
Abstract
Background
Leucine-rich repeat receptor-like protein kinase (LRR-RLK) is the largest gene family of receptor-like protein kinases (RLKs) and actively participates in regulating the growth, development, signal transduction, immunity, and stress responses of plants. However, the patterns of LRR-RLK gene family evolution in the five main Rosaceae species for which genome sequences are available have not yet been reported. In this study, we performed a comprehensive analysis of LRR-RLK genes for five Rosaceae species: Fragaria vesca (strawberry), Malus domestica (apple), Pyrus bretschneideri (Chinese white pear), Prunus mume (mei), and Prunus persica (peach), which contained 201, 244, 427, 267, and 258 LRR-RLK genes, respectively.
Results
All LRR-RLK genes were further grouped into 23 subfamilies based on the hidden Markov models approach. RLK-Pelle_LRR-XII-1, RLK-Pelle_LRR-XI-1, and RLK-Pelle_LRR-III were the three largest subfamilies. Synteny analysis indicated that there were 236 tandem duplicated genes in the five Rosaceae species, among which subfamilies XII-1 (82 genes) and XI-1 (80 genes) comprised 68.6%.
Conclusions
Our results indicate that tandem duplication made a large contribution to the expansion of the subfamilies. The gene expression, tissue-specific expression, and subcellular localization data revealed that LRR-RLK genes were differentially expressed in various organs and tissues, and the largest subfamily XI-1 was highly expressed in all five Rosaceae species, suggesting that LRR-RLKs play important roles in each stage of plant growth and development. Taken together, our results provide an overview of the LRR-RLK family in Rosaceae genomes and the basis for further functional studies.
Similar content being viewed by others

Background
During growth and development, plants are affected by many different kinds of signal stimulation from cell-cell and cell-environment interactions, which trigger a series of signal transductions. The regulatory mechanism of reversible phosphorylation that involves protein kinases (PKs) plays an important role in cellular signal transduction processes and all living cells can perceive and process external or internal signals via cell surface PKs, which mostly belong to the receptor-like kinase (RLK) family [1, 2]. RLKs are ubiquitous in the plant kingdom. Since the first leucine-rich repeat receptor-like kinase (LRR-RLK) gene was isolated in maize (Zea mays) [3], more and more LRR-RLK genes have been confirmed [2, 4]. However, the function of most RLKs has not been confirmed and the natural ligands of most receptor proteins have not been identified.
LRR-RLK is the largest and most highly conserved group of the plant RLK gene family [5]. Previous studies have shown that the approximately 225 LRR-RLK gene members in Arabidopsis can be grouped into 23 subfamilies based on the sequence similarity in their kinase domains [2, 6, 7]. A typical LRR-RLK contains an extracellular tandem arrayed leucine-rich repeat domain, a single-pass transmembrane domain, and an intracellular functional protein kinase domain. The major class of extracellular domain is 20–30 amino acids long. With regards to tertiary structure, each LRR domain forms an α/ß helix and is located on the surface of the protein, enabling it to participate in protein-protein interactions [2, 8]. Because of their structural characteristics, LRR-RLKs play essential roles in mediating cell signal transduction pathways and critically control plant growth, development, responses to stresses, and immunity. In the Brassinosteroid (BR) and abscisic acid (ABA) signal pathways, LRR-RLKs play a pivotal role. BRs are important regulatory substances in plant development. The receptor kinase Brassinosteroid-insensitive 1 (BRI1), which has 25 LRR tandem structures in its extracellular portion, can perceive and deliver BR signaling [9]. BRI1-associated kinase 1 (BAK1) is another LRR-RLK that has five LRR tandem structures in its extracellular portion. The cross-talk between BRI1 and BAK1 makes up a dimer, the key step in the BR signal transduction [10]. In Arabidopsis, receptor-like protein kinase 1 (RPK1) is an LRR-RLK on the surface of the plasma membrane, the LRR domain of which is key to regulating ABA sensitivity [11]. Previous research has shown that the rpk1 mutant exhibits decreased sensitivity to ABA-induced senescence in Arabidopsis leaves, but does not exhibit a change in jasmonic acid- and ethylene-induced senescence [12]. BAK1 not only plays a role in the process of BR signal transmission, but also forms a dimer with the resistance gene flagellin-sensitive 2 (FLS2), and is involved in plant immune responses. BAK1 may also combine with other ligands and participate in other LRR-RLK signaling pathways [13].
There are a large number of LRR-RLKs in plants, but intensive studies on their functions have only been performed for a few genes. Because of the complexity of plant signal transduction pathways and complementary functions between different proteins, very few LRR-RLK ligands and complex LRR-RLK signaling pathways have been found. Thus, there is a great need for system analyses of the evolution and expression differentiation of the LRR-RLK gene family using bioinformatics tools, as well as to identify genes that have potential functions in plant responses to environmental factors; this will also help us to improve our understanding of LRR-RLK functions in regulating networks during plant growth. Recently, LRR-RLK gene family have been analyzed in soybean [14], two Citrus species [15], Amborella trichopoda [16], and evolutionary studies in various species [17, 18]. Specifically, Fischer et al. investigated the involvement of selection during the expansion of the LRR-RLK gene family among angiosperms by analyzing the phylogeny of 7554 LRR-RLK genes from 31 fully sequenced flowering plant genomes, including two genomes of Rosaceae, apple (Malus domestica) and peach (Prunus persica) [18]. However, a more comprehensive investigation of LRR-RLK genes in a wider range of Rosaceae genomes is needed. In this study, we aimed to elucidate the structure, expression, and evolution of Rosaceae LRR-RLK genes. To this end, we analyzed a total of 1622 LRR-RLK genes in 23 LRR-RLK subfamilies in five Rosaceae species [strawberry (Fragaria vesca), peach, mei (Prunus mume), apple and Chinese white pear (Pyrus bretschneideri)] and Arabidopsis, including their chromosomal location, gene structure, duplication events, expansion, expression, and co-expression relationships, which should provide genome-level insights on LRR-RLK genes.
Results
Genome-wide identification and classification of LRR-RLK genes
We searched all annotated genes in the five Rosaceae species (strawberry, peach, mei, apple and Chinese white pear as shown in Table 1 and Fig. 1) for putative PKs and identified 6680 typical PKs with the number of typical PKs in each species ranged from 856 in strawberry to 1614 in Chinese white pear (Additional file 1: Tables S1 and S2). PKs were then classified into families and subfamilies using the HMM search approach, as described in the Methods section, which resulted in 1622 genes being classified into the LRR-RLK family, ranging from 201 genes in strawberry to 427 genes in Chinese white pear (Fig. 1, Table 2, Additional file 1: Table S3). The LRR-RLK genes identified in Arabidopsis and Rosaceae were further classified into 23 subfamilies and showed different patterns of gene subfamily size. Of these subfamilies, XII-1 (also named RLK-Pelle_LRR-XII-1), XI-1, and III were the top three largest subfamilies with an average number of members of 47.2, 66.0, and 44.0. The other subfamilies consisted of no more than 24 members, of which Xb-2 was the smallest subfamily with only one or two members.

The phylogenetic tree of the species used for identifying LRR-RLK genes with whole genome duplication event marked in green star. The number in the parentheses indicate number of LRR-RLK genes in each species
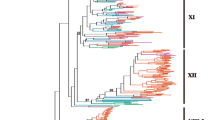
To confirm the accuracy of identification results, we used the method above to identify the LRR-RLK genes in Arabidopsis as a reference (Additional file 1: Tables S1 and S2) and compared the identified LRR-RLK genes in Arabidopsis in this study with previously reported results [6]. The list of LRR-RLK genes in Arabidopsis in this study was identical as reported previously [6], indicating that our gene identification procedure was reliable. We also constructed a phylogenetic tree for LRR-RLK genes in Arabidopsis and the five Rosaceae species (Fig. 2), which showed that most genes in the same subfamily were classified together.

