
Abstract
Background
To date, mitochondrial genomes of more than one hundred flatworms (Platyhelminthes) have been sequenced. They show a high degree of similarity and a strong taxonomic bias towards parasitic lineages. The mitochondrial gene atp8 has not been confidently annotated in any flatworm sequenced to date. However, sampling of free-living flatworm lineages is incomplete. We addressed this by sequencing the mitochondrial genomes of the two small-bodied (about 1 mm in length) free-living flatworms Stenostomum sthenum and Macrostomum lignano as the first representatives of the earliest branching flatworm taxa Catenulida and Macrostomorpha respectively.
Results
We have used high-throughput DNA and RNA sequence data and PCR to establish the mitochondrial genome sequences and gene orders of S. sthenum and M. lignano. The mitochondrial genome of S. sthenum is 16,944 bp long and includes a 1,884 bp long inverted repeat region containing the complete sequences of nad3, rrnS, and nine tRNA genes. The model flatworm M. lignano has the smallest known mitochondrial genome among free-living flatworms, with a length of 14,193 bp. The mitochondrial genome of M. lignano lacks duplicated genes, however, tandem repeats were detected in a non-coding region.
Mitochondrial gene order is poorly conserved in flatworms, only a single pair of adjacent ribosomal or protein-coding genes – nad4l-nad4 – was found in S. sthenum and M. lignano that also occurs in other published flatworm mitochondrial genomes. Unexpectedly, we unambiguously identified the full metazoan mitochondrial protein-coding gene complement including atp8 in S. sthenum and M. lignano. A subsequent search detected atp8 in all mitochondrial genomes of polyclad flatworms published to date, although the gene wasn’t previously annotated in these species.
Conclusions
Manual, but not automated genome annotation revealed the presence of atp8 in basally branching free-living flatworms, signifying both the importance of manual data curation and of diverse taxon sampling. We conclude that the loss of atp8 within flatworms is restricted to the parasitic taxon Neodermata.
Similar content being viewed by others


Background
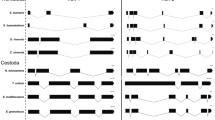
Flatworms represent a group with more than 100 complete mitochondrial genomes sequenced at present and thus it could be assumed that the group is well sampled. However, previous sampling strategies have focussed on the parasitic Neodermata and have not been comprehensive in free-living (turbellarian) flatworms. In particular, 97 mitochondrial genomes are available from the Neodermata, and only 12 from free-living flatworms: four species of Polycladida [1, 2] and 8 species of Tricladida [3,4,5], covering 2 of 11 recognised turbellarian orders (Fig. 1a). Recent phylogenomic studies have shown that the Catenulida and Macrostomorpha are the earliest branching taxa of Platyhelminthes [6, 7], but only a relatively short mitochondrial genome fragment (6,881 bp) from Microstomum lineare (Macrostomorpha) has been characterised [8] from either of these two groups.

(a) Phylogenetic tree of Platyhelminthes with Gastrotricha as sister group reconstructed from several sources [1, 6, 7]. Squares with grey background indicate the number of species with published mitochondrial genomes in the respective taxon. Pink background indicates newly sequenced species in this study. (b) Mitochondrial gene orders representative for the taxa displayed on the phylogenetic tree on the left. Only ribosomal and protein-coding genes are considered. Colours follow the widely conserved gene cartridges indicated by Mwinyi et al. [18]. Putative lophotrochozoan ground pattern after Bernt et al. [14]. Possible gastrotrich ground pattern with the single sequenced species, Lepidodermella squamata [1]. Possible catenulid ground pattern with the single newly sequenced species, Stenostomum sthenum. Duplicated genes in red, transcription from the minus strand is indicated by rectangle outlined in red. Possible macrostomorphan ground pattern with the single complete newly sequenced species, Macrostomum lignano. Possible polyclad ground pattern represented by Prosthiostomum siphunculus [2]. Triclad ground pattern after several sources [3,4,5]. Neodermatan ground patterns after Wey-Fabrizius et al. [9]. Putative flatworm ground pattern calculated with TreeREx (present study)
Until recently, the gene order of Platyhelminthes was considered to be highly conserved, with few exceptions [9]. This notion was largely influenced by taxonomically biased sampling, as only a single complete mitochondrial genome of a free-living flatworm, Dugesia japonica [3], was available. When the first mitochondrial genomes of polyclads became available, a third gene order pattern within flatworms was identified [1, 2].
All previously published free-living flatworm mitogenomes represent large species, which imposes a further sampling bias. Here, we provide the first mitochondrial genomes from microscopic turbellarians representing the orders Catenulida and Macrostomorpha to shed light on the ground plan of mitochondrial genomes in Platyhelminthes. Stenostomum sthenum is a freshwater species from a genus with a worldwide distribution, and which predominantly reproduces asexually by fission [10]. The marine species Macrostomum lignano is considered a model organism whose genome has recently been sequenced [11].
Results
Assemblies and tandem repeats
Bioinformatic assembly delivered mitochondrial genomes with an average coverage of 1,523× (268,745 mapped DNA reads; ca. 0.9% of the original reads) in Stenostomum sthenum, and 440× (63,938 mapped DNA reads; ca. 0.1% of the original reads) in Macrostomum lignano.
