
Abstract
Background
Heat stress is the primary abiotic stress limiting growth of cool-season grass species. The objective of this study was to determine molecular factors and metabolic pathways associated with superior heat tolerance in thermal bentgrass (Agrostis scabra) by comparative analysis of transcriptomic profiles with its co-generic heat-sensitive species creeping bentgrass (A. stolonifera).
Results
Transcriptomic profiling by RNA-seq in both heat-sensitive A. stolonifera (cv. ‘Penncross’) and heat-tolerant A. scabra exposed to heat stress found 1393 (675 up- and 718 down-regulated) and 1508 (777 up- and 731 down-regulated) differentially-expressed genes, respectively. The superior heat tolerance in A. scabra was associated with more up-regulation of genes in oxidative protection, proline biosynthesis, lipid hydrolysis, hemicellulose and lignin biosynthesis, compared to heat-sensitive A. stolonifera. Several transcriptional factors (TFs), such as high mobility group B protein 7 (HMGB7), dehydration-responsive element-binding factor 1a (DREB1a), multiprotein-bridging factor 1c (MBF1c), CCCH-domain containing protein 47 (CCCH47), were also found to be up-regulated in A. scabra under heat stress.
Conclusions
The unique TFs and genes identified in thermal A. scabra could be potential candidate genes for genetic modification of cultivated grass species for improving heat tolerance, and the associated pathways could contribute to the transcriptional regulation for superior heat tolerance in bentgrass species.
Similar content being viewed by others


Background
Heat stress is one of the major environmental stresses limiting plant growth for cool-season plant species. Extensive effort has been taken to investigate physiology and molecular mechanisms of heat tolerance in various plant species (for review, see Wahid et al. [1]). Further studies to determine physiological basis, phenotypic flexibility, and molecular factors modulating plant heat tolerance are essential. Furthermore, it is also imperative to apply genomic, proteomic, and transcriptomic approaches to better understand the molecular basis of plant response to heat stress and heat tolerance.
RNA sequencing has been widely used to investigate plant molecular responses to stress conditions on the scale of the entire transcriptome [2]. The information obtained could further be used to guide plant molecular engineering or marker development. The transcriptomic profiling for heat-responsive genes has been conducted in a large variety of plant species, including model plant species, such as Arabidopsis [3], annual crops, such as rice [4, 5], wheat [6], barley [7], and perennial grass species, such as switchgrass [8] and tall fescue [9]. Previous work on transcriptomic analysis related to heat stress have mainly reported heat-responsive genes involved in various metabolic processes, such as those in respiration (glycolysis and tricarboxylic acid cycle), photosynthesis (light reactions) [4], protein modification [8], antioxidant metabolism [7], and lipid metabolism [10]. In addition, some transcription factor families, such as heat shock factor (HSF), APETALA2/ethylene-responsive element binding factor (AP2/ERF), dehydration-responsive element binding factor (DREB), myeloblastosis factor (MYB), WRKY-domain factor (WRKY), and zinc finger protein, were activated upon heat stress [3, 4, 7, 10, 11]. Although numerous heat-responsive genes have been identified, transcriptional factors and genes uniquely associated with heat tolerance should be further explored for in-depth understanding of molecular mechanisms conferring heat tolerance.
One approach to unraveling mechanisms of plant tolerance to stresses is to examine plants adapted to extremely stressful environments. A temperate (C3) perennial grass species, thermal bentgrass (A. scabra) endemic to geothermal areas of Yellowstone National Park, exhibits superior heat tolerance to other C3 grass species, as it is able to survive at soil temperature up to 45 °C [12, 13], while soil temperature over 18 °C or air temperature over 24 °C is detrimental for most C3 grass species [14]. Physiological, proteomic, and metabolic analysis with thermal bentgrass have found that superior heat tolerance of A. scabra was associated with the adjustment of various metabolic processes, including lowering respiratory consumption of carbohydrates, increases of alternative respiration and carbon use efficiency [15,16,17,18], activation of antioxidant metabolism, induction of stress-protective proteins, such as heat shock proteins [19,20,21] and the accumulation of osmoprotectants, such as soluble sugars and proline [22]. However, the molecular factors underlying the superior heat tolerance of the thermal grass species are not well documented, but such information is useful for improving heat tolerance in cultivated grass species.
The objective of this study was to identify unique transcriptional factors and genes, as well as the associated metabolic pathways accounting for the superior heat tolerance of the wild grass species, thermal A. scabra, by comparative analysis of the transcriptomic changes in response to heat stress between thermal A. scabra and its co-generic heat-sensitive species (A. stolonifera).
Methods
Plant materials and growth conditions
Tillers (30 per individual plant) of A. stolonifera (‘Penncross’) or A. scabra (‘NTAS’) were collected from stock plants and transferred to plastic containers (57 × 44 × 30 cm, 12 drainage holes) filled with fritted clay medium (Profile Products, Deerfield, IL). Plants were established for 35 d in a greenhouse with average temperature of 23/20 °C (day/night), 60% relative humidity (RH), and 750 μmol m−2 s−1 photosynthetically active radiation (PAR) from natural sunlight and supplemental lighting. Plants were irrigated daily, fertilized twice per week with half-strength Hoagland’s nutrient solution [23], and trimmed to 2 cm once per week during establishment. Plants were not trimmed during the final week of establishment to allow for sufficient regrowth prior to stress imposition, after which time all plants were transferred to controlled-environment growth chambers (Environmental Growth Chamber, Chagrin Falls, Ohio).
Heat stress treatments and experimental design
Plants were maintained in controlled-environment growth chambers controlled at 22/18 °C (day/night), 600 μmol m−2 s−1 PAR, 60% RH, and 14-h photoperiod for one week prior to stress imposition, and then air temperature was raised to 35/30 °C to impose heat stress for 21 d. During stress treatment, plants were irrigated daily, and fertilized twice per week with half-strength Hoagland’s nutrient solution. The experiment was arranged in a split-plot design with temperature treatment (control or heat) as the main plots and grass species (A. scabra or A. stolonifera) as subplots. Each species was replicated in four containers and each temperature treatment was repeated in four growth chambers. Plants under the same temperature were relocated across growth chambers every 3 d to avoid possible confounding effects of chamber environmental variations.
Physiological measurements
Leaf relative water content (RWC), chlorophyll content (Chl) and electrolyte leakage (EL) were measured at 0 and 21 d of heat stress to assess differential physiological responses of the two plant species under both control and heat stress conditions. Approximately 0.8 g fresh leaf tissue was collected from four individual plants per line per container, and then pooled for RWC, EL, and Chl measurements. For RWC, 0.2 g of leaf blades were first weighed for fresh weight (FW), soaked in water for 12 h and again weighed for turgid weight (TW), dried in an oven at 80 °C for 3 d, and finally weighed for dry weight (DW). RWC was calculated using the formula (%) = ([FW - DW] / [TW - DW]) × 100 [24]. For Chl, approximately 0.2 g fresh leaf tissue was submerged in 10 ml dimethyl sulphoxide for 3 d to extract total chlorophyll. The absorbance of the leaf extract was measured at 663 nm and 645 nm with a spectrophotometer (Spectronic Genesys 2; Spectronic Instruments, Rochester, NY) and Chl calculated using the formula described in [25]. For EL, approximately 0.2 g of fresh leaf tissue was rinsed with deionized water, placed in a test tube containing 30 mL deionized water, agitated on a conical shaker for 12 h, and initial conductance (Ci) measured using a conductivity meter (YSI Model 32, Yellow Springs, OH). Tubes containing leaf tissue were then autoclaved at 121 °C for 20 min and again agitated for 12 h. The maximal conductance (Cmax) of incubation solution was then measured and EL (%) was calculated as ((Ci/Cmax) × 100) [26]. Four biological replicates (n = 4) of each species were performed for each parameter under either control or heat stress condition, respectively. Statistical differences between treatment means were separated by Student’s t-test at the P level of 0.05.