The phylogenetic tree for LRR-RLK genes identified in Arabidopsis and Rosaceae. A total of 1622 LRR-RLK genes were classified into 23 subfamilies and are distinguished by different colors
Protein domain and intron number analyses revealed divergent evolution for different subfamilies
The total of 1622 LRR-RLK genes identified from Arabidopsis and the five Rosaceae species were scanned against the Pfam database version 28.0 to identify additional protein domains besides Pkinase that may be common among subfamilies. The protein domains identified for each gene are listed in Additional file 1: Table S4. The basic composition of the LRR-RLK gene family consisted of a PK domain and an LRR domain; however, the results revealed that the frequency of Pkinase and LRR domains in each subfamily were different, and the preferred Pkinase and LRR domains were different (Additional file 1: Table S5). For instance, II preferentially included Pkinase, VI-1 preferentially included Pkinase_Tyr, while the result for VII-1 was mixed. In addition, XIIIa preferentially included LRRNT_2 (leucine-rich repeat N-terminal 2) and XIIIb preferentially included LRR_8 (leucine-rich repeat 8). And most genes have more than one type of LRR. These results suggest that different subfamilies have different compositions of protein domains, but genes in the same subfamily share common arrangements, indicating the common evolutionary history within the subfamilies.
To obtain further insights into the structural diversity of the Arabidopsis and Rosaceae LRR-RLK genes, we analyzed their intron numbers (Table 3; Additional file 1: Table S6). These were found to vary widely, ranging from 0 to 42 (an LRR-RLK gene of mei, Pm019437). Most genes (965 or 59.5%, including 114 Arabidopsis genes) contained fewer than three introns (see Additional file 1: Table S6 for a summary and Additional file 1: Table S3 for a detailed list) and there were 250 genes with more than ten introns. Based on the frequency of LRR-RLK genes with different intron sizes (Fig. 3), we found that the frequency of Rosaceae genes with fewer than three introns was greater than that in Arabidopsis; in the latter, this proportion was 50%, compared with proportions ranging from 56% in apple to 68% in Chinese white pear. Similarly, the frequency of genes with an intron number between three and ten was higher in the five Rosaceae species than that in Arabidopsis. The average number of intron numbers in XIIIb was 27.2, significantly higher than other subfamilies. The average number of intron numbers in Xb-2, Xb-1, XV, Xa, VII-3 was lower than 1. These findings suggest that different subfamilies have different patterns of intron number and intron number could be used to represent the course of evolution in these subfamilies, and also verify our previous classification process.

The frequency of genes with different intron sizes in the LRR-RLK gene family in Arabidopsis, strawberry, apple, Chinese white pear, mei, and peach
Segmental duplication
Genome duplications are believed to have had a significant impact on the expansion of certain gene families in plants [19]. A previous study [20] suggested that the apple and Chinese white pear genomes had expanded through two rounds of duplication, named recent duplication and paleohexaploidization. The recent duplication occurred approximately 30–45 million years ago (MYA) [synonymous substitution rate (Ks) between 0.15 and 0.30] in the apple and Chinese white pear genomes, while the paleohexaploidization occurred approximately 140 MYA (Ks between 1.5 and 1.8). To trace which genes originated from these two duplication events, intra-genome scanning of collinearity blocks was performed (Additional file 1: Table S7, Fig. 4) and Ks values were calculated for each gene pair. Gene pairs traced to the recent duplication were only found in apple and Chinese white pear, while gene pairs traced to the paleohexaploidization were found in all five Rosaceae species. However, considering a large number of genes of the LRR-RLK gene family in each species, only a few gene pairs were traced to the paleohexaploidization (Arabidopsis thaliana: 2, Malus domestica: 3, Fragaria vesca: 6, Pbr: 7, Prunus mume: 6, Prunus persica: 8). Among these gene pairs that were traced to the paleohexaploidization event, III was the only subfamily found in all five Rosaceae species, indicating that this subfamily is essential for the Rosaceae, given that it is ancient and conserved. On the other hand, 20 gene pairs in apple and 68 pairs in Chinese white pear were found to be associated with the recent duplication event, indicating that this event contributed more to the expansion of the LRR-RLK gene family in these two species. The proportion of LRR-RLK genes in each species associated with whole genome duplication (WGD)/segmental duplication (Additional file 1: Table S8) had a wide range, from 16% in mei to 51% in Chinese white pear. Although duplicated gene pairs could be found in all subfamilies, some of them were lineage-specific. For instance, XIIIa and VII-2 showed WGD/segmental duplication only in Chinese white pear, Xb-1 showed it only in apple and Chinese white pear, and XIV showed it only in Rosaceae, but not in Arabidopsis; these findings provide clues for how different subfamilies evolved in Rosaceae.

The circos figure for chromosome locations with segmental duplication links. a Arabidopsis thaliana, b Fragaria vesca, c Malus domestica, d Pyrus bretschneideri, e Prunus mume, f Prunus persica. The blue lines indicate segmented duplicated gene pairs that were traced to the paleohexaploidization event (~140 million years ago). The yellow lines indicate segmented duplicated gene pairs that were tranced to recent duplication event (30~45 million years ago)
Tandem duplication
Tandem duplication is another source of gene family expansion as it increases gene numbers and genome complexity [21,22,23,24]. Thus, to examine how tandem duplication contributed to LRR-RLK gene family expansion, we compared the number of tandem duplicated genes in each species (Additional file 1: Table S9) and found that there were 12 tandem arrays in Arabidopsis, 11 in strawberry, 14 in apple, 33 in Chinese white pear, 25 in mei, 27 in peach, and 37 tandem duplicated genes in Arabidopsis. These involved 24 genes in strawberry, 32 genes in apple, 67 genes in Chinese white pear, 51 genes in mei, and 62 genes in peach (Additional file 1: Table S10). In Arabidopsis, we found that most tandem duplication events (28/37 genes) had occurred in the RLK-Pelle_LRR-I-1 subfamily. Similarly, tandem duplications in strawberry, apple, Chinese white pear, mei, and peach seem to have contributed to the generation of approximately 50.0% (12/24 genes), 50.0% (16/32 genes), 25.4% (17/67 genes), 29.4% (15/51 genes), and 32.3% (20/62 genes) of the XI-1 subfamily, respectively (Additional file 1: Table S10). The XII-1 subfamily was also shown to have many genes generated through tandem duplications in Chinese white pear, mei, and peach.
Expression pattern analysis by transcriptome data
To explore the expression patterns of LRR-RLK genes, we analyzed publicly available RNA-seq (RNA sequencing) or transcriptome data from the NCBI (National Center for Biotechnology Information) SRA (Sequence Read Archive) database, for a total of 108 samples from 18 experiments, including pooled organs, biotic stress, abiotic stress, tissue-specific expression, fruit development, and developmental biology (Additional file 1: Tables S11–S15, Additional file 2: Figures S1–S5). In general, we found that not all genes were expressed. The number of LRR-RLK genes in any tissue with FPKM >1 was 97 in strawberry (45.3%), 216 (88.5%) for apple, 338 (79.2%) for pear, 195 (73.0%) for mei and 58 (22.5%) for peach. These expressed (FPKM >1 in any tissue) genes were found in all LRR-RLK subfamilies.
