sensitivity of their juveniles to pollutants such as heavy metals [26], organic pesticides [27] and organotins [28]. Mortality and deficiencies of growth or development have typically been considered to be informative toxicity end-points. Nevertheless, the lack of extensive genomic resources hinders the documentation of molecular pathways in toxicological studies of apple snails.
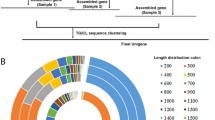
To facilitate molecular-oriented studies on apple snails, we sequenced the transcriptomes of seven species of apple snails: Lanistes nyassanus; Pila ampullacea; Asolene platae; Marisa cornuarietis; Pomacea diffusa; Pomacea scalaris and Pomacea canaliculata. Together with our previously generated RNA-Seq data for P. canaliculata [29] and P. maculata [25], we cover eight species which represent five genera (Fig. 1a) and both the New World and Old World clades. Among the Old World species are L. nyassanus, a species endemic to Lake Malawi in the East African Rift [9, 30]; and Pila ampullacea, a common species in the paddy fields and irrigation channels of northern Thailand [31]. Among the New World species, A. platae is restricted to the La Plata River basin and has a distribution range from Bolivia to the northern Buenos Aires province of Argentina [32]; this species has a slower growth rate and smaller reproductive output than other ampullariids and probably less invasive [33]. The other five species have been introduced from South America to various freshwater ecosystems in North America, Asia and Pacific islands including Hawaii [10, 13, 34, 35]. Following their introduction, two species of Pomacea (i.e., P. canaliculata and P. maculata) have become widely distributed and they are regarded as some of the most notorious invasive species in freshwater habitats [7, 34, 36, 37]. Our species selection thus covers the various phylogenetic lineages, the diversity of reproductive strategies, the most important invaders, and members that are commonly adopted in ecotoxicology. Fig. 1b shows the phylogenetic relationships among the species used in this study, whereas a phylogeny featuring more extensive taxon sampling is presented in Additional files 1 and 2.

Geographical distribution and phylogeny of apple snails used in the present study. a Rough native distribution ranges of the Old World (Lanistes and Pila) and New World (Asolene, Marisa and Pomacea) genera/species [7, 56]. b A maximum likelihood tree showing the phylogenetic relationship among the eight species of ampullariids based on sequences of three genes used in previous phylogenetic studies of ampullariids [6, 52]. Methodological details for the phylogenetic analysis can be found in Additional file 1. Bootstrap support values are shown, as is a scale bar of 0.05 substitution per site. Photo credit: L. nyassanus, Pila ampullacea and M. cornuarietis (JCHI); A. platae and P. scalaris (SI); P. canaliculata, P. maculata and P. diffusa (HM)
Construction and content
Sample collection and preparation
Adult snails were collected from the field in various regions of South America, Africa and Asia, or purchased from an aquarium shop in Hong Kong (Table 1). All snails were reared in aquaria filled with tap water and acclimated for at least one month at 26 ± 1 °C and a photoperiod of 14 h light/ 10 h dark. Snails were fed with a mixed diet of lettuce, carrot and fish meal once a day and the water was renewed twice a week. For most of the species, four to five female and male snails were chosen for dissection to obtain various tissues. For L. nyassanus, however, due to limited individuals available, only a female was used for dissection. Dissected tissues were immediately fixed in RNAlater™ (Invitrogen, USA) and then stored at − 20 °C until they were subjected to RNA extraction.