RNA extraction, library preparation, and RNA sequencing
Total RNA was extracted from 200 mg of leaf samples collected at 21 d of heat stress using TRIzol reagent (Life Technologies, Grand Island, NY), then treated with TURBO DNA-free kit (Life Technologies, Grand Island, NY). The quality and quantity of RNA was assessed in a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). A total of 12 libraries (2 plant species × 2 temperature treatment × 3 biological replicates) were prepared for RNA-seq. Total RNA (2 μg) was used for construction of each library using the Illumina TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA) according to the Low Sample (LS) protocol. LS protocol was amended to lower the Elute 2-Fragment-Prime 94 °C incubation time from 8 min to 1 min to generate larger RNA fragments. Indexes were chosen to allow for library multiplexing per run and libraries were pooled in an equimolar fashion. Pooled libraries were prepared for MiSeq run according to Illumina recommendations and loaded into a 600-cycle MiSeq Reagent Kit v3 cartridge (Illumina, San Diego, CA) at a concentration of 20 pM. Each run was set as pair-end (PE) 2 × 300 bp, fastq format only, and no adapter trimming.
Read alignment, counting, gene expression and functional analysis
Raw reads from MiSeq sequencing were downloaded and analyzed using samtools command flagstat [27]. Reads were then assembled using Trinity [28], with quality trimming using Trimmomatic option. The parameters were set as follows: “Trinity --max_memory 64G, --CPU 8, --bflyCPU 2, --bflyHeapSpaceMax 64G, --trimmomatic ILLUMINACLIP::2:30:15:8:TRUE SLIDINGWINDOW:4:20 LEADING:20 TRAILING:20 MINLEN:60 HEADCROP:6 CROP: 275”. Transcripts obtained were clustered using CDHITEST [29], with the following parameters: “cd-hit-est -c 0.9, −n 8”. The transcripts were then quantified using RSEM [30], which was incorporated as the “align_and_estimate_abundance.pl” script in Trinity program, using default parameters. Differential expression analysis of transcripts were performed using edgeR [31], which was also nested in the “run_DE_analysis.pl” script in Trinity, using default parameters. The ratios of transcript abundances under heat stress to control condition for each species were filtered with threshold of |log2 fold change (log2 FC)| > 1 and false discovery rate (FDR) < 0.01, in order to get differentially expressed genes (DEGs). In addition, the coding regions of transcript assemblies were identified using TransDecoder [28], and then annotated using Trinotate [28], with the options of blastx, blastp, HMMER, signalP, and TMHMM.
Gene ontology (GO) term classification was performed by CateGOrizer [32], using “GO_slim2” method. The GO enrichment analysis for DEGs was performed using GOEAST [33], by first implementing Customized Result Analysis for up- and down-regulated DEGs in each species, respectively, and then comparing between two species in Multi-GOEAST, using default parameters. KEGG pathway enrichment analysis was performed using DAVID v6.8 [34], by using UniProt IDs for the entire transcriptome background and DEGs in both species.
The transcriptome shotgun assembly of both A. stolonifera and A. scabra were deposited at GenBank Transcriptome Shotgun Assembly (TSA) database, under the accession of GFJH00000000 and GFIW00000000, respectively. The version described in this paper is the first version, GFJH01000000 and GFIW01000000.
Validation of gene expression levels
Gene expression analysis was performed by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). Total RNA was isolated from ground leaf powder using TRIzol reagent (Life Technologies, Grand Island, NY) and treated with DNase (TURBO DNA-free kit; Life Technologies, Grand Island, NY) to remove contaminating genomic DNA. Total RNA (2 μg) was reverse-transcribed using a high-capacity cDNA reverse transcription kit (Life Technologies, Grand Island, NY). The synthesized cDNA was amplified in a StepOnePlus Real-Time PCR system (Life Technologies, Grand Island, NY) using the following parameters: pre-heat cycle of 95 °C for 3 min, 40 cycles of 95 °C denaturation for 30 s per cycle, and 60 °C annealing/extension for 30 s per cycle. Power SYBR Green PCR Master Mix (Life Technologies, Grand Island, NY) was the intercalating dye used to detect gene expression level. Gene name, accession number, forward and reverse primer sequences are provided in Table 1. A melting curve analysis was performed for each primer set to confirm its specificity. Actin was used as the reference gene, since its expression was consistent throughout treatments. A ΔΔCt method was used to calculate the relative expression level between genes of interest and reference gene, respectively [35]. Four biological replicates (n = 4) from each species were performed for each gene under either control or heat stress condition, respectively. Statistical differences between treatment means were separated by Student’s t-test at the P level of 0.05.
Results
Physiological responses to heat stress
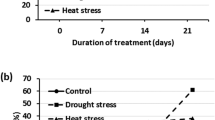
Under control conditions, leaf relative water content (RWC) did not differ significantly between A. stolonifera and A. scabra. Heat treatment caused significantly decline in RWC at 21 d in both A. stolonifera and A. scabra, by 13.1% and 5.8%, respectively. However, RWC in A. scabra was significantly higher than that in A. stolonifera (Fig. 1a). No significant differences in leaf chlorophyll content (Chl) were found between A. stolonifera and A. scabra under control conditions. At 21 d of heat treatment, Chl content decreased significantly in both A. stolonifera and A. scabra, by 17.8% and 9.6%, respectively; leaf Chl in A. scabra was significantly higher than that in A. stolonifera (Fig. 1b). For electrolyte leakage (EL), there was no significant difference found between A. stolonifera and A. scabra under control conditions. Heat stress at 21 d resulted in significantly increases in EL in both A. stolonifera and A. scabra, by 63.7% and 47.6%, respectively. Leaf EL in A. scabra was significantly lower than that in A. stolonifera (Fig. 1c).

Leaf relative water content (RWC) (a), chlorophyll content (Chl) (b), and electrolyte leakage (EL) (c) of A. stolonifera and A. scabra under control and heat stress conditions. Data shown are the means of four biological replicates (n = 4). Bar represents standard error (SE) for each mean value. Different letters atop bars indicate that significant differences exist at P level < 0.05
Next-generation sequencing of A. stolonifera and A. scabra
The RNA sequencing yielded more than 19 million reads per library of A. stolonifera and A. scabra plants exposed to non-stress control and heat stress conditions, providing over 5× coverage of the estimated genome of A. stolonifera (Table 2). The de novo transcript assembly by Trinity algorithm had good alignment rate, indicating that the assembled transcripts were largely representing transcriptome in these two species (Table 2). In addition, transcript qualities were also confirmed by long N50 numbers, contig lengths and similar GC contents (Table 3). It is therefore indicated that the Illumina RNA-seq was successfully performed to obtain transcriptional profiles for A. stolonifera and A. scabra under heat stress.