The expression profiles of genes in pollen, pistil, or stigma are an important issue in developmental biology. In Chinese white pear, the expression of LRR-RLK genes was examined in four growth stages of pollen: mature pollen grain (MP), hydrated pollen grain (HP), growing pollen tube (PT), and stopped-growth pollen tube (SPT) [25]. We found that a cluster of genes was relatively higher expressed in pear pollen (Fig. 5), among which 13 genes (names highlighted in red in Fig. 5) were relative higher expressed during pollen growth. These genes distributed in subfamilies III, V, VI-2, and XII-1 (Additional file 2: Figure S3, Additional file 1: Table S13). To confirm the expression patterns, we verified the expression by qRT-PCR as shown in Fig. 6. The results showed that 10 of these genes had similar expression patterns (Pearson correlation coefficiency (PCC) > 0.2, as labeled in Fig. 6) as transcriptomic data. Specifically, the top expressed gene Pbr011934.4 (subfamily III) was an ortholog of Arabidopsis gene PRK1 (AT5G35390), which accumulated in the plasma membrane of the apical growing tip of pollen tube through the process of exocytosis [26]. Similarly, the subfamily III was also found highly expressed in the pollen of mei. But in the pistil of mei, the highly expressed genes distributed in a variety of subfamilies, including subfamily Xa, II, VI-2, XI-1, etc. One of the top highly expressed genes in the pistil of mei was Pm00409.1 (Subfamily II), an ortholog of AtSERK2 (AT1G34210). And in the stigma of peach, subfamilies XI-1 and Xb-1 were highly expressed. The highest expressed gene in the stigma of peach was ppa001010m, a homolog of Arabidopsis gene AT1G09970.2 (Additional file 1: Table S15), which is involved in the control of germination speed and tolerance to oxidative stress [27]. These indicate spatial expression differences of LRR-RLK gene family in the development of flower.

The heatmap of the expression of a cluster of pear genes that are highly expressed in pear pollens. The ID of the 13 genes that highly expressed in pollens are marked in red. The FPKM values are log2 transformed

qRT-PCR verification of 13 pear genes in four developmental stages of pear pollens, including mature pollen grain (MP), hydrated pollen grain (HP), growing pollen tube (PT), and stopped-growth pollen tube (SPT). The Pearson Correlation Coefficient (PCC) of the expression patterns with their corresponding transcriptome data as presented in Fig. 5 are labeled for each gene
Strawberry and apple are important fruit-producing crops, although the edible fruit in strawberry is different from apple in that it is actually enlarged receptacle tissue [28]. Expression in the apple mature fruit and developmental stages of strawberry fruits revealed that LRR-RLK genes were also involved in fruit development. Among these genes, the top three expressed genes in strawberry were mrna21119.1, mrna17482.1 and mrna05604.1, belonging to subfamilies Xb-1, Xa, and XI-1, respectively. These genes are orthologs of the Arabidopsis genes AtBRI1, BIR1 and HSL2 (Additional file 1: Table S11, Additional file 2: Figure S1). In apple mature fruits, the top three expressed genes in apple mature fruits were MDP0000148501, MDP0000280908 and MDP0000266980, belonging to the subfamilies XI-1, III and XIIIa, respectively, are orthologs of Arabidopsis genes BAM1, RKL1 and FEI1 (Additional file 1: Table S12, Additional file 2: Figure S2).
Additionally, LRR-RLK genes are also involved in the response to biotic and abiotic stresses. The top three highly expressed genes in the Chinese white pear samples that were treated with Alternaria alternata (black spot), salt and GA were Pbr024019.1 (subfamily XI-2), Pbr018826.1 (subfamily XI-1) and Pbr019916.1 (subfamily XIIIb), respectively. Furthermore, Pbr024019.1 is the ortholog of Arabidopsis gene SOBIR gene (AT2G31880.1), which is expressed in response to the bacteria Pseudomonas syringae and regulates cell death and innate immunity [29]. This indicates the potential roles in stress response of these top expressed genes.
Co-expression network of LRR-RLK subfamilies
The gene expression patterns analyzed above showed that some genes have potential co-expression relationships. Thus, we created co-expression networks for LRR-RLK genes in each species using the available datasets as mentioned above. To construct these co-expression networks, we performed pairwise Pearson correlation coefficient (PCC) analyses for each species and used a threshold of PCC > 0.95 (Additional file 2: Figures S6–S10, Additional file 1: Table S16). In strawberry, ten LRR-RLK genes formed four small sub-network with two or three nodes. In apple, 66 LRR-RLK genes with 130 co-expression events formed three bigger co-expression sub-networks (12 to 14 nodes) and 11 small sub-networks (two to five nodes). In pear, 293 LRR-RLK genes with 735 edges formed the main sub-network (182 nodes) and 24 small sub-networks (two to 30 nodes). In mei, 197 LRR-RLK genes with 1515 co-expression events formed the main sub-network (165 nodes) and 11 small sub-networks (two to eight nodes). In peach, 161 LRR-RLK genes with 1085 co-expression events formed two bigger sub-networks (57 and 49 nodes) and 17 small sub-networks (two to five nodes). The frequency distribution of co-expression events for each gene showed that some genes are more highly connected, exhibiting a higher number of edges, while others are less connected. These genes could be considered as hubs with importance in plant growth, development, and stress signaling. Comparing the top connected genes in each species revealed that genes in the subfamily XI-1 were in four of the five Rosaceae species. Specifically, mrna22117.1 (subfamily XI-1) and mrna23069.1 (subfamily I-1) for strawberry (number of edges = 2), MDP0000284907 (subfamily III), MDP0000166578 (subfamily XI-1), MDP0000309850 (subfamily XII-1) and MDP0000229275 (subfamily XI-1) for apple (number of edges = 10), Pm019513 (subfamily XI-1) for mei (number of edges = 46), ppa023423m (subfamily XII-1) for peach (number of edges = 38) and Pbr002506.1 (subfamily XI-1) for Chinese white pear (number of edges = 17).
Tissue-specific expression and subcellular localization of Chinese white pear LRR-RLK genes
AtBRI1 and AtBAK1 (also named AtSERK3) play key regulatory roles in BR signaling processes in Arabidopsis. In Chinese white pear, we identified two BRI1 orthologs (PbrBRI1a and PbrBRI1b), three BRI1-Like (BRL) genes (PbrBRL1, PbrBRL2 and PbrBRL3, orthologs of AtBRL1, AtBRL2 and AtBRL3) (Fig. 7a) and five Somatic Embryogenesis Receptor-like Kinase (SERK) genes (Fig. 7b) that belong to the Xb-1 and II subfamilies, respectively. To study the gene regulatory network that controls pollen tube growth, expression pattern analyses of these genes were performed using reverse transcriptase PCR (RT-PCR) with RNA isolated from seven tissues (roots, stems, leaves, fruit flesh, pollen tubes, styles and ovaries) of Pyrus bretschneideri cv. Dangshansuli as shown in Fig. 7c–d. In roots, stems, leaves, fruit flesh, styles and ovaries, the expression of all PbrBRLs and PbrSERKs was detected. However, in pollen tubes, only three pear BRI1/BRLs genes (PbrBRI1a, PbrBRL1 and PbrBRL3) and four PbrSERKs (PbrSERK1, 2, 3, and 4) were expressed. Specifically, PbrSERK2 was expressed in a very higher level compared with other PbrSERKs. Based on the results of RT-PCR, we tested the seven expressed genes (PbrBRI1a, PbrBRL1, and PbrBRL3; PbrSERK1, 2, 3, and 4) for BR treatment experiment. The results revealed that exogenous BR could significantly increase the expression of these gene (Fig. 7e). Then, we tested the subcellular localization of PbrSERK2 by the overexpression of 35S::PbrSERK2-YFP in Arabidopsis leaf protoplast stained with the FM4–64 dye were used as a positive contrast for cytoplasmic membrane. We found the extensive accumulation of yellow fluorescent protein (YFP) signals and red fluorescent (FM4–64) signals overlapping at the cell membrane (Fig. 7f), indicating that PbrSERK2 was located on the cell membrane. These results reconfirmed the accuracy and reliability of the methods of this study and indicated that PbrSERK2 might participate in BR signaling pathway in Chinese white pear.