RNA isolation and sequencing
Total RNA was extracted separately from each tissue sample using TRIzol® reagent (Invitrogen, MA, USA) following the manufacturer’s protocol. In general, two RNA samples, including one of the albumen gland (AG), and one of other tissues (OT), which contained equal amounts of RNA extracted from three to four tissue types, were prepared for sequencing (Table 1). AG was always processed separately, because this organ, which secrets the perivitelline fluid that protects and nourishes the embryo, is expected to play a crucial role in the reproduction and evolution of ampullariids [24, 25, 38]. More tissue types of P. canaliculata were sequenced due to the need for producing tissue-specific gene expression data in another project for this species. To enhance the comprehensiveness of the transcripts for P. canaliculata, we combined our new data with the transcriptome data we generated from a previous study [29] for assembly. The transcriptome data of P. canaliculata from another study [39] were not included here because of uncertainty of sample preparation, and because more data would not likely improve the assembly metrics [40]. Raw reads of P. maculata were obtained from a recent publication [25], and re-assembled as described below. In P. scalaris, only AG was sequenced due to the lack of high quality RNA in OT preserved in RNAlater. For all samples, the quality of extracted RNA was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Germany). Samples with an RNA Integrity Number ≥ 8 were used for cDNA library construction using a TruSeq RNA Sample Prep Kit v2 (Illumina, California, USA), and sequenced in paired-end mode on an Illumina HiSeq sequencer (Illumina, California, USA). Library construction and sequencing were conducted by BGI Hong Kong as a commercial service.
Transcriptome assembly and annotation
Illumina raw reads were cleaned by removing adaptor sequences, reads with > 5% unknown “N” bases or > 20% bases with a quality score ≤ 10 (Table 1). Trimmomatic v0.33 was then used to further remove low quality reads with a quality score < 20 and a length < 40 base pairs (bp) [41]. For each species, clean reads from different tissue samples were pooled for de novo assembly using Trinity 2.2.0 under default settings [42]. The assembled transcripts (ranging from 126,582 to 388,329; Table 2) were clustered with CD-HIT-EST 4.6.6 to reduce redundancy using a threshold of 95% sequence similarity [43]. Open reading frames (ORFs) were predicted with TransDecoder 3.0.0 (https://transdecoder.github.io/) using a threshold of ≥100 amino acids. Only the single best ORF per transcript was retained. The longest ORF in each gene cluster was selected as the unigene. Expression levels were estimated as transcripts per kilobase million read (TPM) using Salmon 0.7.2 [44], and unigenes with TPM less than 0.5 were considered as non-expressed [25]. The level of completeness of our eight assembled transcriptomes was evaluated using BUSCO (benchmarking universal single-copy orthologs) v1.1b [45].
Predicted protein sequences were annotated using BLASTp 2.4.0+ [46] against NCBI’s non-redundant (nr) database with an E-value of 1 × 10− 5. Gene Ontology (GO) function for each unigene was assigned using Blast2GO [47] with BLASTp nr input. Sequences were also submitted to the Kyoto Encyclopedia of Genes and Genomes (KEGG) Automatic Annotation Sever (http://www.genome.jp/kegg/kaas/) to determine their functional relationships using the bi-directional best-hit method. References for the KEGG annotation included the default representative eukaryotic genomes as well as the genomes of several invertebrates: Helobdella robusta, Lottia gigantea, Crassostrea gigas, Octopus bimaculoides, Schistosoma mansoni, Nematostella vectensis and Hydra vulgaris. The annotation results are summarized in Table 2.
AmpuBase database construction
AmpuBase is a relational database that provides public access to these newly assembled ampullariid transcriptomes and annotations. The database structure and layout are similar to those of PcarnBase [48], except that data from several species can be searched at the same time and that the GO and KEGG search pages are integrated. In brief, for each species, a relational database was developed using MySQL v5.6.34 and hosted on an Apache HTTP server. The BLAST search function is powered by ViroBLAST [49] using the PHP programming language. The database consists of DNA and protein sequences of all unigenes that are linked with associated NCBI nr, GO and KEGG annotations through unigene ID. The database consists of five entity tables (“NCBI annotation”, “Proteins”, “DNAs”, “Gene Ontology” and “KEGG”) and two relation tables (“NCBI_GO_relation” and “NCBI_KEGG_relation”).