After transcript clustering and annotation, a total of 75,253 and 81,597 UniGenes were obtained by BlastX against NCBI protein NR database (Table 4). Further annotation with GO, KEGG, COG and Pfam also had similar results among them. The components of annotation were mainly from Arabidopsis and rice (Table 5). GO term classification showed that the functional distributions of UniGenes were similar between A. stolonifera and A. scabra (Fig. 2).

GO term classification of total transcripts in A. stolonifera and A. scabra. BP: Biological process; MF: Molecular function; CC: Cellular Component
GO term enrichment analysis
Using the threshold of |log2FC| > 1, and FDR < 0.01, we identified 675 and 777 up-regulated DEGs, and 718 and 731 down-regulated DEGs in A. stolonifera and A. scabra, respectively, by heat stress (Fig. 3). In order to find out specific molecular factors for the superior heat tolerance in A. scabra, up- and down-regulated DEGs due to heat tress were analyzed by GO term enrichment analysis in the two species separately (Figs. 4, 5, 6, 7, 8 and 9; For heat map, see Additional files 1 and 2). In the up-regulated DEGs, several functional categories were enriched only in A. scabra, including hemicellulose metabolic process, cell wall biogenesis, L-proline biosynthetic process, lipid catabolic process, lipid transport, lignan biosynthetic process for Biological Process (BP) terms (Fig. 4); In Molecular Function (MF) terms, monooxygenase activity, oxidoreductase activity, several glucosidase activity, and several monosaccharidase activity, such as arabinosidase activity, mannosidase activity, galactosidase activity, fucosidase activity were also uniquely enriched in A. scabra (Fig. 5). The uniquely enriched DEGs of A. scabra in Cellular Component (CC) terms were mainly at anchored component of membrane and apoplast region (Fig. 6). Some down-regulated DEGs were found to be enriched only in A. stolonifera, including DNA-templated transcription, glucose metabolic process, several amino acid metabolic process, such as L-serine, cysteine, and glycine, pentose-phosphate shunt, hydrogen peroxide catabolic process, chloroplast organization, regulation of photosynthesis, positive regulation of translation, and response to oxidative stress in BP terms (Fig. 7). Several cofactor binding functions, such as poly(U) binding, NAD binding, NADP binding, FMN binding, beta-glucosidase activity, cis-trans isomerase activity, several transaminase activity, sulfate adenyltransferase (ATP) activity, adenylate kinase activity, transketolase activity, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity, glycolate oxidase activity, glucose-6-phosphate dehydrogenase activity, monooxygenase activity and peroxidase activity were also uniquely enriched in A. stolonifera in MF terms (Fig. 8). The CC terms further showed that down-regulated transcripts uniquely enriched in A. stolonifera were located in oxidoreductase complex, apoplast, NAD(P)H dehydrogenase complex, peroxisome, and chloroplast membrane (Fig. 9).

Number of differentially expressed genes (DEGs) under heat stress in A. stolonifera and A. scabra, using the threshold of |log2 fold change (log2 FC)| > 1 and FDR > 0.01

Biological Process (BP) of GO term enrichment for up-regulated DEGs in A. stolonifera and A. scabra. Green color indicates GO terms that were specifically enriched in A. stolonifera. Red color indicates GO terms that were specifically enriched in A. scabra. Yellow color indicates GO terms that were commonly enriched in both A. stolonifera and A. scabra. The density of color was proportional to statistical significance, which was shown as p1 for P-value of A. stolonifera and p2 for P-value of A. scabra

Molecular Function (MF) of GO term enrichment for up-regulated DEGs in A. stolonifera and A. scabra. Green color indicates GO terms that were specifically enriched in A. stolonifera. Red color indicates GO terms that were specifically enriched in A. scabra. Yellow color indicates GO terms that were commonly enriched in both A. stolonifera and A. scabra. The density of color was proportional to statistical significance, which was shown as p1 for P-value of A. stolonifera and p2 for P-value of A. scabra

Cellular Component (CC) of GO term enrichment for up-regulated DEGs in A. stolonifera and A. scabra. Green color indicates GO terms that were specifically enriched in A. stolonifera. Red color indicates GO terms that were specifically enriched in A. scabra. Yellow color indicates GO terms that were commonly enriched in both A. stolonifera and A. scabra. The density of color was proportional to statistical significance, which was shown as p1 for P-value of A. stolonifera and p2 for P-value of A. scabra

Biological Process (BP) of GO term enrichment for down-regulated DEGs in A. stolonifera and A. scabra. Green color indicates GO terms that were specifically enriched in A. stolonifera. Red color indicates GO terms that were specifically enriched in A. scabra. Yellow color indicates GO terms that were commonly enriched in both A. stolonifera and A. scabra. The density of color was proportional to statistical significance, which was shown as p1 for P-value of A. stolonifera and p2 for P-value of A. scabra

Molecular Function (MF) of GO term enrichment for down-regulated DEGs in A. stolonifera and A. scabra. Green color indicates GO terms that were specifically enriched in A. stolonifera. Red color indicates GO terms that were specifically enriched in A. scabra. Yellow color indicates GO terms that were commonly enriched in both A. stolonifera and A. scabra. The density of color was proportional to statistical significance, which was shown as p1 for P-value of A. stolonifera and p2 for P-value of A. scabra

Cellular Component (CC) of GO term enrichment for down-regulated DEGs in A. stolonifera and A. scabra. Green color indicates GO terms that were specifically enriched in A. stolonifera. Red color indicates GO terms that were specifically enriched in A. scabra. Yellow color indicates GO terms that were commonly enriched in both A. stolonifera and A. scabra. The density of color was proportional to statistical significance, which was shown as p1 for P-value of A. stolonifera and p2 for P-value of A. scabra
The biological process and molecular functions of GO terms in up-regulated DEGs showing specific enrichment to A. scabra, and the GO terms in down-regulated DEGs showing specific enrichment to A. stolonifera were identified, and the individual transcripts in each category were also analyzed (Tables 6 and 7). In the up-regulated DEGs, those related to cell wall biogenesis, lipid metabolism, proline biosynthesis, lignan biosynthesis, oxidoreductase activity and glucosidase activity, were uniquely enriched in A. scabra (Table 6). The down-regulated DEGs found only in A. stolonifera included dhurrin biosynthetic process, amino acid metabolism, glucose metabolic process, pentose phosphate shunt, peroxidase activity, beta-glucosidase activity, cis-trans isomerase activity, aminotransferase activity, sulfate adenylyltranferase activity, transketolase activity, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) activity, glycolate oxidase activity, chloroplast organization, regulation of transcription and translation, energy metabolism and monooxygenase activity (Table 7).