Phylogenetic trees, tissue-specific expression, and subcellular localization for the pear BRI1/BRL and SERK genes. a The phylogenetic tree of the BRI1/BRL genes identified in Arabidopsis and Rosaceae. b The phylogenetic tree of the BAK1/SERK genes identified in Arabidopsis and Rosaceae. c Tissue-specific expression of pear BRI1/BRL genes. d Tissue-specific expression of pear SERK genes. e The expression of seven selected pear BRI1/BRL and SERK genes in pollen tubes before and after BR treatment. All these seven genes expression has increased after exogenous BR treatment. f Subcellular localization analysis of PbrSERK2 in the membranes of the Arabidopsis mesophyll protoplasts. YFP and FM4–64 fluorescence were localized exclusively to the membranes. Bar = 2 μm
Discussion
In previous research, RLK genes were divided into six groups according to differences in the structures of their RLK extracellular domain, as follows: LRR [30,31,32], pathogenesis-related protein 5-like receptor kinase (PR5K) [33, 34], epidermal growth factor-like repeats (EGF) [35, 36], lectin-binding domain (LB) [37, 38], tumor necrosis factor receptor-like (TNFR) [39, 40], and S-domain [3, 41]. LRR-RLKs constitute more than half of the total number of RLKs. This group is one of the largest gene families in plants, playing important roles in plant growth and responses to pathogens [42,43,44]. In this study, we identified 1397 LRR-RLK genes in Rosaceae; of these, 201 genes were in strawberry, 244 in apple, 427 in Chinese white pear, 267 in mei, and 258 in peach (Table 2). These numbers are far greater than the average gene family size, which ranges from 1.71 to 2.17 in Arabidopsis, Chinese white pear, apple, and strawberry [20]. It was also noted that strawberry, apple, Chinese white pear, mei, and peach LRR-RLK genes represented 23.5%, 22.8%, 26.5%, 24.1%, and 25.2% of their total typical PK genes, respectively. These proportions are all higher than that in Arabidopsis (22.3%). In a previous study, it was asserted that LRR-RLK gene family expansion is mainly a consequence of high rates of duplicate retention [45], and that segmental and tandem duplication are two major sources of this expansion. In this study, we found that approximately one-third to two-thirds of LRR-RLK genes in Rosaceae could be explained by segmental duplication or tandem duplication, confirming this hypothesis. Considering the high ratio of LRR-RLK genes compared to PK genes in Rosaceae, the expansion of LRR-RLK genes could partially explain why the PK superfamily is larger in these species than in Arabidopsis. In Rosaceae, the Pyreae tribe was reported to have undergone one recent duplication [46] and it was found that 57% of apple LRR-RLK genes attributed to segmental duplication could be explained by this recent duplication, while the corresponding proportion was 54% in Chinese white pear. This explains why the proportions of total segmental duplicated genes in the Pyreae tribe are higher than in other studied species. We also noted that the genome and proteome sizes in the Pyreae tribe are larger than in other studied species, which could be another reason for the larger gene family size of LRR-RLK in these species. However, there is one exception to this in the apple genome, where the number of LRR-RLK genes identified were much less than in Chinese white pear, because we used the apple genome version 1.0 pseudo-haplotype assembly primary resource in the Genome Database for Rosaceae (GDR) [47]. The total number of genes in this version of the apple genome is 30,294, which is approximately half of that in the apple genome version 1.0. Nevertheless, we still observed a higher proportion of genes attributed to recent duplication.
Novel gene functions are acquired in four processes of genetic evolution: chromosome doubling, chromosome fragment insertion mutation, tandem duplication, and transposition [19, 48, 49]. Duplications can help species resist stress and to adapt to diverse environments by providing abundant genetic materials for evolution and producing numerous genetic variations. As described above, duplications could have driven the lineage-specific expansion of the LRR-RLK gene family, especially in the Pyreae tribe. We also noted that duplications had different effects on specific subfamilies, especially for tandem duplications. For instance, in Arabidopsis, 37 genes assigned to tandem duplication arrays were distributed in only five subfamilies, which represented approximately 16% of the 225 LRR-RLK genes; in addition, in Chinese white pear, 67 genes assigned to tandem duplication arrays were distributed in six subfamilies. Furthermore, the proportions of genes in tandem duplication arrays against the total LRR-RLK genes in each species in Rosaceae were not consistent, from 12% in strawberry and 13% in apple, less than in Arabidopsis, to 16% in Chinese white pear, 19% in mei, and 24% in peach, greater than or equal to that in Arabidopsis (Additional file 1: Table S10). Although different species had different numbers of subfamilies that expanded through tandem duplication, the observation that some subfamilies were consistently expanded through tandem duplication in all studied species (I-1, XI-1, and XII-1) while others were not indicates the preference for tandem duplication. Those subfamilies that expanded markedly through tandem duplication were partly consistent with those subfamilies with the greatest number of members, except subfamily III, which showed tandem duplication events only in apple. In contrast, the expansion of subfamily III was mainly the consequence of segmental duplication.
Many LRR-RLKs have been reported to play roles in plant disease resistance. For example, dependent on the suppressor of BIR1–1 (SOBIR1), BAK1 negatively regulated Arabidopsis defenses interacted with the RLK BIR1 (BAK1-interacting receptor-like kinase 1) [50]. Moreover, the steroidal hormones BRs were shown to promote pollen tube growth in Arabidopsis [51]. Interaction of BRI1 with BAK1 also play a key role in this promoted signaling network. In addition, BAK1 acts not only as the second receptor of BRs, but is also a co-receptor of LRR-RLK FLS2 and EF-Tu receptor (EFR) [42, 52, 53]. In the processes of plant immune response, BAK1 and FLS2/EFR form a dynamic receptor and work with Botrytis-induced kinase 1 (BIK1). As a member of the Catharanthus roseus receptor-like kinase (CrRLK) family, FERONIA (FER) plays two important roles. First, it works as a pollen tube guidance protein during plant fertilization. Second, it plays a negative role in resistance to powdery mildew fungus [54,55,56]. The functionally redundant genes ANXUR (ANX1 and ANX2) are close homologs of FER and expressed in pollen. Overexpressed ANXUR could negatively regulate pollen tube growth. However, more research is required to determine whether FER and ANXUR are involved in the process of BR promotion of pollen tube growth and plant immune responses and what roles they play. Although LRR-RLKs and CrRLKs, two important subfamilies of the RLK family, have evolved in different directions, they retain many similar biological functions in the process of plant growth.
According to the analyses of expression data presented in this work, we found that genes in the same subfamily have no consistent expression patterns; some genes in a subfamily might be highly expressed, whereas others are expressed at a low level or not at all. This pattern differs from an analysis of the soybean kinome, where it was found that gene members belonging to the same PK subfamilies showed similar expression patterns [57]. The reason for this difference could be the limitation of available expression data for soybean PKs, just 38%. By examining the expression of some duplicated genes within a subfamily, we found different expression patterns, which could be the result of the sub-functionalization or neo-functionalization of duplicated genes. Considering that some LRR-RLK subfamilies are very large, with over 100 members, the use of averaged expression data for such subfamilies could result in the loss of substantial information. LRR-RLK genes have been proven to have various functions in multiple tissues. Previous studies showed that LRR-RLK genes play important roles in root development; for example, OsRPK1 in rice was shown to affect the root system architecture by negatively regulating polar auxin transport [58]. In this study, tissue expression analyses also showed that LRR-RLK genes were relatively highly expressed in root and stem compared with their levels in other examined tissues.