The mitochondrial genome assembly of S. sthenum revealed an inverted region of 1,884 bp length, which is otherwise identical to its non-inverted counterpart (Fig. 2). The bioinformatically predicted flanking regions of the duplicated region were confirmed by sequencing several different PCR products. No tandem repeats were detected in the mitochondrial genome of S. sthenum.

Circular mitochondrial genome of Stenostomum sthenum. Outermost circle displays logarithmically mapped RNAseq reads. Red lines below represent sequenced amplicons from PCR experiments. Bold line in innermost circle indicates transcription from the minus strand. tRNA genes are abbreviated with single capital letter and a number if applicable. Colours indicate the widely conserved gene cartridges proposed by Mwinyi et al. [18]. Dark grey colour indicates an overlap between genes. Unlabeled and uncoloured regions are intergenic spacers. Pictured is an adult specimen of S. sthenum with three zooids, anterior is to the right
The assembled mitogenome of M. lignano (Fig. 3) was compared to the published genome assembly [11], and a matching contig of 15,041 bp length (Additional file 1) with non-overlapping ends was detected. This contig allowed us to determine the length and sequence of a tandem repeat region consisting of eight 61 bp repeats. The sequence of the repeat units is GGGGGGAATAAATATTCCCCCCTATATTACTAATGTAATATAAGGAGAAATATAATATATA (Additional file 1). The 15,041 bp long genome contig showed a leading (G)5 sequence in repeats #4, 5 and 8, but PCR verification suggested that all 8 repeats have a leading (G)6 sequence.

Circular mitochondrial genome of Macrostomum lignano. Outermost circle displays logarithmically mapped RNAseq reads. Red line below represents sequenced amplicons from PCR experiments. tRNA genes are abbreviated with single capital letter and a number if applicable. Colours indicate the widely conserved gene cartridges proposed by Mwinyi et al. [18]. Dark grey colour indicates an overlap between genes. Unlabeled and uncoloured regions are intergenic spacers. Pictured is an adult specimen of M. lignano, anterior is to the right
Tandemly repeated elements are common in the mitochondrial genomes of flatworms, and a re-analysis of the published mitochondrial genome of Prosthiostomum siphunculus [2] revealed several large, previously undetected overlapping tandem repeats (positions 14,695-15,122) in an intergenic region.
RNAseq read mapping on assembled mitochondrial genomes
We logarithmically mapped 21.47 million RNAseq reads onto the mitochondrial genome of S. sthenum with an average coverage of 126,712× (Fig. 2). For M. lignano, 0.80 million RNAseq reads mapped onto the mitochondrial genome with an average coverage of 5,637× (Fig. 3), and for P. siphunculus 0.66 million RNAseq reads resulted in an average coverage of 4,348× (Additional file 2). In all species, the highest expression levels (by a factor of about 4-50 as compared to protein coding genes) were found for rrnL, and all genes were found to be transcriptionally active. In S. sthenum, even the large non-coding regions between positions 3,307 and 4,140, and between positions 4,277 and 4,424 (Additional file 3) appear to be transcribed at high levels (Fig. 2), while in M. lignano the non-coding region between positions 9,444 and 10,165 (Additional file 3) is only weakly expressed (Fig. 3). In P. siphunculus, a large non-coding region between rrnL and nad6 [2] is quite strongly expressed (Additional file 2). Generally, tRNAs show the lowest level of mapped RNAseq reads (Figs. 2 and 3, Additional file 2).
Non-coding regions and tRNAs
The largest non-coding region in S. sthenum is located between tRNA I and atp8, and has a length of 840 bp (Fig. 2, Additional file 3). This region includes a small (110 bp) part of the duplicated piece containing nad3 and rrnS.
We found the full complement of 22 tRNAs in S. sthenum, in addition to 8 duplicated (namely tRNAs A, E, H, I, K, L1, L2 and V) and one triplicated (tRNA M) (Fig. 2,Additional file 3). In contrast, the mitochondrial genome of M. lignano encodes only 21 tRNAs, as a homologue of tRNA L1 was not detected by either MITOS or ARWEN (Figs. 3, Additional file 3). Secondary structures of all tRNAs predicted by MITOS are shown in Additional files 4 and 5.
Protein-coding genes
We detected all 13 protein-coding genes in the mitochondrial genomes of S. sthenum and M. lignano. This set included atp8 both in newly sequenced species and also in the four published polyclads [1, 2]. In all but one polyclad, the starting amino acid sequence is MPQM, while in Hoploplana elisabelloi it is MPHM. In S. sthenum, the atp8 amino acid sequence starts with MYQM, in M. lignano with the widely conserved MPQL (Fig. 4). We performed BLASTP searches with all 6 identified flatworm atp8 amino acid sequences against NCBI's non-redundant protein sequences database, and recovered e-values between 0.027 and 0.22 for atp8 hits (except for the atp8 sequence of S. sthenum, which did not return any e-values below 1). To provide context for these e-values, we used BLASTP with the published derived atp8 sequences of the gastropod Ascobulla fragilis and the gnathostomulid Gnathostomula armata, where the lowest e-values compared to other atp8 sequences are 0.22 and 0.23, respectively. Similarly, the atp8 sequence of Mytilus edulis is only similar to other Mytilus species, but not to any other atp8 sequences.