KEGG pathway enrichment analysis
KEGG pathway enrichment analysis compared DEGs between A. stolonifera and A. scabra, and identified pathways in the degree of enrichment upon heat stress (Tables 8 and 9). In the up-regulated DEGs by heat stress, the top six enriched pathways in A. scabra were cutin, suberine and wax biosynthesis, biosynthesis of secondary metabolites, metabolic pathways, fatty acid elongation, phenylpropanoid biosynthesis, ABC transporters, and those in A. stolonifera were biosynthesis of secondary metabolites, arginine and proline metabolism, alpha-linolenic acid metabolism, galactose metabolism, beta-alanine metabolism and plant-pathogen interaction (Table 8). In the down-regulated DEGs, the top six enriched pathways were the same in both A. stolonifera and A. scabra, including metabolic pathways, biosynthesis of secondary metabolites, glyoxylate and dicarboxylate metabolism, carbon metabolism, glycine, serine and threonine metabolism and biosynthesis of antibiotics (Table 9).
Transcription factors related to heat tolerance
Transcription factors (TFs) responsive to heat stress showed high similarity between A. stolonifera and A. scabra, including up-regulation of ABA-inducible, basic Helix-Loop-Helix (bHLH), ethylene-responsive factor (ERF), protein FD, G-box-binding factor, heat stress factor (HSF), homeobox-leucine zipper, MYB, NAC, nuclear transcription factor Y, WRKY, and down-regulation of APG, PHL1-like, RNA-polymerase sigma factor, zinc-finger protein (Table 10). However, some TFs were uniquely regulated by heat stress in A. scabra, such as the up-regulation of high mobility group B protein 7 (HMGB7), dehydration-responsive element-binding factor 1A (DREB1A), multiprotein-bridging factor 1c, CCCH-domain containing protein 47, and down-regulation of GLK1, GATA transcription factor 21 and 26, protein REVEILLE, ASR3, HY5 (Table 11).
Validation of RNA-seq with qRT-PCR
The differential expressions of several DEGs in the RNA-seq data were validated using qRT-PCR. Heat stress significantly increased gene expression levels of xyloglucan endo-transferase 25 (XET25), GDSL esterase, Dirigent protein 5, pyrroline-5-carboxylate reductase (P5CR), Cytochrome P450 77A3, HMGB7, and DREB1A in both A. stolonifera and A. scabra. However, expression levels for these genes in A. scabra under heat stress were significantly higher than those in A. stolonifera (Fig. 10a-g). Heat stress significantly decreased gene expression levels of glycine cleavage system H protein, GAPDH A, peroxidase 4, and beta-glucosidase 3 in both A. stolonifera and A. scabra. However, the expression levels for these genes in A. scabra under heat stress were significantly higher than those in A. stolonifera (Fig. 10h-k). Heat stress also significantly increased expression level of transcription factor DIVARICATA in both A. stolonifera and A. scabra, but the expression level in A. stolonifera under heat stress was significantly higher than that in A. scabra (Fig. 10l). Furthermore, the fold changes of these genes obtained by qRT-PCR analysis were compared with RNA-seq data. The Person’s correlation coefficient between data from RNA-seq and qRT-PCR was 0.95 for A. scabra, and 0.93 for A. stolonifera, indicating that the transcriptional regulations under heat stress in these two species were valid regardless of detecting methods (Table 12).

Relative gene expression levels of selected transcripts, including XET25 (a), GDSL esterase (b), Dirigent protein 5 (c), P5CR (d), Cytochrome P450 77A3 (e), HMGB7 (f), DREB1A (g), Glycine cleavage system H protein (h), GAPDH (i), Peroxidase 4 (j), Beta-glucosidase 3 (k), and DIVARICATA (l) in A. stolonifera and A. scabra under control and heat stress conditions by qRT-PCR. Data shown are the means of four biological replicates (n = 4). Bar represents standard error (SE) for each mean value. Different letters atop bars indicate that significant differences exist at P level < 0.05
Discussion
The comparative analysis of transcriptome profiles between A. stolonifera and A. scabra exposed to heat stress found that metabolic processes involved in heat responses were similar in the two species, but some TFs and genes uniquely enriched in A. scabra, which could account for its superior heat tolerance. The following sections focus on the discussion of uniquely up-regulated TFs and genes in A. scabra and uniquely down-regulated TFs and genes in A. stolonifera regarding their functions and roles in heat tolerance.
Previous studies with A. scabra found that accumulation of carbohydrates and amino acids play major roles in heat tolerance for this heat-tolerant grass species [20, 22, 36, 37]. The GO term and KEGG analysis identified some specific pathways of genes involved in carbohydrate, amino acid, and energy metabolism, including unique up-regulation of glucosidases and monosaccharidases activity in A. scabra (Fig. 5, Table 6), and down-regulation of aminotransferase activity, glucose metabolic process, pentose phosphate shunt, transketolase, cis-trans isomerase, GAPDH, glycolate oxidase activity, cofactor binding (NAD, NADP, FMN), glucose-6-phosphate dehydrogenase and chloroplast organization in A. stolonifera (Fig. 8, Table 7). In addition, serine hydroxymethyltransferase 1 (SHMT1) was significantly down-regulated only in A. stolonifera (Table 7). SHMT catalyzes the interconversion between serine and glycine [38]. Previous study of root proteomic profiles between A. stolonifera and A. scabra under heat stress showed that one SHMT protein spot was decreased only in A. stolonifera, which agreed our transcriptional observation [19, 20]. In contrast to transcript responses of heat-sensitive A. stolonifera, lack of down-regulation of some genes mentioned above in A. scabra suggest that the maintenance of transcriptional levels of genes in carbohydrate, and amino acid, and energy metabolism may be associated with the corresponding metabolite accumulation under heat stress, contributing to the superior heat tolerance.
Under heat stress, A. scabra showed up-regulation of several functional categories that were related in antioxidative responses and antioxidant protection, while many of the functional categories related to oxidative protection, such as peroxidase activity, peroxisome, were down-regulated in A. stolonifera (Figs. 8 and 9). Most of the up-regulated antioxidant-related genes were Cytochrome P450s (Table 6). The cytochrome P450 is a superfamily catalyzing various oxidative reactions, including biosynthesis of lipophilic compounds (fatty acids, sterols, cutin, suberine and wax, phenylpropanoids, brassinolides and gibberellins [39]. The microarray analysis of cytochrome P450 family in Arabidopsis showed that they are highly responsive to both abiotic and biotic stresses [40]. Other up-regulated genes involved in antioxidant defense included oxidoreductase and monooxygenase activity found in A. scabra under heat stress, which were also mainly involved in plant antioxidative response. ROS content in A. scabra root tissue under heat stress was significantly lower than that in A. stolonifera [41], suggesting that the ROS scavenging capacity was better maintained in A. scabra under heat stress. It is also worthy to point out the 4-hydroxyphenylacetaldehyde oxime monooxygenase that were uniquely down-regulated in A. stolonifera was also a Cytochrom P450 gene (CYP71E1), which is involved in the oxime-metabolizing step in biosynthesis of dhurrin [42]. Little information was known about dhurrin and its relation to heat response in plants, which deserves further investigation.