In our study, subfamily XI-1 was not only the largest subfamily, but was also highly expressed in all five Rosaceae species. It is worth mentioning that a total of 80 tandem duplicated genes were found only in the XI-1 subfamily, representing approximately 33.9% of the 236 tandem duplicated genes in the five Rosaceae species (Additional file 1: Table S10). This indicates that the gene expansion of this subfamily mainly occurred through fragment replication and tandem copies. CLAVATA (CLV), a member of the XI-1 subfamily in Arabidopsis, plays an important role in maintaining stem cell numbers in the apical meristem. CLV1 encodes a typical LRR-RLK domain, the mutation of which leads to increases in the stem cell number of shoot apical meristem, flower number, and stem tip size [59]. Another example is provided by the LRR receptor kinase HAESA (HAE), which belongs to the XI-1 subfamily and is expressed in sepals, petals, the abscission zone of stamens, the bottom of the peduncle, and the base of the petiole. Decreased expression of this gene delays the withering of flower organs [60]. These results indicate that the Rosaceae XI-1 subfamily plays a very important role in growth and development. However, the functions of numerous LRR-RLK genes still remain unclear.
According to the expression analysis results presented here, we found that some genes from the III, V, VI-2, and LRR-XII-1 subfamilies were highly expressed during Chinese white pear pollen growth. Interestingly, a pattern was identified of most of these genes being up-regulated in the SPT stage, while some LRR-RLK genes were shown to promote pollen tube growth. Therefore, these highly expressed genes may function in the process of plant sperm cell release. In addition, there were 40 tandem duplicated genes in the XII-1 subfamily, which represented approximately 60% of the 67 tandem duplicated genes in Chinese white pear. This suggested that the gene tandem copies are one of the main causes of high expression of the XII-1 subfamily in pollen. Notably, the expression data showed that the Xb-1 and II subfamilies were not highly expressed in Chinese white pear pollen, but the RT-PCR experiment revealed that the genes PbrBRL2 and PbrSERK2, which belong to these two subfamilies, were highly expressed in pollen tissues. This features might be associated with the age, location, and biotic and abiotic stresses of the tested samples.
Conclusions
Our analyses of LRR-RLK genes in five Rosaceae species revealed their diversity in terms of member number, gene structure, tissue expression patterns, and expression patterns in response to stress. Expression patterns of these LRR-RLK genes should promote our understanding of the involvement of this gene family in plant growth, development, signal transduction, immunity, and stress responses.
Methods
Identification and classification of LRR-RLK genes
LRR-RLK genes were identified in five Rosaceae species: Chinese white pear (Pyrus bretschneideri), apple (Malus domestica), peach (Prunus persica), mei (Prunus mume), and strawberry (Fragaria vesca). To confirm the reliability of the identification process, which is similar to that used by [6], the model eudicot plant, Arabidopsis, was used as a reference, including in the identification process. Genome resources of the Chinese white pear were obtained from GigaDB (http://gigadb.org/dataset/100083). Genome resources of Arabidopsis, strawberry, and peach were obtained from Phytozome version 9.0 (http://phytozome.jgi.doe.gov/). Genome resources of mei (version 1.0) were obtained from the Prunus mume Genome Project website (https://github.com/lileiting/prunusmumegenome). Genome resources of apple (version 1.0 primary) were obtained from GDR (https://www.rosaceae.org). A summary of the genome information of Arabidopsis and the five Rosaceae species is provided in Table 1.
To identify LRR-RLK genes, putative PKs were initially obtained by searching hidden Markov models of the “typical” Pkinase clan [Pkinase (PF00069) and Pkinase_Tyr (PF07714)], obtained from the Pfam database version 28.0 [61], against the proteome in each studied species using HMMER v3.1 [62] with an E-value cut-off of 0.0001. After initial screening, typical PKs were identified with coverage of Pfam domain models of at least 50% [6]; otherwise, genes were designated as atypical PKs and excluded from further analysis. Furthermore, previously defined HMMs of different PK subfamilies (https://github.com/lileiting/Plant_Pkinase_fam.hmm) [6] were used to classify the identified typical PKs into families and subfamilies (genes were assigned to their best matched subfamily HMMs with an E-value cutoff of 0.0001). The subfamily HMMs were built from four plant model species [A. thaliana (dicotyledon), Oryza sativa (monocotyledon), Physcomitrella patens (moss), Chlamydomonas reinhardtii (a green alga)] with a phylogenetic approach as described by Lehti-Shiu and Shiu [6]. Briefly, a maximum likelihood phylogenetic tree for protein kinases of the four plant models and nine well-annotated eukaryote species (Caenorhabiditis elegans, Dictyostelium discoideum, Drosophila melanogaster, Homo sapiens, Monosiga brevicollis, Mus musculus, Saccharomyces cerevisiae, Strongylocentrotus purpuratus and Tetrahymena thermophila) was constructed and used for the classification.
To confirm this classification, a maximum likelihood phylogenetic tree was constructed with full-length protein sequences using RAxML [63]. Multiple sequence alignment of full-length protein sequences was performed using MUSCLE software [64]. Furthermore, phylogenetic trees for genes in subfamilies II and Xb-1 (fewer than 100 genes for each subfamily) were constructed using RAxML software [63]. The number of bootstrap replicates was automatically determined by the bootstrapping option provided by RAxML with the parameter “-N autoMRE” [65]. The online tool iTol [66] and Figtree software (version 1.4.2, http://tree.bio.ed.ac.uk/) were used to visualize the phylogenetic tree.
Conserved domains and intron number
Conserved domains for each gene were obtained by scanning against the Pfam database version 28.0 using HMMER v3.1. Results were filtered with coverage for each domain of not less than 50%, and overlapping domains were resolved by comparing their E-values and retaining the best-matched domain. Intron number and intron length were calculated based on the gene feature files from the genome resources. Statistical analysis was performed using R programming language [67].
Chromosome location and synteny analysis of Chinese white pear LRR-RLK genes
Chromosome locations of each LRR-RLK gene were obtained from their genome resources. Genome regions that showed synteny relationships were identified using the Multiple Collinearity Scan Toolkit (MCScanX) [68] with an E-value of 1e−5. Synonymous and non-synonymous rates were calculated using the “add_ka_and_ks_to_collinearity.pl” script in the MCScanX package. Tandem repeated genes were searched by comparing LRR-RLK genes in their corresponding positions in chromosome/scaffold sequences, and adjacent genes were designated as tandem duplicated genes.
Expression analysis
To analyze the expression patterns of LRR-RLK genes in the five species, transcriptome or RNAseq data were retrieved from the NCBI SRA database [69], and accession numbers for these data are presented in the data availability section. In total, 13 datasets for Chinese white pear, 19 for apple, seven for peach, ten for mei, and 59 for strawberry were selected for expression analysis. The data retrieved from the SRA database were first decompressed into the FASTQ format using the SRA toolkit [69]. Each dataset was first mapped to its corresponding template genome using Tophat v2.1.0 [70] with default parameters; then, the expression level was normalized to fragments per kilobase of exon per million reads (FPKM) using Cufflinks v2.2.1 with the default parameters [71]. Additionally, the best matched Arabidopsis gene for each Rosaceae LRR-RLK genes were obtained using BLASTP [72] with E-value cutoff of 1e-5 and the function annotation for the Arabiodpsis genes were performed using R programming language [67].