Amino acid alignment of atp8 in flatworms and other lophotrochozoans: a gastrotrich (Lepidodermella squamata), annelids (Platynereis dumerilii and Sipunculus nudus), molluscs (Sepiella japonica and Ascobulla fragilis), a phoronid (Phoronopsis harmeri), a kamptozoan (Loxosomella aloxiata), a bryozoan (Bugula neritina), a brachiopod (Terebratalia transversa) and a nemertean (Micrura bella). Transmembrane regions are underlaid in light green. Putative triclad atp8 sequences were omitted because of their derivedness (for inclusion of triclad sequences, see Additional file 9)
Using SMART [12], we detected transmembrane regions in putative and published atp8 sequences (Fig. 4). SMART also reported the detection of protein family (Pfam) ATP-synt_8 in the putative atp8 sequences of the polyclads P. siphunculus and S. maculata, with e-values of 0.0073 and 0.72, respectively. For comparison, the published atp8 sequences of the gastrotrich Lepidodermella squamata and of the nemertean Micrura bella scored Pfam ATP-synt_8 e-values of 0.035 and 0.022, respectively.
The hydrophobicity profiles between the putative flatworm and two published atp8 amino acid sequences provide very similar graphs, with positive hydrophobicity scores at the N-terminus and mostly negative scores at the C-terminus (Additional file 6).
We found the identified atp8 sequences of S. sthenum, M. lignano and P. siphunculus also by BLASTN searches in their respective transcriptomes, providing additional evidence that these sequences are expressed.
A manual search for atp8 in non-coding regions, as well as using BLAST with the putative atp8 sequences of free-living flatworms in published mitochondrial genomes from Neodermata species did not detect any homologous genes in this group of flatworms.
Gene order
The mitochondrial ribosomal and protein-coding gene orders of the two newly sequenced species and several published species are shown in Fig. 1b.
An analysis of gene order rearrangements with CREx [13] suggests that at least four steps (all inversions, called 'reversals' in CREx) are required to change from the putative lophotrochozoan ground pattern [14] to the gene order of the single published representative of Gastrotricha, Lepidodermella squamata [1] (Additional file 7). Similarly, it takes four steps from the putative lophotrochozoan ground pattern to M. lignano, and seven steps to the S. sthenum gene arrangement. The fewest steps from the lophotrochozoan ground pattern to any known free-living flatworm mitochondrial genome is two in the Tricladida (both tandem duplication, random loss events) (Additional file 7).
TreeREx calculates likely ancestral gene orders in nodes of a given phylogenetic tree [15], which we provided (Fig. 1a) for the taxa where mitochondrial gene order data is available. TreeREx suggests an ancestral gene order for Platyhelminthes (Fig. 1b), which is identical to the ancestral gene order of Rhabditophora (Additional file 7). According to the TreeREx analysis, the gene order of the polyclad Prosthiostomum siphunculus used in Fig. 1b is the same as the putative ancestral gene order of Polycladida as well as of Trepaxonemata (all flatworms except Catenulida and Macrostomorpha) (Additional file 7). TreeREx and CREx only accept gene matrices with a full complement of genes in all taxa as input, consequently we were not able to include Neodermata (which lack atp8) in these analyses.
Discussion
Mitochondrial genome sizes and nucleotide composition
Mitogenome sizes of polyclads vary between 14,651 bp (Enchiridium sp.) and 15,329 bp (Stylochoplana maculata) [1, 2], while those of triclads are generally larger and range between 14,909 bp (Obama sp.) and 27,133 bp (Schmidtea mediterranea, sexual biotype) [4, 5]. Macrostomum lignano, with 14,193 bp, has the smallest mitochondrial genome of a free-living flatworm studied so far. Only neodermatans are smaller, in particular the mitochondrial genomes of the cestodes rarely exceed 14,000 bp [9]. A survey of published mitochondrial genomes shows that the sizes of cestode mitogenomes are within a narrow range between 13,387 bp (Taenia pisiformis) and 13,900 bp (Hymenolepis diminuta), with only two exceptions larger than 14,000 bp, (Pseudanoplocephala crawfordi 14,192 bp and Anoplocephala perfoliata 14,459 bp). Trematode mitogenomes are on average slightly larger, ranging between 13,796 bp (Clinostomum complanatum) and 15,258 bp (Metagonimus yokogawai) in size, with one notable exception of 16,901 bp (Schistosoma spindale). In polyopisthocotyleans and monopisthocotyleans, mitogenome sizes are between 13,392 bp (Tetrancistrum nebulosi) and 15,527 bp (Polyabris halichoeres).
Gene order
The mitochondrial gene order differs between the two basally branching flatworms in this study, lacking any shared pairs of protein-coding or ribosomal gene boundaries other than nad4l-nad4 (although nad4l is transcribed from the minus strand in S. sthenum) and a transposed rrnL-nad1 gene pair (Fig. 1b).
While in gastrotrichs the putative lophotrochozoan ancestral gene order is largely recognisable (Fig. 1b), the extensive gene duplication in Stenostomum makes comparisons difficult. In fact, the only remaining adjacent gene pairs between the gastrotrich Lepidodermella squamata and S. sthenum are nad4l-nad4 and cob-nad6. The gene pair nad4l-nad4 is shared throughout the flatworms (Fig. 1b).