Transcripts in proline biosynthesis, mainly pyrroline-5-carboxylate reductase (P5CR), were also up-regulated in A. scabra under heat stress (Fig. 4, Table 6). P5CR is the final step in proline biosynthesis pathway, which reduces proline-5-carboxylate to proline [43]. It is generally accepted that proline acts as a cellular osmolyte, and thus its accelerated biosynthesis indicates enhanced plant osmotic stress resistance [44]. In addition, proline is also involved in maintenance of redox balance and ROS scavenging [45,46,47]. Higher levels of proline were identified in heat-stressed cotton (Gossypium hirsutum L.) [48], and its positive role in heat tolerance was confirmed in various plant species [1, 49, 50]. Our previous study found that proline content was significantly higher in A. scabra root tissues under heat stress than that in A. stolonifera [22]. However, there is little information regarding to P5CR expression regulation under heat stress. This study found the up-regulation of pyrroline-5-carboxylate reductase in A. scabra, which could play positive roles in the maintenance of proline synthesis in the heat-tolerant species under heat stress.
Most of the transcripts up-regulated in lipid catabolic process were GDSL esterases and Phospholipase A1 (Table 6). The GDSL-motif enzyme is a newly discovered lipase family that shares the highly conserved motif Gly-X-Ser-X-Gly (X means any amino acid) in the sequence [51, 52]. The number of GDSL esterase/lipase family members ranged from 57 to 130 in several plant organisms [53, 54]. The GDSL esterases/lipases might play an important role in plant development and morphogenesis [52]. Some of the GDSL esterases were reported to confer plant abiotic stress tolerance, such as drought and salt stress [55, 56]. Phospholipase A1 is one of the multigene family of phospholipases, hydrolyzing the sn-1 acylester bond of phospholipids to free fatty acids and 2-acyl-1-lysophospholipids [57]. Compared to mammalian phospholipase A1s, only a few genes were discovered in plants. A phospholipase A1 homolog in Arabidopsis, AtDAD1, was placed in the initial step of jasmonic acid biosynthesis, making it important for plant responses to abiotic stress, tendril coiling, fruit ripening and developmental maturation of stamens and pollens [58]. Another phospholipase A1 in hot pepper (Capsicum annuum) showed high sequence similarity to Arabidopsis [59]. Another phospholipase A1 homolog in Arabidopsis, AtLCAT3, was determined to have in vitro enzymatic activity, although its molecular function has yet to be assigned [60]. Therefore, the GDSL esterases and phospholipase A1s found in the up-regulated transcripts in A. scabra were also considered to be involved in lipid catalysis, possibly through jasmonic acid signal transduction pathway. This is first report of GDSL esterases and phospholipase A1s related to heat tolerance. Further studies regarding to their functions and regulation of heat tolerance in plants are needed.
The transcripts involved in cell wall structure and properties were up-regulated in A. scabra under heat stress, including xyloglucan endo-transglycosylases (XETs), and cellulose synthase (Table 6). XETs make nonhydrolytic cleavage and ligation of xyloglucan chains, which is involved in cell wall loosening [61]. Cellulose synthase family is also well-defined, and involved in the formation of plant primary and secondary cell wall [62] Plant cell wall structure undergoes reassembly that involves biosynthesis of major cell wall components during plant responses to abiotic stress [63,64,65]. Xu et al. [66] reported that transcript levels of XETs in tall fescue root tissues were decreased under water stress, and exogenous application of ascorbic acid could mitigate the reduction. Little information was known regarding to genes for cell wall biosynthesis and properties related to heat tolerance.
Several transcripts involved in lignan biosynthetic process were also up-regulated in A. scabra, including dirigent protein 5, aureusidin synthase 1, and (+)-larreatricin hydroxylase 1 (Table 6). Dirigent proteins play an important role in monolignol coupling to both lignin and lignan formations [67]. Dirigent protein family was reported to participate in defense responses, secondary metabolism, temperature, and salinity stress [68,69,70]. Aureusidin synthase is a binuclear copper enzyme, and a homolog of plant phenol oxidase [71]. It is proposed to be a chalcone-specific plant phenol oxidase for aurone biosynthesis [72]. Larreatricin hydroxylase is an enantio-specific polyphenol oxidase [73], but their physiological roles in plant adaptation to abiotic stress are unknown. Results in our study indicated that the up-regulation of genes involved in secondary cell-wall materials could contribute to the maintenance of cell wall structure and functional properties for A. scabra to maintain growth under heat stress.
Several transcription factors were uniquely up-regulated under heat stress, including high mobility group B protein (HMGB) 7, dehydration-responsive element-binding factor (DREB) 1a, Multiprotein-bridging factor (MBF) 1c, and CCCH-type zinc finger protein 47 (Table 11). The high mobility group B protein (HMGB) belongs to chromatin-associated proteins, and acts primarily as architectural facilitator in nucleoprotein complex assembly and transcriptional regulation and recombination [74, 75]. Little is known about its function in plant stress responses, except that Arabidopsis HMGBs showed induced expression levels under cold stress [76]. Dehydration-responsive element-binding factor (DREB) is one of the sub-groups in AP2/EREBP family, and activates target genes that have dehydration-responsive elements (DREs) [77, 78]. DREB1 and DREB2 were reported to confer plant drought, salinity and low-temperature tolerance [79,80,81], while only Arabidopsis DREB2A has dual functions in water-stress and heat-stress response [82]. Multiprotein-bridging factor 1 (MBF1) is a conserved transcriptional coactivator that bridges a basic region/leucine zipper (bZIP) type coactivator and a TATA-box binding protein [83, 84]. The Arabidopsis MBF1c was induced in response to heat stress [84, 85]. The transgenic Arabidopsis constitutively expressing MBF1c enhanced plant heat tolerance by perturbing ethylene response signal transduction pathway [86]. Plant CCCH-type zinc finger proteins were shown to be involved in embryo formation, floral reproductive organ formation, delay of leaf senescence, and calmodulin-mediated RNA processing [87,88,89,90]. Two CCCH-type zinc finger proteins, AtSZF1 and AtSZF2, were induced upon salt stress, and negatively regulate salt-responsive genes in Arabidopsis [91]. Our results suggest that the up-regulation of HMGB7, DREB1a, MBF1c, and CCCH-type zinc finger protein 47 could active the related down-stream genes regulating heat tolerance. Further research is needed to identify the down-stream genes in order to unravel the molecular roles of those transcriptional factors in heat tolerance.
Conclusions
In summary, our comparative analysis of transcriptomic changes in response to heat stress for heat-tolerant thermal A. scabra and heat-sensitive A. stolonifera showed divergent transcriptional regulations of heat tolerance in perennial grass species, which complemented to previous findings of physiological traits and proteins conferring the superior heat tolerance of the thermal species adapted to extremely high soil temperature. The potential novel transcriptional regulatory mechanisms for the superior heat tolerance in thermal A. scabra plants are proposed based on the results described above (Fig. 11). Heat stress could trigger molecular responses in A. scabra by up-regulating TFs, such as high mobility group B protein 7 (HMGB7), dehydration-responsive element-binding factor 1a (DREB1a), multiprotein-bridging factor 1c (MBF1c), CCCH-domain containing protein 47 (CCCH47), and downstream genes involved in serine metabolism (serine hydroxymethyltransferase, SHMT1), oxidative protection (cytochrome P450s), proline biosynthesis (pyrroline-5-carboxylate reductase, P5CR), lipid hydrolysis (GDSL estarases, phospholipase A1), hemicellulose and lignan biosynthesis (xyloglucan endo-transglycosylases, XETs and dirigent protein 5, aureusidin synthase 1, and (+)-larreatricin hydroxylase 1). The direct relationship and roles of those uniquely-expressed TFs and genes in heat-tolerant A. scabra than those in heat-sensitive A. stolonifera requires further confirmation.