Utility and discussion
Transcriptome assembly metrics
There were between 72,341,892 to 462,231,634 bp of clean data, corresponding to between 20,730 and 35,828 unigenes with ORFs in each of the eight species (Table 2; Fig. 1a). The mean N50 value (shortest sequence length at 50% of the unigenes; 1576 bp) and the percentage of annotated unigenes (average 75.9%) in our study are higher than the corresponding values from previously published ampullariid transcriptomes (P. canaliculata, N50: 283 bp, 29.2% unigenes annotated [29]; P. maculata, N50: 1332 bp, 36.6% unigenes annotated [25]). Our transcriptome assembly metrics are comparable to those of recently published transcriptomes from other families of mollusks (Table 3), indicating the overall robustness of our transcriptome sequencing, assembly and annotation pipeline.
To further evaluate the completeness of transcriptomes, we examined the proportions of complete as well as partial homologs of 843 conserved metazoan genes within the eight coding unigene sets. The transcriptomes contain 77.46 to 92.41% of the complete conserved metazoan genes, and 87.54 to 96.09% of the genes if fragmented BUSCO hits are also included (Table 2). These BUSCO metrics are comparable with those of other mollusc transcriptomes published in recent years (Table 3).
AmpuBase: Functions and applications
AmpuBase is available online via web interface at http://www.comp.hkbu.edu.hk/~db/AmpuBase/. The database can be searched with BLAST or other query terms. The BLAST search function allows users to blast query sequence(s) or fasta files against single or multiple DNA/protein datasets with default settings (under Basic Search option) or customizable settings (under Advance Search option) (Fig. 2a). Upon submitting a BLAST search, matched sequences are returned with their E-value and similarity score, and information on the corresponding annotation can be obtained by clicking “Unigene ID” (Fig. 2b).

The web interface of AmpuBase. a Illustration of the Basic and Advanced BLAST search options. b An example of the search result of a BLAST search, showing matched sequences, each with their BLAST statistics. c Illustration of the search functions in AmpuBase based on annotation
Apart from BLAST search, the transcriptome data can be searched in two other ways (Fig. 2c). General Annotation Search allows one to query the relevant annotations (i.e., NCBI annotation, GO and KEGG) either by using the unigene ID or unigene name (e.g., perivitellin ovorubin). Each successful query returns a table that contains Unigene ID, NCBI’s nr, GO and KEGG description (if available). The resultant sequences can be downloaded by selecting the Unigene ID and clicking “Submit” for further analysis, for example, phylogenetic analysis of perivitellin ovorubins, major and multiple functional proteins in PVF [20, 24, 25]. In addition, GO and KEGG Annotation Search is also provided for searching GO and KEGG information using GO ID, KEGG ID or a keyword. All sequence data for these ampullariid transcriptomes are available for download under the “Downloads” menu, for transcriptome wide data mining and analysis of a specific gene.
Conclusions
In this study, we have generated a large set of transcriptome data for eight species that represent five genera of Ampullariidae. These data are compiled in a relational database, AmpuBase, which greatly enhances the publicly available genomic resources for ampullariids. The database provides tools for sequence- or keyword-based query functions, which will facilitate in-depth ecological and evolutionary studies on ampullariids, and comparative studies with other invertebrates. AmpuBase will be updated when more genomic data become available in the future.
Abbreviations
- 16S:
-
16S rRNA
- 18S:
-
18S rRNA
- AG:
-
Albumen gland
- bp:
-
Base pair
- BUSCO:
-
Benchmarking universal single-copy orthologs
- COI:
-
Cytochrome c oxidase I
- DG:
-
Digestive gland
- F:
-
Foot
- G:
-
Gill
- GO:
-
Gene Ontology
- K:
-
Kidney
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- L:
-
Lung
- M:
-
Mantle
- N50:
-
Shortest sequence length at 50% of the unigenes
- nr:
-
Non-redundant
- ORFs:
-
Open reading frames
- OT:
-
Other tissues
- PVF:
-
Perivitelline fluid
- S:
-
Stomach
- T:
-
Testis
- TPM:
-
Transcripts per kilobase million read
References
Bouchet P, Rocroi JP, Frýda J, Hausdorf B, Ponder W, Valdés Á, Classification WA. Nomenclator of gastropod families. Malacologia. 2005;47:1–397.