Construction of co-expression networks
Co-expression networks were generated for each of the five Rosaceae species for all identified members of the LRR-RLK gene family using the expression data analyzed above. The Pearson correlation coefficient (PCC) of FPKM values for each gene pair were calculated using R programming language [67]. The PCC threshold were 0.95 for all species. Co-expression network was visualized using Cytoscape v3.3.0 and analyzed using the NetworkAnalyzer plugin in Cytoscape [73].
RT-PCR, qRT-PCR and subcellular localization
RT-PCR
Total RNA was extracted from seven Pyrus bretschneideri tissues (roots, stems, fruit flesh, pollen tubes, styles, and ovaries) using a total RNA extraction kit (Foregene, Chengdu, China). First-strand cDNA was synthesized using a reverse transcriptase kit (TaKaRa, Dalian, China). Actin was used as an internal control for RT-PCR. Amplifications were carried out with 35 cycles for all programs. The PCR products were detected with 1.5% agarose gel electrophoresis and visualized under UV light.
qRT-PCR
The mature ‘Dangshansuli’ (Pyrus bretschneideri) pollen was incubated in pollen germination medium (PGM) [74] at 25 °C for germination and growth with pollens and tubes collected after 0 h (mature pollen grain, MP), 40 min (hydrated pollen grain, HP), 6 h (growing pollen tube, PT) and 16 h (stopped-growth pollen tube, SPT). Additionally, the mature pollens were also incubated in PGM with the treatment of 0.01 μM epibrassinolide (epiBL; Sigma-Aldrich, E1641) at 25 °C after 3 h. Total RNA was then extracted for the synthesis of the first-strand cDNA. PCR reactions were carried out with 45 cycles using the SYBR Green Master Mix (SYBR Premix EX Taq™, TaKaRa). The pear tubulin (TUB) gene was used as the housekeeping gene for the normalization of the gene expression. The primer sequences used for amplifying the genes as shown in Fig. 6 are presented in Additional file 1: Table S17.
Subcellular localization
The 35S::PbrSERK2-YFP fusion constructs were infiltrated into Arabidopsis mesophyll protoplast [75]. After fusion, protoplasts were incubated at 25 °C for 24 h in the dark. After 24 h, 2 μM FM4–64 dye (T13320; Thermo Fisher Scientific) was added in the medium for 30 min to stain the cytoplasmic membrane of the protoplasts as positive control. The subcellular localization of the fused proteins was monitored using a confocal laser scanning microscope (LSM780; Zeiss, Oberkochen, Germany).
Abbreviations
- BAK1:
-
BRI1-associated kinase 1
- BIK1:
-
Botrytis-induced kinase 1
- BIR1:
-
BAK1-interacting receptor-like kinase 1
- BR:
-
Brassinosteroid
- BRI1:
-
Brassinosteroid-insensitive 1
- BRL:
-
BRI1-Like
- CLV:
-
CLAVATA
- CrRLKs:
-
Catharanthus roseus receptor-like kinases
- EFR:
-
EF-Tu receptor
- EGF:
-
Epidermal growth factor-like repeats
- FER:
-
FERONIA
- FLS2:
-
Flagellin-sensitive 2
- FPKM:
-
Fragments per kilobases exons million reads
- GDR:
-
Database for Rosaceae
- HAE:
-
HAESA
- HMM:
-
Hidden Markov models
- HP:
-
Hydrated pollen grains
- JA:
-
Jasmonic acid
- Ks :
-
Synonymous substitution rate
- LB:
-
Lectin-binding domain
- LRR_8:
-
Leucine rich repeat 8
- LRRNT_2:
-
Leucine rich repeat N-terminal 2
- LRR-RLK:
-
Leucine-rich repeat receptor-like kinases
- MCScanX:
-
Multiple Collinearity Scan Toolkit
- MP:
-
Mature pollen grains
- MYA:
-
Million years ago
- NCBI:
-
National Center for Biotechnology Information
- PCC:
-
Pearson correlation coefficient
- Pfam:
-
Protein family (http://pfam.xfam.org)
- PK:
-
Protein kinase
- PR5K:
-
Pathogenesis-related protein 5-like receptor kinase
- PT:
-
Growing pollen tubes
- p-value:
-
Probability value
- RLK:
-
Receptor-like kinase
- RNA-seq:
-
RNA sequencing
- RPK1:
-
Receptor-like protein kinase 1
- RT-PCR:
-
Reverse transcriptase PCR
- SERK:
-
Somatic Embryogenesis Receptor-like Kinase
- SOBIR1:
-
Suppressor of BIR1–1
- SPT:
-
Stopped-growth pollen tubes
- SRA:
-
Sequence Read Archive
- TNFR:
-
Tumor necrosis factor receptor-like
- WGD:
-
Whole genome duplication
- XII-1:
-
RLK-Pelle_LRR-XII-1
- YFP:
-
Yellow fluorescent protein
References
Gou X, He K, Yang H, Yuan T, Lin H, Clouse SD, Li J. Genome-wide cloning and sequence analysis of leucine-rich repeat receptor-like protein kinase genes in Arabidopsis thaliana. BMC Genomics. 2010;11:19.
Shiu SH, Bleecker AB. Plant receptor-like kinase gene family: diversity, function, and signaling. Sci STKE 2001. 2001;113(re22):1–13.
Walker JC, Zhang R. Relationship of a putative receptor protein kinase from maize to the S-locus glycoproteins of Brassica. Nature. 1990;345:743–6.
Gao LL, Xue HW. Global analysis of expression profiles of rice receptor-like kinase genes. Mol Plant. 2012;5:143–53.
Shiu SH, Bleecker AB. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc Natl Acad Sci USA. 2001;98:10763–8.
Lehti-Shiu MD, Shiu SH. Diversity, classification and function of the plant protein kinase superfamily. Philos Trans R Soc Lond Ser B Biol Sci. 2012;367:2619–39.
Lehti-Shiu MD, Zou C, Hanada K, Shiu SH. Evolutionary history and stress regulation of plant receptor-like kinase/pelle genes. Plant Physiol. 2009;150:12–26.
Kobe B, Deisenhofer J. The leucine-rich repeat: a versatile binding motif. Trends Biochem Sci. 1994;19:415–21.
Wang ZY, Seto H, Fujioka S, Yoshida S, Chory J. BRI1 is a critical component of a plasma-membrane receptor for plant steroids. Nature. 2001;410:380–3.
Clouse SD. Brassinosteroid signal transduction: from receptor kinase activation to transcriptional networks regulating plant development. Plant Cell. 2011;23:1219–30.
Osakabe Y, Maruyama K, Seki M, Satou M, Shinozaki K, Yamaguchi-Shinozaki K. Leucine-rich repeat receptor-like kinase1 is a key membrane-bound regulator of abscisic acid early signaling in Arabidopsis. Plant Cell. 2005;17:1105–19.
Lee IC, Hong SW, Whang SS, Lim PO, Nam HG, Koo JC. Age-dependent action of an ABA-inducible receptor kinase, RPK1, as a positive regulator of senescence in Arabidopsis leaves. Plant Cell Physiol. 2011;52:651–62.
Chinchilla D, Shan L, He P, de Vries S, Kemmerling B. One for all: the receptor-associated kinase BAK1. Trends Plant Sci. 2009;14:535–41.
Zhou F, Guo Y, Qiu LJ. Genome-wide identification and evolutionary analysis of leucine-rich repeat receptor-like protein kinase genes in soybean. BMC Plant Biol. 2016;16:58.
Magalhães DM, Scholte LL, Silva NV, Oliveira GC, Zipfel C, Takita MA, De Souza AA. LRR-RLK family from two citrus species: genome-wide identification and evolutionary aspects. BMC Genomics. 2016;17:623.