Summary and proposed pathways for transcriptional regulation of heat tolerance in Agrostis grass species
Abbreviations
- AP2/ERF:
-
APETALA2/ethylene-responsive element binding factor
- CCCH:
-
CCCH-domain containing protein
- DEG:
-
Differentially expressed gene
- DREB:
-
Dehydration-responsive element binding factor
- DW:
-
Dry weight
- EL:
-
Electrolyte leakage
- FDR:
-
False discovery rate
- FW:
-
Fresh weight
- GO:
-
Gene ontology
- HMGB:
-
High mobility group B protein
- HSF:
-
Heat shock factor
- MBF:
-
Miltiprotein-bridging factor
- MYB:
-
Myeloblastosis factor
- P5CR:
-
Pyrroline-5-carboxylate reductase
- PAR:
-
Photosynthetically active radiation
- RH:
-
Relative humidity
- RWC:
-
Relative water content
- SHMT:
-
Serine hydroxymethyltransferase
- TF:
-
Transcription factor
- TW:
-
Turgid weight
- WRKY:
-
WRKY-domain factor
- XET:
-
Xyloglucan endo-transferase
References
Wahid A, Gelani S, Ashraf M, Foolad M. Heat tolerance in plants: an overview. Environ Exp Bot. 2007;61(3):199–223.
Martin L, Fei Z, Giovannoni J, Rose JKC. Catalyzing plant science research with RNA-seq. Front Plant Sci. 2013;4:66.
Zeller G, Henz SR, Widmer CK, Sachsenberg T, Rätsch G, Weigel D, Laubinger S. Stress-induced changes in the Arabidopsis thaliana transcriptome analyzed using whole-genome tiling arrays. Plant J. 2009;58(6):1068–82.
Zhang X, Rerksiri W, Liu A, Zhou X, Xiong H, Xiang J, Chen X, Xiong X. Transcriptome profile reveals heat response mechanism at molecular and metabolic levels in rice flag leaf. Gene. 2013;530(2):185–92.
González-Schain N, Dreni L, Lawas LM, Galbiati M, Colombo L, Heuer S, Jagadish KS, Kater MM. Genome-wide transcriptome analysis during anthesis reveals new insights into the molecular basis of heat stress responses in tolerant and sensitive rice varieties. Plant Cell Physiol. 2016;57(1):57–68.
Qin D, Wu H, Peng H, Yao Y, Ni Z, Li Z, Zhou C, Sun Q. Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using wheat genome array. BMC Genomics. 2008;9(1):432.
Mangelsen E, Kilian J, Harter K, Jansson C, Wanke D, Sundberg E. Transcriptome analysis of high-temperature stress in developing barley caryopses: early stress responses and effects on storage compound biosynthesis. Mol Plant. 2011;4(1):97–115.
Li Y-F, Wang Y, Tang Y, Kakani VG, Mahalingam R. Transcriptome analysis of heat stress response in switchgrass (Panicum virgatum L.). BMC Plant Biol. 2013;13(1):153.
Hu T, Sun X, Zhang X, Nevo E, Fu J. An RNA sequencing transcriptome analysis of the high-temperature stressed tall fescue reveals novel insights into plant thermotolerance. BMC Genomics. 2014;15(1):1147.
Frey FP, Urbany C, Hüttel B, Reinhardt R, Stich B. Genome-wide expression profiling and phenotypic evaluation of European maize inbreds at seedling stage in response to heat stress. BMC Genomics. 2015;16(1):123.
Li T, Xu X, Li Y, Wang H, Li Z, Li Z. Comparative transcriptome analysis reveals differential transcription in heat-susceptible and heat-tolerant pepper (Capsicum annum L.) cultivars under heat stress. J Plant Biol. 2015;58(6):411–24.
Stout RG, AL-NIEMI TS. Heat-tolerant flowering plants of active geothermal areas in Yellowstone National Park. Ann Bot. 2002;90(2):259–67.
Tercek MT, Hauber DP, Darwin SP. Genetic and historical relationships among geothermally adapted Agrostis (bentgrass) of North America and Kamchatka: evidence for a previously unrecognized, thermally adapted taxon. Am J Bot. 2003;90(9):1306–12.
Fry J, Huang B. Applied turfgrass science and physiology. Hoboken, NJ: John Wiley & Sons; 2004.
Rachmilevitch S, Huang B, Lambers H. Assimilation and allocation of carbon and nitrogen of thermal and nonthermal Agrostis species in response to high soil temperature. New Phytol. 2006;170(3):479–90.
Rachmilevitch S, Lambers H, Huang B. Root respiratory characteristics associated with plant adaptation to high soil temperature for geothermal and turf-type Agrostis species. J Exp Bot. 2006;57(3):623–31.
Rachmilevitch S, Xu Y, Gonzalez-Meler MA, Huang B, Lambers H. Cytochrome and alternative pathway activity in roots of thermal and non-thermal Agrostis species in response to high soil temperature. Physiol Plant. 2007;129(1):163–74.
Lyons EM, Pote J, DaCosta M, Huang B. Whole-plant carbon relations and root respiration associated with root tolerance to high soil temperature for Agrostis grasses. Environ Exp Bot. 2007;59(3):307–13.
Xu C, Huang B. Root proteomic responses to heat stress in two Agrostis grass species contrasting in heat tolerance. J Exp Bot. 2008;59(15):4183–94.
Xu C, Huang B. Differential proteomic response to heat stress in thermal Agrostis scabra and heat-sensitive Agrostis stolonifera. Physiol Plant. 2010;139(2):192–204.
Huang B, Rachmilevitch S, Xu J. Root carbon and protein metabolism associated with heat tolerance. J Exp Bot. 2012;63(9):3455–65.
Xu Y, HM D, Huang BR. Identification of metabolites associated with superior heat tolerance in thermal bentgrass through metabolic profiling. Crop Sci. 2013;53(4):1626–35.
Hoagland DR, Arnon DI. The water-culture method for growing plants without soil. Circ Calif Agric Exp Sta. 1950;347–461.
Barrs H, Weatherley P. A re-examination of the relative turgidity technique for estimating water deficits in leaves. Austr J Biol Sci. 1962;15(3):413–28.
Arnon DI. Copper enzymes in isolated chloroplasts. Polyphenoloxidase in Beta vulgaris. Plant Physiol. 1949;24(1):1.