Within Macrostomorpha, the only full-length mitochondrial genome available is that of M. lignano (Fig. 3). The partial mitochondrial genome of Microstomum lineare is notably different from M. lignano, however (Additional file 8), hinting at non-conserved mitochondrial gene orders within Macrostomorpha, similar to the situation found in Polycladida, where Stylochoplana maculata has a completely different gene order from that of other polyclad representatives [1, 2].
Between M. lignano and two of the four published polyclads (the cotyleans Prosthiostomum siphunculus and Enchiridium sp.), the only identical adjacent gene pairs are nad3-cox3, nad4l-nad4, and nad1-atp6. Not surprisingly, the gene order in M. lignano requires the lowest number of rearrangements from the inferred ancestral gene order in flatworms, while the gene order of P. siphunculus may closely reflect the ancestral gene order of trepaxonematans (Additional file 7).
We have noticed some confusion about gene content and order in the partial mitochondrial genome of Microstomum lineare, originally published by Ruiz-Trillo et al. [8]. In the study of Golombek et al. [1], an additional gene, nad6, was shown in their Figure 7. Our own analysis suggests this may be an artifact of an inaccurate MITOS annotation, which weakly supports nad6 in the genome fragment, but further BLAST searches show this to be a very unlikely nad6 candidate. In the study of Aguado et al. [2], the gene order of M. lineare is shown inverted in their Fig. 2. In our own figure (Additional file 8), we depict the original gene order and content [8].
The gene block nad6, nad5 and cox3 was suggested to be a possible plesiomorphy for the Rhabditophora [2], but it is not found in both studied macrostomorphans or the catenulid S. sthenum (Fig. 1b, Additional file 8) and is thus more likely an apomorphy for Trepaxonemata, which is supported by the TreeREx analysis (Additional file 7). Another possible trepaxonematan apomorphy is the block cob, nad4l and nad4 (Fig. 1b), also corroborated by TreeREx (Additional file 7). The gene pair cox3 and nad3 is ancestral in bilaterians, in lophotrochozoans and also in gastrotrichs (Fig. 1b) [1, 14] the likely sister group of Platyhelminthes [6, 16, 17]. Within flatworms, a close association between cox3 and nad3 - either as cox3-nad3 or nad3-cox3 is conserved within most polyclads and also in Macrostomum, and is thus likely a plesiomorphy in these groups (Fig. 1b, Additional file 7). In all other flatworms, including S. sthenum and M. lineare, cox3 and nad3 are not adjacent, suggesting an independently derived character state.
Ribosomal gene adjacency (rrnS and rrnL) is very common in bilaterians and lophotrochozoans [14] and appears to be conserved in M. lineare, in triclads and neodermatans (Fig. 1b, Additional file 8). As noted by Aguado et al. [2], the two ribosomal genes are not adjacent in polyclads, and this is also true for S. sthenum and M. lignano (Figs. 1b, 2 and 3). A parsimonious explanation for these findings is multiple, independent loss of ribosomal gene adjacency. This solution is supported by the relative vicinity of rrnS and rrnL in S. sthenum, only interrupted by nad3 (Fig. 2). Alternatively, the adjacency of ribosomal genes was lost for all flatworms and was regained in M. lineare and in Trepaxonemata.
The location of tRNA V between rrnS and rrnL is an ancestral and widespread condition [18] and commonly found also in lophotrochozoans, including the gastrotrich Lepidodermella squamata [1]. This feature was lost in the common ancestor of the Platyhelminthes, as it is not found in any of the 109 flatworms sequenced so far, including those flatworm groups where rrnS and rrnL are still adjacent. In S. sthenum, both rrnS and tRNA V are duplicated and adjacent to each other (Fig. 2, Additional file 3), which is likely a plesiomorphy. In M. lignano tRNA V is located downstream of rrnL (Fig. 3, Additional file 3), probably the result of a transposition. In the other sequenced macrostomorphan, M. lineare, both ribosomal genes are present in the published mitochondrial genome fragment, but tRNA V was not identified between the ribosomal genes [8].
Our CREx and TreeREx analyses (Additional file 7) suggest that the gene order of S. sthenum is highly derived; possibly representatives of other Catenulida families could shed light on the ancestral mitochondrial gene order of Catenulida. Similarly, the ancestral gene order of Macrostomorpha cannot be deduced from the available data, thus more representatives of early branching flatworms should be analysed.
Atp8 is in the ground pattern of flatworms
Atp8 has been noted as missing from the mitochondrial genomes of flatworms [1, 2, 4, 9, 19], but it was annotated in the single sequenced gastrotrich, Lepidodermella squamata [1]. Annotation of atp8 is particularly difficult, as its amino acid sequence - apart from the first four amino acids MPQL - is not well conserved, and it has been reported as lacking in nematodes, chaetognaths, rotiferans and bivalve molluscs [19]. Recently it was suggested that an extremely derived atp8 may be present in triclad flatworms (planarians) [5]. The authors based their argument on the fact that their putative atp8 was expressed in RNAseq reads and that the sequence codes for a protein with putative transmembrane domains, like atp8. However, no pronounced similarity with atp8 sequences from other animals was found (see also Additional files 9, 10 and 11), and the putative atp8 sequences were annotated with a single question mark instead of a specific gene name [5].