Ponder WF, Colgen D, Healy J, Nützel A, Simone LRL, Caenogastropoda SEE. In: Ponder WF, Lindberg DR, editors. Phylogeny and evolution of the Mollusca. Berkeley: University of California Press; 2008. p. 331–83.
Strong EE, Gargominy O, Ponder WF, Bouchet P. Global diversity of gastropods (Gastropoda; Mollusca) in freshwater. Hydrobiologia. 2008;595:149–66.
Hayes KA, Cowie RH, Jørgensen A, Schultheiß R, Albrecht C, Thiengo SC. Molluscan models in evolutionary biology: apple snails (Gastropoda: Ampullariidae) as a system for addressing fundamental questions. Am Malacol Bull. 2009;27:47–58.
Van Damme D, Bogan AE, Dierick M. A revision of the Mesozoic naiads (Unionoida) of Africa and the biogeographic implications. Earth-Sci Rev. 2015;147:141–200.
Jørgensen A, Kristensen TK, Madsen H. A molecular phylogeny of apple snails (Gastropoda, Caenogastropoda, Ampullariidae) with an emphasis on African species. Zool Scripta. 2008;37:245–52.
Hayes KA, Burks RL, Castro-Vazquez A, Darby PC, Heras H, Martín PR, Qiu JW, Thiengo SC, Vega IA, Wada T. Insights from an integrated view of the biology of apple snails (Caenogastropoda: Ampullariidae). Malacologia. 2015;58:245–302.
Berthold T. Phylogenetic relationships, adaptations and biogeographic origin of the Ampullariidae (Mollusca, Gastropoda) endemic to Lake Malawi. Africa Abh Naturwiss Ver Hamburg. 1990;31:47–84.
Van Bocxlaer B. Hierarchical structure of ecological and non-ecological processes of differentiation shaped ongoing gastropod radiation in the Malawi Basin. Proc R Soc London B Biol Sci. 2017;284:20171494.
Cowie HR. Apple snails (Ampullariidae) as agricultural pests: their biology, impacts and management. In: Baker GM, editor. Molluscs as crop pests. CABI, Wallingford; 2002. p. 145–92.
Seuffert ME, Martín PR. Dependence on aerial respiration and its influence on microdistribution in the invasive freshwater snail Pomacea canaliculata (Caenogastropoda, Ampullariidae). Biol Invasions. 2010;12:1695–708.
Berthold T. Vergleichende Anatomie, Phylogenie und Historische Biogeographie der Ampullariidae (Mollusca, Gastropoda). Abhandlungen des Naturwissenschaftlichen Vereins in Hamburg(NF). 1991;29:1–256 [In German].
Joshi RC, Cowie RH, Sebastian LS. Biology and Management of Invasive Apple Snails. Nueva Ecija: Philippine Rice Research Institute; 2017.
Wong PK, Liang Y, Liu NY, QIU JW. Palatability of macrophytes to the invasive freshwater snail Pomacea canaliculata: differential effects of multiple plant traits. Freshw Biol. 2010;55:2023–31.
Fang L, Wong PK, Lin L, Lan C, Qiu JW. Impact of invasive apple snails in Hong Kong on wetland macrophytes, nutrients, phytoplankton and filamentous algae. Freshw Biol. 2010;55:1191–204.
Giraud-Billoud M, Vega IA, Tosi MER, Abud MA, Calderón ML, Castro-Vazquez A. Antioxidant and molecular chaperone defences during estivation and arousal in the south American apple snail Pomacea canaliculata. J Exp Biol. 2013;216:614–22.