Blum A, Ebercon A. Cell-membrane stability as a measure of drought and heat tolerance in wheat. Crop Sci. 1981;21(1):43–7.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–512.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–9.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Z-L H, Bao J, Reecy JM. CateGOrizer: a web-based program to batch analyze gene ontology classification categories. Online J Bioinform. 2008;9(2):108–12.
Zheng Q, Wang X-J. GOEAST: a web-based software toolkit for gene ontology enrichment analysis. Nucleic Acids Res. 2008;36(suppl 2):W358–W63.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25(4):402–8.
Xu Y, Huang B. Heat-induced leaf senescence and hormonal changes for thermal bentgrass and turf-type bentgrass species differing in heat tolerance. J Am Soc Hortic Sci. 2007;132(2):185–92.
Tian J, Belanger FC, Huang B. Identification of heat stress-responsive genes in heat-adapted thermal Agrostis scabra by suppression subtractive hybridization. J Plant Physiol. 2009;166(6):588–601.
Hanson AD, Roje S. One-carbon metabolism in higher plants. Annu Rev Plant Biol. 2001;52(1):119–37.
Bolwell GP, Bozak K, Zimmerlin A. Plant cytochrome P450. Phytochemistry. 1994;37(6):1491–506.
Narusaka M, Seki M, Umezawa T, Ishida J, Nakajima M, Enju A, Shinozaki K. Crosstalk in the responses to abiotic and biotic stresses in Arabidopsis: analysis of gene expression in cytochrome P450 gene superfamily by cDNA microarray. Plant Mol Biol. 2004;55(3):327–42.
Xu Y, Burgess P, Huang B. Root antioxidant mechanisms in relation to root thermotolerance in perennial grass species contrasting in heat tolerance. PLoS One. 2015;10(9):e0138268.
Bak S, Kahn RA, Nielsen HL, Møller BL, Halkier BA. Cloning of three A-type cytochromes P450, CYP71E1, CYP98, and CYP99 from Sorghum bicolor (L.) Moench by a PCR approach and identification by expression in Escherichia Coli of CYP71E1 as a multifunctional cytochrome P450 in the biosynthesis of the cyanogenic glucoside dhurrin. Plant Mol Biol. 1998;36(3):393–405.
Delauney AJ, Verma DPS. Proline biosynthesis and osmoregulation in plants. Plant J. 1993;4(2):215–23.
Kishor PK, Sangam S, Amrutha R, Laxmi PS, Naidu K, Rao K, Rao S, Reddy K, Theriappan P, Sreenivasulu N. Regulation of proline biosynthesis, degradation, uptake and transport in higher plants: its implications in plant growth and abiotic stress tolerance. Curr Sci. 2005;88(3):424–38.
Matysik J, Bhalu B, Mohanty P. Molecular mechanisms of quenching of reactive oxygen species by proline under stress in plants. Curr Sci. 2002;82(5):525–32.
Shao H-B, Chu L-Y, Z-H L, Kang C-M. Primary antioxidant free radical scavenging and redox signaling pathways in higher plant cells. Int J Biol Sci. 2008;4(1):8–14.
Szabados L, Savoure A. Proline: a multifunctional amino acid. Trends Plant Sci. 2010;15(2):89–97.
Ashraf M, Saeed M, Qureshi M. Tolerance to high temperature in cotton (Gossypium hirsutum L.) at initial growth stages. Environ Exp Bot. 1994;34(3):275–83.
Jiang Y, Huang B. Osmotic adjustment and root growth associated with drought preconditioning-enhanced heat tolerance in Kentucky bluegrass. Crop Sci. 2001;41(4):1168–73.
Cvikrová M, Gemperlová L, Dobrá J, Martincová O, Prásil IT, Gubis J, Vanková R. Effect of heat stress on polyamine metabolism in proline-over-producing tobacco plants. Plant Sci. 2012;182:49–58.
Brick DJ, Brumlik MJ, Buckley JT, Cao J-X, Davies PC, Misra S, Tranbarger TJ, Upton C. A new family of lipolytic plant enzymes with members in rice, Arabidopsis and maize. FEBS Lett. 1995;377(3):475–80.
Akoh CC, Lee G-C, Liaw Y-C, Huang T-H, Shaw J-F. GDSL family of serine esterases/lipases. Prog Lipid Res. 2004;43(6):534–52.
Ling H. Sequence analysis of GDSL lipase gene family in Arabidopsis thaliana. Pak J Biol Sci. 2008;11(5):763.
Volokita M, Rosilio-Brami T, Rivkin N, Zik M. Combining comparative sequence and genomic data to ascertain phylogenetic relationships and explore the evolution of the large GDSL-lipase family in land plants. Mol Biol Evol. 2011;28(1):551–65.
Naranjo MA, Forment J, RoldÁN M, Serrano R, Vicente O. Overexpression of Arabidopsis thaliana LTL1, a salt-induced gene encoding a GDSL-motif lipase, increases salt tolerance in yeast and transgenic plants. Plant Cell Environ. 2006;29(10):1890–900.
Hong JK, Choi HW, Hwang IS, Kim DS, Kim NH, Choi DS, Kim YJ, Hwang BK. Function of a novel GDSL-type pepper lipase gene, CaGLIP1, in disease susceptibility and abiotic stress tolerance. Planta. 2008;227(3):539–58.
Wang X. Plant phospholipases. Annu Rev Plant Biol. 2001;52(1):211–31.
Ishiguro S, Kawai-Oda A, Ueda J, Nishida I, Okada K. The DEFECTIVE IN ANTHER DEHISCENCE1 gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell. 2001;13(10):2191–209.
Seo YS, Kim EY, Mang HG, Kim WT. Heterologous expression, and biochemical and cellular characterization of CaPLA1 encoding a hot pepper phospholipase A1 homolog. Plant J. 2008;53(6):895–908.
Noiriel A, Benveniste P, Banas A, Stymne S, Bouvier-Navé P. Expression in yeast of a novel phospholipase A1 cDNA from Arabidopsis thaliana. Eur J Biochem. 2004;271(18):3752–64.
Eklöf JM, Brumer H. The XTH gene family: an update on enzyme structure, function, and phylogeny in xyloglucan remodeling. Plant Physiol. 2010;153(2):456–66.
Cosgrove DJ. Growth of the plant cell wall. Nat Rev Mol Cell Bio. 2005;6(11):850–61.
Sasidharan R, Voesenek LA, Pierik R. Cell wall modifying proteins mediate plant acclimatization to biotic and abiotic stresses. Crit Rev Plant Sci. 2011;30(6):548–62.
Tenhaken R. Cell wall remodeling under abiotic stress. Front Plant Sci. 2014;5:771.
Le Gall H, Philippe F, Domon J-M, Gillet F, Pelloux J, Rayon C. Cell wall metabolism in response to abiotic stress. Plants. 2015;4(1):112–66.
Xu Y, Xu Q, Huang B. Ascorbic acid mitigation of water stress-inhibition of root growth in association with oxidative defense in tall fescue (Festuca arundinacea Schreb.). Front Plant Sci. 2015;6:807.
Gang DR, Costa MA, Fujita M, Dinkova-Kostova AT, Wang H-B, Burlat V, Martin W, Sarkanen S, Davin LB, Lewis NG. Regiochemical control of monolignol radical coupling: a new paradigm for lignin and lignan biosynthesis. Chem Biol. 1999;6(3):143–51.