As automated gene annotation with MITOS [20] could not identify atp8 neither in our two basally branching flatworms nor in three of the four published polyclad mitochondrial genomes (only in P. siphunculus does MITOS correctly annotate atp8, but it was not annotated by the original authors [2]), we have manually searched for atp8 candidates in all six mitochondrial genomes. In this way, we were able to identify atp8 in Stenostomum, Macrostomum and in all four published polyclads (Fig. 4). The presence of atp8 in these mitochondrial genomes is supported by RNAseq read mapping (Figs. 2, 3 and 4, Additional file 2), transmembrane regions at the N-terminus (Fig. 4), hydrophobicity profiles (Additional file 6) and, weakly, by BLAST and Pfam e-values. This implies that atp8 has not been lost in all flatworms, but only in the parasitic Neodermata, and it also supports the notion that the extremely derived atp8 candidate found in triclads may be a functional atp8 [5]. This case also highlights the importance of manual curation following automated annotation, and that observation bias (“no atp8 in flatworms”) can lead to oversight.
Multiple gene duplications and transcription from the minus strand in Stenostomum
With S. sthenum, we present the first mitochondrial genome of a flatworm with both extensive duplication (including triplication of tRNA genes, duplication of a ribosomal and a protein-coding gene) and transcription from the minus strand of a substantial number of genes (Figs. 1b, 2). So far, only tRNA genes have been reported as being duplicated in mitochondrial genomes of some flatworms, namely tRNA N in the triclads Dugesia japonica and D. ryukyuensis [3] and tRNA C in the two parasitic flatworms Schistosoma mansoni and S. japonicum [9]. The highly unusual features of S. sthenum most likely represent a derived condition, as neither gastrotrichs, nor the inferred lophotrochozoan ground pattern show any similarity to Stenostomum in this regard (Fig. 1b).
Large non-coding RNA
RNAseq reads were mapped onto the mitochondrial genome of several triclad flatworms [5], providing evidence for large (>100 bp) regions that were transcriptionally active, but not coding. Here we have mapped RNAseq reads to large non-coding regions of S. sthenum, M. lignano and P. siphunculus (Figs. 2 and 3, Additional file 2), supporting the idea that large non-coding RNA is not restricted to vertebrates [5].
Tandem repeats in both M. lignano and P. siphunculus occur in their large non-coding regions and such sequences have been hypothesised to represent control regions and/or potential origins of replication [3]. Tandem repeats in non-coding regions are also very common in parasitic flatworms [21]. Interestingly, in S. sthenum there are no tandem repeats, suggesting that the putative control region is not uniformly structured in flatworms, and that tandem repeats were either lost in catenulids or are an apomorphy for Rhabditophora.
Conclusions
We have sequenced two mitochondrial genomes from the earliest branching flatworm taxa, Catenulida (Stenostomum sthenum) and Macrostomorpha (Macrostomum lignano). S. sthenum shows a highly unusual duplication pattern including a ribosomal, a protein-coding and several tRNA genes, and has several genes transcribed from the minus strand. M. lignano has a markedly different gene order from that known from the partial mitochondrial genome of another macrostomorphan, Microstomum lineare. Even with our newly sequenced species, mitochondrial genome data are still lacking from half of flatworm orders (Fig. 1a). With the currently available information on flatworm mitochondrial genomes, only a preliminary ancestral mitochondrial gene order for flatworms can be inferred. More representatives of early divergent, non-parasitic taxa such as Catenulida and Macrostomorpha, but also from the 7 as yet unsequenced flatworm orders are required to obtain a comprehensive picture of mitochondrial genome evolution in flatworms. Another major conclusion of the present study is that atp8 is present in several and possibly all free-living flatworms, and that atp8 has been lost only in the parasitic Neodermata.
Manual curation of automatically annotated mitochondrial genomes is still very important. Without meticulous analysis of new and published mitochondrial genomes of free-living flatworms, and the combined use of RNA and DNA data, atp8 may not have been found and annotated in flatworm mitochondrial genomes.
A very recent paper on the mitochondrial genome of the closely related catenulid Stenostomum leucops [22] has annotated a divergent atp8 gene, which supports the findings reported here for S. sthenum.
Methods
Animals
Stenostomum sthenum and Macrostomum lignano have been kept as laboratory cultures since 2009 and 1995, respectively. For S. sthenum, a clonal line started from a single animal collected in Innsbruck, Austria, and for M. lignano, non-inbred culture animals [23] have been used for generating the RNAseq reads as described by Egger et al. [6] as well as for total genomic DNA extraction for next-generation sequencing (NGS) and PCR experiments.
DNA extraction and Illumina sequencing
About 100 specimens of clonal S. sthenum and M. lignano, respectively, were pooled for total genomic DNA extraction using the E.Z.N.A. Tissue DNA kit (Omega Bio-Tek) following the Tissue DNA-Spin protocol. Next-generation sequencing was performed on an Illumina HiSeq 2000 instrument delivering 33,362,560 (S. sthenum) and 54,410,350 (M. lignano) 100 bp paired-end reads that were subsequently trimmed and QC filtered using the fastx-toolkit (hannonlab.cshl.edu/fastx_toolkit/), leaving 29,990,734 (S. sthenum) and 52,268,272 (M. lignano) 100 bp paired-end reads for assembly. The custom sequencing service was provided by the Norwegian Sequencing Centre (www.sequencing.uio.no).