Mu H, Sun J, Fang L, Luan T, Williams GA, Cheung SG, Wong CK, Qiu JW. Genetic basis of differential heat resistance between two species of congeneric freshwater snails: insights from quantitative proteomics and base substitution rate analysis. J Proteome Res. 2015;14:4296–308.
Yusa Y, Sugiura N, Wada T. Predatory potential of freshwater animals on an invasive agricultural pest, the apple snail Pomacea canaliculata (Gastropoda: Ampullariidae), in southern Japan. Biol Invasions. 2006;8:137–47.
Ip KK, Liang Y, Lin L, Wu H, Xue J, Qiu J-W. Biological control of invasive apple snails by two species of carp: effects on non-target species matter. Biol Control. 2014;71:16–22.
Heras H, Dreon M, Ituarte S, Pollero R. Egg carotenoproteins in neotropical Ampullariidae (Gastropoda: Arquitaenioglossa). Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 2007;146:158–67.
Heras H, Frassa MV, Fernandez PE, Galosi CM, Gimeno EJ, Dreon MS. First egg protein with a neurotoxic effect on mice. Toxicon. 2008;52:481–8.
Dreon MS, Ituarte S, Heras H. The role of the proteinase inhibitor ovorubin in apple snail eggs resembles plant embryo defense against predation. PLoS One. 2010;5:e15059.
Dreon MS, Frassa MV, Ceolín M, Ituarte S, Qiu JW, Sun J, Fernández PE, Heras H. Novel animal defenses against predation: a snail egg neurotoxin combining lectin and pore-forming chains that resembles plant defense and bacteria attack toxins. PLoS One. 2013;8:e63782.
Sun J, Zhang H, Wang H, Heras H, Dreon MS, Ituarte S, Ravasi T, Qian PY, Qiu JW. First proteome of the egg perivitelline fluid of a freshwater gastropod with aerial oviposition. J Proteome Res. 2012;11:4240–8.
Mu H, Sun J, Heras H, Chu KH, Qiu JW. An integrated proteomic and transcriptomic analysis of perivitelline fluid proteins in a freshwater gastropod laying aerial eggs. J Proteome. 2017;155:22–30.
Rogevich E, Hoang T, Rand G. The effects of water quality and age on the acute toxicity of copper to the Florida apple snail, Pomacea paludosa. Arch Environ Contam Toxicol. 2008;54:690–6.
Sawasdee B, Köhler HR. Embryo toxicity of pesticides and heavy metals to the ramshorn snail, Marisa cornuarietis (Prosobranchia). Chemosphere. 2009;75:1539–47.
Schulte-Oehlmann U, Tillmann M, Markert B, Oehlmann J, Watermann B, Scherf S. Effects of endocrine disruptors on prosobranch snails (Mollusca: Gastropoda) in the laboratory. Part II: Triphenyltin as a xeno-androgen. Ecotoxicology. 2000;9:399–412.
Sun J, Wang M, Wang H, Zhang H, Zhang X, Thiyagarajan V, Qian P, Qiu JW. De novo assembly of the transcriptome of an invasive snail and its multiple ecological applications. Mol Ecol Resour. 2012;12:1133–44.
Schultheiß R, Van Bocxlaer B, Wilke T, Albrecht C. Old fossils–young species: evolutionary history of an endemic gastropod assemblage in Lake Malawi. Proc R Soc Lond B Biol Sci. 2009;276:2837–46.
Thaewnon-Ngiw B, Klinbunga S, Phanwichien K, Sangduen N, Lauhachinda N, Menasveta P. Genetic diversity and molecular markers in introduced and Thai native apple snails (Pomacea and Pila). BMB Rep. 2004;37:493–502.
Cowie R, Thiengo S. The apple snails of the Americas (Mollusca: Gastropoda: Ampullariidae: Asolene, Felipponea, Marisa, Pomacea, Pomella): a nomenclatural and type catalog. Malacologia. 2003;45:41–100.