Ralph SG, Jancsik S, Bohlmann J. Dirigent proteins in conifer defense II: extended gene discovery, phylogeny, and constitutive and stress-induced gene expression in spruce (Picea spp.). Phytochemistry. 2007;68(14):1975–91.
Wu R, Wang L, Wang Z, Shang H, Liu X, Zhu Y, Qi D, Deng X. Cloning and expression analysis of a dirigent protein gene from the resurrection plant Boea hygrometrica. Prog Nat Sci. 2009;19(3):347–52.
Gao C, Liu G, Wang Y, Jiang J, Yang C. Cloning and analysis of dirigent-like protein in gene from Tamarix androssowii. Bull Bot Res. 2010;30(1):81–6.
Nakayama T, Yonekura-Sakakibara K, Sato T, Kikuchi S, Fukui Y, Fukuchi-Mizutani M, Ueda T, Nakao M, Tanaka Y, Kusumi T. Aureusidin synthase: a polyphenol oxidase homolog responsible for flower coloration. Science. 2000;290(5494):1163–6.
Nakayama T, Sato T, Fukui Y, Yonekura-Sakakibara K, Hayashi H, Tanaka Y, Kusumi T, Nishino T. Specificity analysis and mechanism of aurone synthesis catalyzed by aureusidin synthase, a polyphenol oxidase homolog responsible for flower coloration. FEBS Lett. 2001;499(1–2):107–11.
Cho M-H, Moinuddin SG, Helms GL, Hishiyama S, Eichinger D, Davin LB, Lewis NG. (+)-Larreatricin hydroxylase, an enantio-specific polyphenol oxidase from the creosote bush (Larrea tridentata). Proc Natl Acad Sci. 2003;100(19):10641–6.
Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19(8):5237–46.
Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem Sci. 2001;26(3):167–74.
Kwak KJ, Kim JY, Kim YO, Kang H. Characterization of transgenic Arabidopsis plants overexpressing high mobility group B proteins under high salinity, drought or cold stress. Plant Cell Physiol. 2007;48(2):221–31.
Sakuma Y, Liu Q, Dubouzet JG, Abe H, Shinozaki K, Yamaguchi-Shinozaki K. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration-and cold-inducible gene expression. Biochem Biophys Res Commun. 2002;290(3):998–1009.
Agarwal PK, Agarwal P, Reddy M, Sopory SK. Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep. 2006;25(12):1263–74.
Liu Q, Kasuga M, Sakuma Y, Abe H, Miura S, Yamaguchi-Shinozaki K, Shinozaki K. Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought-and low-temperature-responsive gene expression, respectively, in Arabidopsis. Plant Cell. 1998;10(8):1391–406.
Kasuga M, Miura S, Shinozaki K, Yamaguchi-Shinozaki K. A combination of the Arabidopsis DREB1A gene and stress-inducible rd29A promoter improved drought-and low-temperature stress tolerance in tobacco by gene transfer. Plant Cell Physiol. 2004;45(3):346–50.
Z-S X, Ni Z-Y, Li Z-Y, Li L-C, Chen M, Gao D-Y, X-D Y, Liu P, Ma Y-Z. Isolation and functional characterization of HvDREB1 - a gene encoding a dehydration-responsive element binding protein in Hordeum vulgare. J Plant Res. 2009;122(1):121–30.
Sakuma Y, Maruyama K, Osakabe Y, Qin F, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell. 2006;18(5):1292–309.
Takemaru K-i, Harashima S, Ueda H, Hirose S. Yeast coactivator MBF1 mediates GCN4-dependent transcriptional activation. Mol Cell Biol. 1998;18(9):4971–6.
Tsuda K, Tsuji T, Hirose S, Yamazaki K-i. Three Arabidopsis MBF1 homologs with distinct expression profiles play roles as transcriptional co-activators. Plant Cell Physiol. 2004;45(2):225–31.
Rizhsky L, Liang H, Shuman J, Shulaev V, Davletova S, Mittler R. When defense pathways collide. The response of Arabidopsis to a combination of drought and heat stress. Plant Physiol. 2004;134(4):1683–96.
Suzuki N, Rizhsky L, Liang H, Shuman J, Shulaev V, Mittler R. Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol. 2005;139(3):1313–22.
Li J, Jia D, Chen X. HUA1, a regulator of stamen and carpel identities in Arabidopsis, codes for a nuclear RNA binding protein. Plant Cell. 2001;13(10):2269–81.
Schmitz RJ, Hong L, Michaels S, Amasino RM. FRIGIDA-ESSENTIAL 1 interacts genetically with FRIGIDA and FRIGIDA-LIKE 1 to promote the winter-annual habit of Arabidopsis thaliana. Development. 2005;132(24):5471–8.
Delaney KJ, Xu R, Zhang J, Li QQ, Yun K-Y, Falcone DL, Hunt AG. Calmodulin interacts with and regulates the RNA-binding activity of an Arabidopsis polyadenylation factor subunit. Plant Physiol. 2006;140(4):1507–21.
Kong Z, Li M, Yang W, Xu W, Xue Y. A novel nuclear-localized CCCH-type zinc finger protein, OsDOS, is involved in delaying leaf senescence in rice. Plant Physiol. 2006;141(4):1376–88.
Sun J, Jiang H, Xu Y, Li H, Wu X, Xie Q, Li C. The CCCH-type zinc finger proteins AtSZF1 and AtSZF2 regulate salt stress responses in Arabidopsis. Plant Cell Physiol. 2007;48(8):1148–58.
Acknowledgements
Authors wish to thank Center for Turfgrass Science, Rutgers University and New Jersey Agricultural Experiment Station for funding support of this research project.
Funding
Center for Turfgrass Science, Rutgers University and New jersey Agricultural Experiment Station.
Availability of data and materials
The transcriptome shotgun assembly of both A. stolonifera and A. scabra were deposited at GenBank Transcriptome Shotgun Assembly (TSA) database, under the accession of GFJH00000000 and GFIW00000000, respectively. The version described in this paper is the first version, GFJH01000000 and GFIW01000000. Other than that, all the data is contained within the manuscript.
Author information
Authors and Affiliations
Contributions
YX and BH designed the experiment and wrote the manuscript; YX conducted the experiment and data analysis. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Plant materials were properties of Rutgers University. Our experimental research complies with institutional, national and international guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Heat map of GO term enrichment analysis for up-regulated DEGs in A. stolonifera (P) and A. scabra (N). Scale represents log10 of P-value in the enrichment analysis. (JPEG 575 kb)
Additional file 2:
Heat map of GO term enrichment analysis for down-regulated DEGs in A. stolonifera (P) and A. scabra (N). Scale represents log10 of P-value in the enrichment analysis. (JPEG 3438 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Xu, Y., Huang, B. Transcriptomic analysis reveals unique molecular factors for lipid hydrolysis, secondary cell-walls and oxidative protection associated with thermotolerance in perennial grass. BMC Genomics 19, 70 (2018). https://doi.org/10.1186/s12864-018-4437-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4437-z