Assembly
The mitochondrial genome of each species was reconstructed by directly assembling the NGS reads using the mitochondrial baiting and iterative mapping algorithm MITObim 1.8 [24] with the default settings of the program, and the optional quality trimming option trimreads (see the program’s tutorial at https://github.com/chrishah/MITObim for details). The partial cytochrome oxidase subunit 1 sequences of S. leucops (GenBank accession number AJ405976) and M. lignano (GenBank accession number KP308282), respectively, were used as starting seeds for the assemblies.
BLAST search in transcriptomes
The BLAST+ suite [25] with published protein-coding mitochondrial genes as queries was used to screen the transcriptomes [6] of S. sthenum and M. lignano. Furthermore, all genes annotated from the MITObim assemblies were used for BLAST searches of the respective transcriptomes.
DNA extraction for PCR
DNA was extracted from 100-220 animals per species with phenol:chloroform:isoamylalcohol or a QIAamp DNA Micro Kit (Qiagen), eluted in nuclease-free water or buffer AE. PCRs were done with 15-170 ng of extracted DNA in 50 μl tubes, with varying conditions.
Annotation and gene order
For gene annotation, BLAST searches [26], as well as MITOS [20], ARWEN [27] and DOGMA [28] predictions were employed. For the analysis of putative atp8 sequences, we also used SMART [12] with default parameters to identify domains, and the ExPASy tool ProtScale (http://web.expasy.org/protscale/) to determine hydrophobicity profiles of amino acid sequences. Gene order rearrangements were analysed with CREx [13] and TreeREx [15]. Tandem Repeats Finder was used to identify tandem repeats [29].
Mapping RNAseq reads to the mitochondrial genomes
Bowtie2 [30] and samtools [31] were used with default parameters to map RNAseq reads [6] to the assembled mitochondrial genomes of S. sthenum, M. lignano and the published mitochondrial genome of the polyclad Prosthiostomum siphunculus [2]. Mapped reads were visualised with the Integrative Genomics Viewer [32]. Figures were made with mtviz (pacosy.informatik.uni-leipzig.de/mtviz), Inkscape (www.inkscape.org) and GIMP (www.gimp.org).
Amino acid alignments
For multiple gene alignments of atp8, we have used MAFFT [33] with the parameters “-l -o ' --inputorder --auto ' -a 50 -e 1.0e-10 -s input” on amino acid sequences of our own sequences, downloaded sequences from NCBI of lophotrochozoan atp8 proteins and putative atp8 sequences in triclads [5]. Accession number of used sequences are: AKD00049 (Lepidodermella squamata), AAF02682 (Platynereis dumerilii), ACJ11904 (Sipunculus nudus), BAM11165 (Sepiella japonica), ACE62808 (Ascobulla fragilis), AES86298 (Phoronopsis harmeri), BAG12588 (Loxosomella aloxiata), AJP00039 (Bugula neritina), AAK95499 (Terebratalia transversa) and AKT74029 (Micrura bella). For visualisation of alignments, we have used GeneDoc (http://genedoc.software.informer.com/2.7/).
Abbreviations
- atp6:
-
ATP synthase F0 subunit 6
- atp8:
-
ATP synthase F0 subunit 8
- cob:
-
Cytochrome b
- cox1:
-
Cytochrome c oxidase subunit I
- cox2:
-
Cytochrome c oxidase subunit II
- cox3:
-
Cytochrome c oxidase subunit III
- nad1:
-
NADH dehydrogenase subunit 1
- nad2:
-
NADH dehydrogenase subunit 2
- nad3:
-
NADH dehydrogenase subunit 3
- nad4:
-
NADH dehydrogenase subunit 4
- nad4l:
-
NADH dehydrogenase subunit 4L
- nad5:
-
NADH dehydrogenase subunit 5
- nad6:
-
NADH dehydrogenase subunit 6
- PCR:
-
Polymerase chain reaction
- RNAseq:
-
RNA sequencing.
References
Golombek A, Tobergte S, Struck TH. Elucidating the phylogenetic position of Gnathostomulida and first mitochondrial genomes of Gnathostomulida, Gastrotricha and Polycladida (Platyhelminthes). Mol Phylogenet Evol. 2015;86:49–63. https://doi.org/10.1016/j.ympev.2015.02.013.
Aguado MT, Grande C, Gerth M, Bleidorn C, Norena C. Characterization of the complete mitochondrial genomes from Polycladida (Platyhelminthes) using next-generation sequencing. Gene. 2016;575:199–205. https://doi.org/10.1016/j.gene.2015.08.054.
Sakai M, Sakaizumi M. The complete mitochondrial genome of Dugesia japonica (Platyhelminthes; order Tricladida). Zoolog Sci. 2012;29:672–80. https://doi.org/10.2108/zsj.29.672.
Solà E, Álvarez-Presas M, Frías-López C, Littlewood DTJ, Rozas J, Riutort M. Evolutionary analysis of mitogenomes from parasitic and free-living flatworms. PLoS One. 2015;10:e0120081. https://doi.org/10.1371/journal.pone.0120081.