Tiecher MJ, Burela S, Martín PR. Life cycle of the south American apple snail Asolene platae (Maton, 1811) (Caenogastropoda: Ampullariidae) under laboratory conditions. J Molluscan Stud. 2016;82:432–9.
Horgan FG, Stuart AM, Kudavidanage EP. Impact of invasive apple snails on the functioning and services of natural and managed wetlands. Acta Oecol. 2014;54:90–100.
Wu JY, Meng PJ, Liu MY, Chiu YW, Liu LL. A high incidence of imposex in Pomacea apple snails in Taiwan: a decade after triphenyltin was banned. Zool Stud. 2010;49:85–93.
Hayes K, Joshi R, Thiengo S, Cowie R. Out of South America: multiple origins of non-native apple snails in Asia. Divers Distrib. 2008;14(4):701–12.
Kwong KL, Wong PK, Lau SS, Qiu JW. Determinants of the distribution of apple snails in Hong Kong two decades after their initial invasion. Malacologia. 2008;50:293–302.
Heras H, Garin CF, Pollero RJ. Biochemical composition and energy sources during embryo development and in early juveniles of the snail Pomacea canaliculata (Mollusca: Gastropoda). J Exp Zool. 1998;280:375–83.
Mu X, Hou G, Song H, Xu P, Luo D, Gu D, Xu M, Luo J, Zhang J, Hu Y. Transcriptome analysis between invasive Pomacea canaliculata and indigenous Cipangopaludina cahayensis reveals genomic divergence and diagnostic microsatellite/SSR markers. BMC Genet. 2015;16:12.
Francis WR, Christianson LM, Kiko R, Powers ML, Shaner NC, Haddock SH. A comparison across non-model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC Genomics. 2013;14:167.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9.
Patro R, Duggal G, Kingsford C. Salmon: accurate, versatile and ultrafast quantification from RNA-seq data using lightweight-alignment. bioRxiv 2015;021592.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Sun J, Chen Q, Lun JC, Xu J, Qiu JW. PcarnBase: development of a transcriptomic database for the brain coral Platygyra carnosus. Mar Biotechnol. 2013;15:244–51.
Deng W, Nickle DC, Learn GH, Maust B, Mullins JI. ViroBLAST: a stand-alone BLAST web server for flexible queries of multiple databases and user's datasets. Bioinformatics. 2007;23:2334–6.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I. Ostell J. Sayers EW. GenBank. Nucleic Acids Res: Pruitt KD; 2017.
Hayes KA, Cowie RH, Thiengo SC. A global phylogeny of apple snails: Gondwanan origin, generic relationships, and the influence of outgroup choice (Caenogastropoda: Ampullariidae). Biol J Linnean Soc. 2009;98:61–76.
Colgan D, Ponder W, Beacham E, Macaranas J. Molecular phylogenetics of Caenogastropoda (Gastropoda: Mollusca). Mol Phylogenet Evol. 2007;42:717–37.
Lydeard C, Holznagel WE, Glaubrecht M, Ponder W. F. Molecular phylogeny of a circum-global, diverse gastropod superfamily (Cerithioidea: Mollusca: Caenogastropoda): pushing the deepest phylogenetic limits of mitochondrial LSU rDNA sequences. Mol Phylogenet Evol. 2002;22:399–406.
Giribet G, et al. Evidence for a clade composed of molluscs with serially repeated structures: monoplacophorans are related to chitons. Proc Natl Acad Sci USA. 2006;103:7723–8.
Brown DS. Freshwater snails of Africa and their medical importance. 2nd ed. London: Taylor & Francis; 1994.
Ip JC, Leung PT, Ho KK, Qiu J, Leung KM. De novo transcriptome assembly of the marine gastropod Reishia clavigera for supporting toxic mechanism studies. Aquat Toxicol. 2016;178:39–48.
Bankers LA, Fields P, McElroy KE, Boore JL, Logsdon JM, Neiman M. Genomic evidence for population-specific responses to coevolving parasites in a New Zealand freshwater snail. Mol Ecol. 2016;26:3663–75.