Liu PL, Xie LL, Li PW, Mao JF, Liu H, Gao SM, Shi PH, Gong JQ. Deuplication and divergence of leucine-rich repeat receptor-like protein kinase (LRR-RLK) genes in basal angiosperm. Front Plant Sci. 2016;7:1952.
Liu PL, Du L, Huang Y, Gao SM, Origin YM. Diversification of leucine-rich repeat receptor-like protein kinase (LRR-RLK) genes in plants. BMC Evol Biol. 2017;17:47.
Fischer I, Diévart A, Droc G, Dufayard JF, Chantret N. Evolutionary dynamics of the leucine-rich repeat receptor-like kinase (LRR-RLK) subfamily in angiosperms. Plant Physiol. 2016;170:1595–610.
Freeling M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol. 2009;60:433–53.
Wu J, Wang Z, Shi Z, Zhang S, Ming R, Zhu S, Khan MA, Tao S, Korban SS, Wang H, et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013;23:396–408.
Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–49.
Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, Dodson RJ, Gwinn M, Hickey EK, Peterson JD, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–70.
Li WH, Gu Z, Wang H, Nekrutenko A. Evolutionary analyses of the human genome. Nature. 2001;409:847–9.
Theologis A, Ecker JR, Palm CJ, Federspiel NA, Kaul S, White O, Alonso J, Altafi H, Araujo R, Bowman CL, et al. Sequence and analysis of chromosome 1 of the plant Arabidopsis thaliana. Nature. 2000;408:816–20.
Zhou H, Yin H, Chen J, Liu X, Gao Y, Wu J, Zhang S. Gene-expression profile of developing pollen tube of Pyrus bretschneideri. Gene Expr Patterns. 2016;20:11–21.
Chang F, Gu Y, Ma H, Yang Z. AtPRK2 promotes ROP1 activation via RopGEFs in the control of polarized pollen tube growth. Mol Plant. 2013;6:1187–201.
Pitorre D, Llauro C, Jobet E, Guilleminot J, Brizard JP, Delseny M, Lasserre E. RLK7, a leucine-rich repeat receptor-like kinase, is required for proper germination speed and tolerance to oxidative stress in Arabidopsis thaliana. Planta. 2010;232:1339–53.
Kang C, Darwish O, Geretz A, Shahan R, Alkharouf N, Liu Z. Genome-scale transcriptomic insights into early-stage fruit development in woodland strawberry Fragaria vesca. Plant Cell. 2013;25:1960–78.
Liu Y, Huang X, Li M, He P, Zhang Y. Loss-of-function of Arabidopsis receptor-like kinase BIR1 activates cell death and defense responses mediated by BAK1 and SOBIR1. New Phytol. 2016;212:637–45.
Shiu SH, Karlowski WM, Pan R, Tzeng YH, Mayer KF, Li WH. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell. 2004;16:1220–34.
Song D, Li G, Song F, Zheng Z. Molecular characterization and expression analysis of OsBISERK1, a gene encoding a leucine-rich repeat receptor-like kinase, during disease resistance responses in rice. Mol Biol Rep. 2008;35:275–83.
Torii KU. Leucine-rich repeat receptor kinases in plants: structure, function, and signal transduction pathways. Int Rev Cytol. 2004;234:1–46.
Hu X, Reddy AS. Cloning and expression of a PR5-like protein from Arabidopsis: inhibition of fungal growth by bacterially expressed protein. Plant Mol Biol. 1997;34:949–59.
Wang X, Zafian P, Choudhary M, Lawton M. The PR5K receptor protein kinase from Arabidopsis thaliana is structurally related to a family of plant defense proteins. Proc Natl Acad Sci USA. 1996;93:2598–602.
Decreux A, Thomas A, Spies B, Brasseur R, Van Cutsem P, Messiaen J. Vitro characterization of the homogalacturonan-binding domain of the wall-associated kinase WAK1 using site-directed mutagenesis. Phytochemistry. 2006;67:1068–79.
He ZH, Cheeseman I, He D, Kohorn BD. A cluster of five cell wall-associated receptor kinase genes, Wak1-5, are expressed in specific organs of Arabidopsis. Plant Mol Biol. 1999;39:1189–96.
Chen X, Shang J, Chen D, Lei C, Zou Y, Zhai W, Liu G, Xu J, Ling Z, Cao G, et al. A B-lectin receptor kinase gene conferring rice blast resistance. The. Plant J. 2006;46:794–804.
Navarro-Gochicoa MT, Camut S, Timmers AC, Niebel A, Herve C, Boutet E, Bono JJ, Imberty A, Cullimore JV. Characterization of four lectin-like receptor kinases expressed in roots of Medicago Truncatula. Structure, location, regulation of expression, and potential role in the symbiosis with Sinorhizobium meliloti. Plant Physiol. 2003;133:1893–910.
Becraft PW, Stinard PS, DR MC. CRINKLY4: A TNFR-like receptor kinase involved in maize epidermal differentiation. Science. 1996;273:1406–9.
Jin P, Guo T, Becraft PW. The maize CR4 receptor-like kinase mediates a growth factor-like differentiation response. Genesis. 2000;27:104–16.
Stein JC, Howlett B, Boyes DC, Nasrallah ME, Nasrallah JB. Molecular cloning of a putative receptor protein kinase gene encoded at the self-incompatibility locus of Brassica oleracea. Proc Natl Acad Sci USA. 1991;88:8816–20.
Chinchilla D, Zipfel C, Robatzek S, Kemmerling B, Nurnberger T, Jones JD, Felix G, Boller TA. Flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature. 2007;448:497–500.
Gomez-Gomez L, Boller T. FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol Cell. 2000;5:1003–11.
Wu Y, Zhou JM. Receptor-like kinases in plant innate immunity. J Integr Plant Biol. 2013;55:1271–86.
Lehti-Shiu MD, Zou C, Shiu S-H. Origin, diversity, expansion history, and functional evolution of the plant receptor-like kinase/pelle family. In: Receptor-like kinases in plants. Berlin Heidelberg: Springer; 2012. p. 1–22.
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D, et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat Genet. 2010;42:833–9.
Jung S, Staton M, Lee T, Blenda A, Svancara R, Abbott A, Main DGDR. (Genome database for Rosaceae): integrated web-database for Rosaceae genomics and genetics data. Nucleic Acids Res. 2008;36:D1034–40.
Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc Biol Sci. 1994;256:119–24.
Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–8.
Gao M, Wang X, Wang D, Xu F, Ding X, Zhang Z, Bi D, Cheng YT, Chen S, Li X, et al. Regulation of cell death and innate immunity by two receptor-like kinases in Arabidopsis. Cell Host Microbe. 2009;6:34–44.
Vogler F, Schmalzl C, Englhart M, Bircheneder M, Sprunck S. Brassinosteroids promote Arabidopsis pollen germination and growth. Plant Reprod. 2014;27:153–67.
Heese A, Hann DR, Gimenez-Ibanez S, Jones AM, He K, Li J, Schroeder JI, Peck SC, Rathjen JP. The receptor-like kinase SERK3/BAK1 is a central regulator of innate immunity in plants. Proc Natl Acad Sci USA. 2007;104:12217–22.
Nam KH, Li J. BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell. 2002;110:203–12.
Escobar-Restrepo JM, Huck N, Kessler S, Gagliardini V, Gheyselinck J, Yang WC, Grossniklaus U. The FERONIA receptor-like kinase mediates male-female interactions during pollen tube reception. Science. 2007;317:656–60.
Huck N, Moore JM, Federer M, Grossniklaus U. The Arabidopsis mutant feronia disrupts the female gametophytic control of pollen tube reception. Development. 2003;130:2149–59.