Ross E, Blair D, Guerrero-Hernández C, Sánchez Alvarado A. Comparative and transcriptome analyses uncover key aspects of coding- and long noncoding RNAs in flatworm mitochondrial genomes. G3 (Bethesda). 2016;6:1191–200. https://doi.org/10.1534/g3.116.028175.
Egger B, Lapraz F, Tomiczek B, Müller S, Dessimoz C, Girstmair J, Skunca N, Rawlinson KA, Cameron CB, Beli E, Todaro MA, Gammoudi M, Noreña C, Telford MJ. A transcriptomic-phylogenomic analysis of the evolutionary relationships of flatworms. Curr Biol. 2015;25:1347–53. https://doi.org/10.1016/j.cub.2015.03.034.
Laumer CE, Hejnol A, Giribet G. Nuclear genomic signals of the 'microturbellarian' roots of platyhelminth evolutionary innovation. Elife. 2015;12:4. https://doi.org/10.7554/eLife.05503.
Ruiz-Trillo I, Riutort M, Fourcade HM, Baguñà J, Boore JL. Mitochondrial genome data support the basal position of Acoelomorpha and the polyphyly of the Platyhelminthes. Mol Phylogenet Evol. 2004;33:321–32. https://doi.org/10.1016/j.ympev.2004.06.002.
Wey-Fabrizius AR, Podsiadlowski L, Herlyn H, Hankeln T. Platyzoan mitochondrial genomes. Mol Phylogenet Evol. 2013;69:365–75. https://doi.org/10.1016/j.ympev.2012.12.015.
Egger B, Gschwentner R, Rieger R. Free-living flatworms under the knife: past and present. Dev Genes Evol. 2007;217:89–104. https://doi.org/10.1007/s00427-006-0120-5.
Wasik K, Gurtowski J, Zhou X, Ramos OM, Delás MJ, Battistoni G, El Demerdash O, Falciatori I, Vizoso DB, Smith AD, Ladurner P, Schärer L, McCombie WR, Hannon GJ, Schatz M. Genome and transcriptome of the regeneration-competent flatworm, Macrostomum lignano. Proc Natl Acad Sci U S A. 2015;112:12462–7. https://doi.org/10.1073/pnas.1516718112.
Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucl Acids Res. 2014;43:257–60. https://doi.org/10.1093/nar/gku949.
Bernt M, Merkle D, Ramsch K, Fritzsch G, Perseke M, Bernhard D, Schlegel M, Stadler PF, Middendorf M. CREx. inferring genomic rearrangements based on common intervals. Bioinformatics. 2007;23:2957–8. https://doi.org/10.1093/bioinformatics/btm468.
Bernt M, Bleidorn C, Braband A, Dambach J, Donath A, Fritzsch G, Golombek A, Hadrys H, Jühling F, Meusemann K, Middendorf M, Misof B, Perseke M, Podsiadlowski L, von Reumont B, Schierwater B, Schlegel M, Schrödl M, Simon S, Stadler PF, Stöger I, Struck TH. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol Phylogenet Evol. 2013;69:352–64. https://doi.org/10.1016/j.ympev.2013.05.002.
Bernt M, Merkle D, Middendorf M. An algorithm for inferring mitochondrial genome arrangements in a phylogenetic tree. In: Nelson CE, Vialette S, editors. Comparative Genomics. RECOMB-CG 2008. Lecture Notes in Computer Science, vol. 5267. Berlin, Heidelberg: Springer; 2008. p. 143–57.
Struck TH, Wey-Fabrizius AR, Golombek A, Hering L, Weigert A, Bleidorn C, Klebow S, Iakovenko N, Hausdorf B, Petersen M, Kück P, Herlyn H, Hankeln T. Platyzoan paraphyly based on phylogenomic data supports a noncoelomate ancestry of Spiralia. Mol Biol Evol. 2014;31:1833–49. https://doi.org/10.1093/molbev/msu143.
Laumer CE, Bekkouche N, Kerbl A, Goetz F, Neves RC, Sørensen MV, Kristensen RM, Hejnol A, Dunn CW, Giribet G, Worsaae K. Spiralian phylogeny informs the evolution of microscopic lineages. Curr Biol. 2015;25:2000–6. https://doi.org/10.1016/j.cub.2015.06.068.
Mwinyi A, Meyer A, Bleidorn C, Lieb B, Bartolomaeus T, Podsiadlowski L. Mitochondrial genome sequence and gene order of Sipunculus nudus give additional support for an inclusion of Sipuncula into Annelida. BMC Genomics. 2009;10:27. https://doi.org/10.1186/1471-2164-10-27.
Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity (Edinb). 2008;101:301–20. https://doi.org/10.1038/hdy.2008.62.
Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9. https://doi.org/10.1016/j.ympev.2012.08.023.
Bachmann L, Fromm B, Patella de Azambuja L, Boeger WA. The mitochondrial genome of the egg-laying flatworm Aglaiogyrodactylus forficulatus (Platyhelminthes:Monogenoidea). Parasit Vectors. 2016;9:285. https://doi.org/10.1186/s13071-016-1586-2.