De Oliveira A, Wollesen T, Kristof A, Scherholz M, Redl E, Todt C, Bleidorn C, Wanninger A. Comparative transcriptomics enlarges the toolkit of known developmental genes in mollusks. BMC Genomics. 2016;17:905.
Gerdol M, Fujii Y, Hasan I, Koike T, Shimojo S, Spazzali F, Yamamoto K, Ozeki Y, Pallavicini A, Fujita H. The purplish bifurcate mussel Mytilisepta virgata gene expression atlas reveals a remarkable tissue functional specialization. BMC Genomics. 2017;18:590.
Acknowledgements
The Cultural and Museum Centre Karonga, Harrison Simfukwe and Friedemann Schrenk facilitated fieldwork in Malawi. We thank Prof. Henry Madsen (University of Copenhagen) for suggestions on sampling Lanistes, and Prof. Ka Hou Chu (The Chinese University of Hong Kong) for helpful comments on the manuscript.
Funding
JWQ was supported by Shenzhen Science and Technology Innovation Committee (JCYJ20170307161326613) and General Research Fund of Hong Kong (HKBU 12301415). HH was supported by China and Agencia Nacional de Promoción Científica y Tecnológica, Argentina (0850 and 0122). JCHI received a PhD studentship from Hong Kong Baptist University. BVB was supported by a postdoctoral fellowship of the FWO Vlaanderen (12N3915N) and a grant from the French Agence Nationale de la Recherche (ANR-JCJC-EVOLINK).
Availability and requirements
All clean reads are deposited in the NCBI Sequence Read Archive (SRA) with accession numbers listed in Table 1. The assembled and annotated transcriptomes are available on the AmpuBase website (http://www.comp.hkbu.edu.hk/~db/AmpuBase/). The transcriptome data and phylogeny data are deposited in the Dryad Digital Repository at https://doi.org/10.5061/dryad.117cf.
Author information
Authors and Affiliations
Contributions
JCHI performed the experiments, data analyses and drafted the manuscript. HM and JS coordinated the experiments and revised the manuscript. XH and QC designed and constructed the database website, wrote the database section of the manuscript, and revised the manuscript. SI, HH, BVB and MG collected samples, provided advice on snail culturing and revised the manuscript. JWQ designed and oversaw the study, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Our research adheres to the legislation of the Wild Animals Protection Ordinance of Hong Kong (Cap. 170), the Argentinean provincial Wildlife Hunting Law (Ley 5786, Art. 2) and the Wildlife Preservation and Protection Act (BE 2535) of Thailand. The sampling in Malawi was undertaken under the framework of research cooperation KM/1/1.64 between Ghent University, Belgium and the Karonga Museum, Ministry of Tourism, Wildlife and Culture, Malawi.
Consent for publication
All authors have endorsed the manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Phylogenetic tree of ampullariids based on DNA sequences of cytochrome c oxidase I (COI), 16S rRNA (16S) and 18S rRNA (18S) as listed in Additional file 2. Sequences were aligned and gaps were trimmed with MUSCLE. Phylogenetic analysis was conducted using the concatenated sequences (COI: 502 bp; 16S: 362 bp; 18S: 269 bp). The maximum-likelihood method implemented in MEGA5 [50] was used and the GTR + Γ + I evolutionary model was selected. Members of Viviparidae and Campanilidae served as outgroups. Values at nodes are percentages of 100 bootstrap replicates. Scale bar represents 0.1 substitution per site. Species with transcriptomes assembled in the present study are highlighted in blue. List of taxa and GenBank accession numbers for sequences of COI, 16S and 18S used in phylogenetic analysis. (DOCX 364 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ip, J.C.H., Mu, H., Chen, Q. et al. AmpuBase: a transcriptome database for eight species of apple snails (Gastropoda: Ampullariidae). BMC Genomics 19, 179 (2018). https://doi.org/10.1186/s12864-018-4553-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4553-9