Kessler SA, Shimosato-Asano H, Keinath NF, Wuest SE, Ingram G, Panstruga R, Grossniklaus U. Conserved molecular components for pollen tube reception and fungal invasion. Science. 2010;330:968–71.
Liu J, Chen N, Grant JN, Cheng ZM, Stewart CN Jr, Hewezi T. Soybean kinome: functional classification and gene expression patterns. J Exp Bot. 2015;66:1919–34.
Zou Y, Liu X, Wang Q, Chen Y, Liu C, Qiu Y, Zhang W. OsRPK1, a novel leucine-rich repeat receptor-like kinase, negatively regulates polar auxin transport and root development in rice. Biochim Biophys Acta. 2014;1840:1676–85.
Sijacic P, Liu Z. Novel insights from live-imaging in shoot meristem development. J Integr Plant Biol. 2010;52:393–9.
Jinn TL, Stone JM, Walker JCHAESA. An Arabidopsis leucine-rich repeat receptor kinase, controls floral organ abscission. Genes Dev. 2000;14:108–17.
Finn RD, Clements J, Eddy SRHMMER. Web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Edgar RCMUSCLE. Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Pattengale ND, Alipour M, Bininda-Emonds OR, Moret BM, Stamatakis A. How many bootstrap replicates are necessary? J Comput Biol. 2010;17:337–54.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49.
Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39:D19–21.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562–78.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ, Gapped BLAST. PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–4.
Wu J, Shang Z, Wu J, Jiang X, Moschou P, Sun W, Roubelakis-Angelakis K, Zhang S. Spermidine oxidase-derived H2O2 regulates pollen plasma membrane hyperpolarization-activated Ca2+-permeable channels and pollen tube growth. The. Plant J. 2010;63:1042–53.
Yoo SD, Cho YH, Sheen J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc. 2007;2:1565–72.
Acknowledgements
The authors would like to thank anonymous reviewers for comments on this manuscript.
Availability of data and materials
Expression data for strawberry (Fragaria vesca) used in this study were available in NCBI SRA database with accession numbers of ERR419273, ERR419274, ERR424891 to ERR424893, SRR072202, SRR081562, SRR1930097, SRR1930099, SRR673779 and SRR674026 to SRR674074. Expression data for apple (Malus domestica) used in this study were available in SRA database with accession numbers of SRR767660, SRR767668 to SRR767674 and SRR768127 to SRR768137. Expression data for mei (Prunus mume) used in this study were available in SRA database with accession numbers of DRR002283, DRR002284, DRR013975 to DRR013977, SRR542478 to SRR542482. Expression data for peach (Prunus persica) used in this study were available in SRA database with accession numbers of SRR1556451, SRR1559275, SRR1561576 and SRR531862 to SRR531865. Expression data of Chinese white pear (Pyrus bretschneideri) used in this study were available in pear genome project website and SRA database with accession numbers of PRJNA299117, SRR1260324, SRR1269627, SRR1582086, SRR1582087, SRR1786024, SRR1786028, SRR1786033 and SRR1786036.
Funding
This work was supported by National Key Technology R&D Program of the Ministry of Science and Technology of China (2014BAD16B03–4), National Natural Science Foundation of China (31,522,048, 31,772,256) and the Fundamental Research Funds for the Central Universities (KYTZ201602 and KYLH201502–2). The funding bodies were not involved in the design of the study or in any aspect of the collection, analysis and interpretation of the data or paper writing.
Author information
Authors and Affiliations
Contributions
JS, LL, JW conceived and designed the study. JS, LL conducted the data analyses. JS, PW performed the experiments. JS, LL, JW drafted and revised the manuscript. JW, SZ supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The plant materials used in this study were collected from Jiangpu Horticulture Experiment Station of Nanjing Agricultural University (Nanjing, Jiangsu, China). The experimental research on plants was conducted under the guidelines of the Convention on the Trade in Endangered Species of Wild Fauna and Flora.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Typical protein kinase in Arabidopsis and Rosaceae. Table S2. Atypical protein kinase in Arabidopsis and Rosaceae. Table S3. Subfamily classification of protein kinases and their related information. Table S4. Conserved domains of LRR-RLK subfamily members in Arabidopsis and Rosaceae. Table S5. Statistics of domain numbers and list of conserved domains for LRR-RLK genes in Arabidopsis and Rosaceae. Table S6. Statistics of intron number and intron length for LRR-RLK genes in Arabidopsis and Rosaceae. Table S7. Collinearity events and Ka/Ks values LRR-RLK genes in Arabidopsis and Rosaceae. Table S8. Number of WGD events and number of genes involved in WGD events in 23 LRR-RLK subfamilies in Arabidopsis and Rosaceae. Table S9. List of tandem duplicated LRR-RLK genes in Arabidopsis and Rosaceae. Table S10. Number tandem arrays and tandem duplicated genes in 23 LRR-RLK subfamilies in Arabidopsis and Rosaceae. Table S11. Expression (FPKM) of 201 strawberry LRR-RLK genes. Table S12. Expression (FPKM) of 244 apple LRR-RLK genes. Table S13. Expression (FPKM) of 427 Chinese white pear LRR-RLK genes. Table S14. Expression (FPKM) of 267 mei LRR-RLK genes. Table S15. Expression (FPKM) of 258 peach LRR-RLK genes. Table S16. Co-expression network attributes of LRR-RLK genes in five Rosaceae species. Table S17. Sequences of primers used in qRT-PCR. (XLSX 1945 kb)
Additional file 2: Figure S1.
Heat map of the expression patterns of 201 strawberry LRR-RLK genes in different tissues. Red and green colours correspond to up-regulation and down-regulation, respectively. Normalized gene expression values are provided in Additional file 1: Table S10. Figure S2. Heat map of the expression patterns of 244 apple LRR-RLK genes in different tissues. Red and green colours correspond to up-regulation and down-regulation, respectively. Normalized gene expression values are provided in Additional file 1: Table S11. Figure S3. Heat map of the expression patterns of 427 Chinese white pear LRR-RLK genes in different tissues. Red and green colours correspond to up-regulation and down-regulation, respectively. Normalized gene expression values are provided in Additional file 1: Table S12. Figure S4. Heat map of the expression patterns of 267 mei LRR-RLK genes in different tissues. Red and green colours correspond to up-regulation and down-regulation, respectively. Normalized gene expression values are provided in Additional file 1: Table S13. Figure S5. Heat map of the expression patterns of 258 peach LRR-RLK genes in different tissues. Red and green colours correspond to up-regulation and down-regulation, respectively. Normalized gene expression values are provided in Additional file 1: Table S14. Figure S6. Co-expression network of strawberry LRR-RLK genes. Nodes indicate genes and edges indicate significant co-expression events between genes. Figure S7. Co-expression network of apple LRR-RLK genes. Nodes indicate genes and edges indicate significant co-expression events between genes. Figure S8. Co-expression network of Chinese white pear LRR-RLK genes. Nodes indicate genes and edges indicate significant co-expression events between genes. Figure S9. Co-expression network of mei LRR-RLK genes. Nodes indicate genes and edges indicate significant co-expression events between genes. Figure S10. Co-expression network of peach LRR-RLK genes. Nodes indicate genes and edges indicate significant co-expression events between genes. (PDF 1698 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sun, J., Li, L., Wang, P. et al. Genome-wide characterization, evolution, and expression analysis of the leucine-rich repeat receptor-like protein kinase (LRR-RLK) gene family in Rosaceae genomes. BMC Genomics 18, 763 (2017). https://doi.org/10.1186/s12864-017-4155-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4155-y