Rosa MT, Oliveira DS, Loreto ELS. Characterization of the first mitochondrial genome of a catenulid flatworm: Stenostomum leucops (Platyhelminthes). J Zool Syst Evol Res. https://doi.org/10.1111/jzs.12164
Egger B, Ishida S. Chromosome fission or duplication in Macrostomum lignano (Macrostomida, Plathelminthes) - remarks on chromosome numbers in "archoophoran turbellarians". J Zool Syst Evol Res. 2005;43:127–32. https://doi.org/10.1111/j.1439-0469.2005.00300.x.
Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucl Acids Res. 2013;41:e129. https://doi.org/10.1093/nar/gkt371.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. https://doi.org/10.1186/1471-2105-10-421.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. https://doi.org/10.1016/S0022-2836(05)80360-2.
Laslett D, Canbäck B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–5. https://doi.org/10.1093/bioinformatics/btm573.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–5. https://doi.org/10.1093/bioinformatics/bth352.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucl Acid Res. 1999;27:573–80.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. https://doi.org/10.1038/nmeth.1923.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G. Durbin R and 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative Genomics Viewer. Nat Biotechnol. 2011;29:24–6. https://doi.org/10.1038/nbt.1754.
Kuraku S, Zmasek CM, Nishimura O, Katoh K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucl Acids Res. 2013;41:W22–28. https://doi.org/10.1093/nar/gkt389.
Acknowledgements
We are grateful for having access to the Abel high computing facility of the University of Oslo (UiO) through the Norwegian Metacenter for Computational Science (NOTUR; grant k9201) to LB. We thank the Research Focal Point Scientific Computing at the University of Innsbruck for providing infrastructure for assemblies. Max Telford is gratefully acknowledged for providing opportunity to start this work, and we are indebted to Pia Aanstad and Lena Egger for English corrections.
Funding
"The sequencing service for Stenostomum sthenum and Macrostomum lignano gDNA was provided by the Norwegian Sequencing Centre (www.sequencing.uio.no), a national technology platform hosted by the University of Oslo and supported by the 'Functional Genomics' and 'Infrastructure' programs of the Research Council of Norway and the Southeastern Regional Health Authorities". BE was supported by a Leverhulme Trust grant (F/07 134/DA). Publication costs are supported by the University of Innsbruck. The funding bodies had no role in study design, data collection, analysis and interpretation, decision to publish, or writing of the manuscript.
Availability of data and materials
The assembled and annotated mitochondrial genomes of Stenostomum sthenum and Macrostomum lignano are available on NCBI's Genbank under the accession numbers MF078638 and MF078637, respectively.
Authors’ contributions
BE and BF designed the study, and LB and BF sequenced and assembled DNA reads. BE cultured experimental animals, performed PCRs, mapped RNA reads to the genome and prepared all figures and tables. All authors have analysed the data, contributed to the manuscript and approved the final version.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Flatworms are not subjected to laws governing their usage, but care has been taken to minimise unnecessary harm to the experimental animals.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Mitochondrial genome sequence from the assembled nuclear genome of Macrostomum lignano [11]. (TXT 14 kb)
Additional file 2:
Circular mitochondrial genome of Prosthiostomum siphunculus [2]. Outermost circle displays logarithmically mapped RNAseq reads. tRNA genes are abbreviated with tRNA and three letters and a number if applicable. Colours indicate the widely conserved gene cartridges proposed by Mwinyi et al. [18]. Unlabeled and uncoloured regions are intergenic spacers. (TIF 870 kb)
Additional file 3:
Gene annotation tables of Stenostomum sthenum and Macrostomum lignano with gene name, start and stop positions and codons, length and the direction of transcription (- for the minus strand). (PDF 41 kb)
Additional file 4:
tRNA secondary structures in the mitochondrial genome of Stenostomum sthenum as predicted by MITOS [20]. (PDF 640 kb)
Additional file 5:
tRNA secondary structures in the mitochondrial genome of Macrostomum lignano as predicted by MITOS [20]. (PDF 591 kb)
Additional file 6:
Hydrophobicity profiles of the six putative atp8 amino acid sequences of free-living flatworms (A-F) and of two representative published atp8 amino acid sequences, from a mollusc (G) and an annelid (H). (TIF 101 kb)
Additional file 8:
Mitochondrial gene order in macrostomorphans, showing the newly sequenced complete mitochondrial genome of Macrostomum lignano and the partial mitochondrial genome of Microstomum lineare [8]. The gene order between these two members of Macrostomorpha is not conserved. (TIF 133 kb)
Additional file 9:
Amino acid alignment of atp8 in flatworms and other lophotrochozoans: a gastrotrich (Lepidodermella squamata), annelids (Platynereis dumerilii and Sipunculus nudus), molluscs (Sepiella japonica and Ascobulla fragilis), a phoronid (Phoronopsis harmeri), a kamptozoan (Loxosomella aloxiata), a bryozoan (Bugula neritina), a brachiopod (Terebratalia transversa) and a nemertean (Micrura bella). Putative triclad atp8 sequences were extracted from Ross et al. [5]. (TIF 129 kb)
Additional file 10:
atp8 alignment matrix without triclads (for details see Fig. 4). (TXT 1 kb)
Additional file 11:
atp8 alignment matrix with triclads (for details see Additional file 9). (TXT 4 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Egger, B., Bachmann, L. & Fromm, B. Atp8 is in the ground pattern of flatworm mitochondrial genomes. BMC Genomics 18, 414 (2017). https://doi.org/10.1186/s12864-017-3807-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-3807-2