
Abstract
Background
The African wild dog (Lycaon pictus) is an endangered African canid threatened by severe habitat fragmentation, human-wildlife conflict, and infectious disease. A highly specialized carnivore, it is distinguished by its social structure, dental morphology, absence of dewclaws, and colorful pelage.
Results
We sequenced the genomes of two individuals from populations representing two distinct ecological histories (Laikipia County, Kenya and KwaZulu-Natal Province, South Africa). We reconstructed population demographic histories for the two individuals and scanned the genomes for evidence of selection.
Conclusions
We show that the African wild dog has undergone at least two effective population size reductions in the last 1,000,000 years. We found evidence of Lycaon individual-specific regions of low diversity, suggestive of inbreeding or population-specific selection. Further research is needed to clarify whether these population reductions and low diversity regions are characteristic of the species as a whole. We documented positive selection on the Lycaon mitochondrial genome. Finally, we identified several candidate genes (ASIP, MITF, MLPH, PMEL) that may play a role in the characteristic Lycaon pelage.
Similar content being viewed by others

Background
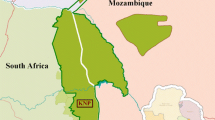
The African wild dog (Lycaon pictus) is an endangered canid species (International Union for Conservation of Nature Red List Classification: C2a (i)) [1]. While the species formerly ranged over most of sub-Saharan Africa, wild dogs suffer from a suite of threats including severe habitat fragmentation, human persecution, and disease epidemics. They are now restricted to less than seven percent of their former range [2], with only small, and frequently declining, remnant populations in fragmented pockets of eastern and southern Africa (Fig. 1). They maintain enormous home ranges (varying between 200 and 2000 km2) and naturally live at very low densities, even compared to other carnivores [3]. Primarily a hunter of antelopes, the African wild dog is a highly distinct canine. Wild dogs are differentiated from other canine species by their anatomical adaptations related to hypercarnivory and cursorial hunting, including high-crowned, sectorial teeth and the lack of dewclaws [4]. They have a highly specialized social structure in which both males and females disperse to form new packs and only a single dominant pair in each pack reproduces [5]. Wild dogs are also noted for their colorful pelage, from which they derive their species name pictus (‘painted’), and the absence of an undercoat.

South African wild dog pack (top) and map of extant and former wild dog range (bottom). The sampling locations of the two individuals are noted on the map. Ranges are modified from Woodroffe and Sillero-Zubiri [1] and Marsden et al. [2]. Extant range data used with permission from the International Union for the Conservation of Nature [Woodroffe R, Sillero-Zubiri C 2012. Lycaon pictus. In: IUCN 2016. IUCN Red List of Threatened Species. Version 2016–2. http://www.iucnredlist.org. Downloaded 12 July 2016]. Photograph by Micaela Szykman Gunther
Eastern and southern populations of wild dogs are genetically and morphologically distinct [6], although there is a large admixture zone covering Botswana, south-eastern Tanzania, and Zimbabwe [7]. Gene flow occurs across the species’ entire range [8], which is unsurprising given the excellent dispersal capabilities documented in Lycaon pictus [1]. Nevertheless, Marsden et al. [2] documented both genetic structuring between extant Lycaon populations (likely the result of habitat fragmentation) and a recent reduction in effective population size (N e) since the 1980s. Furthermore, African wild dogs exhibit very little major histocompatibility complex variation, which may reflect population decline [8].
To better understand African wild dog genomic evolution, genetic variation and population history, we shotgun sequenced whole genomes from two individuals from widely separated populations with very different modern ecological histories: Laikipia County, Kenya and Hluhluwe-Imfolozi Park, KwaZulu-Natal Province, South Africa. Wild dogs disappeared from Laikipia, Kenya in the 1980s and reappeared in 2000 – likely recolonizing from a small population in neighboring Samburu district (~50 km distant). The population in Laikipia alone now numbers more than 150 dogs and 15 packs [9]. We sequenced a female (sampled July 2003) from this recolonized population. In contrast, wild dogs were reintroduced to Hluhluwe-Imfolozi Park, KwaZulu-Natal Province in 1980 and remained as a single pack for many years [10]. Eventually, Lycaon breeding effectively ceased until new animals were introduced in 1997 and afterwards (2001, 2003, etc.) [11]. The KwaZulu-Natal wild dogs are now managed as part of the South African Lycaon “metapopulation” [12, 13]. We sequenced a male (sampled October 2007), born in KwaZulu-Natal to parents that were translocated from Limpopo province in 2003. Therefore, the South African individual’s ancestry represents genes from the northeastern part of the country.
The genomes from these two populations represent some of the first published wild canid genomes and are particularly valuable given the susceptibility of wild dogs to diseases and habitat fragmentation [9, 14]. We used our novel genome sequences to reconstruct the last 1,000,000 years of Lycaon genome demography and population history. We identified over a million polymorphic sequence variants for further population-level study. These variants produced ~35 million predicted genic effects. We identified over 15,000 candidate genes that may have undergone adaptation since the Lycaon/Canis divergence. We found evidence of positive selection on the Lycaon mitochondrial genome. Finally, we examined genes involved in canid coat phenotype to identify candidate genes underlying the characteristic Lycaon pelage.
Results and discussion
Genome sequencing of the African wild dog
Based on alignment with the domestic dog genome, we have sequenced ~90% (5.8× mean read depth) of the Kenyan individual’s genome and ~93% (5.7×) of the South African Lycaon individual’s genome. We identified 16,967,383 autosomal sequence variants (including 14,360,480 single nucleotide polymorphisms [SNPs] and 2,606,903 indels) separating our Lycaon genomes from the domestic dog (Canis familiaris) genome [GenBank: CanFam3.1] (Additional files 1 and 2) [15]. Of these, 1,092,450 (781,329 SNPs and 311,121 indels) were polymorphic in the African wild dog. The remaining 15,874,933 autosomal sequence variants (13,579,151 SNPs and 2,295,782 indels) were monomorphic in the two Lycaon individuals. We identified 717,870 X-chromosomal variants (619,606 SNPs and 98,264 indels), of which 32,801 (23,001 SNPs and 9,800 indels) were polymorphic and 685,069 (596,605 SNPs and 88,464 indels) were monomorphic in the African wild dog. Additionally, we sequenced the Kenyan and South African wild dog mitochondrial genomes to depths of 943× and 1021×, respectively. We annotated the Lycaon autosomal and X-chromosomal sequences using the domestic dog genome annotations [GenBank:CanFam3.1.81] and the mitochondrial genome [GenBank:NC_002008.4] and MSY sequences [GenBank:KP081776.1] using their reference sequence annotations [15–17].
Lycaon demographic history
We analyzed the two wild dogs’ autosomal population histories using PSMC (Fig. 2) [18]. Both genomes exhibited a strong reduction in N e starting 700,000 years ago from maximum N es of ~28,000 (Kenyan) and ~35,000 (South African) individuals and leveling off 200,000 years ago at N es of ~7,000 individuals each. Analysis of the Kenyan individual’s X - chromosomal history using PSMC showed a similar pattern (Fig. 2). N e fell from a maximum of ~40,000 individuals 600,000 years ago to ~7000 individuals 200,000 years ago. This N e reduction may represent a past population bottleneck e.g. [19] or lineage splitting e.g. [20]. Further genomic analysis of individuals from across the Lycaon range, especially those from larger founder populations, would help clarify the cause of this pattern.

Reconstruction of the Lycaon individuals’ autosomal and X-chromosomal demographic history using the pairwise sequentially Markovian coalescent. Initial results are plotted using dark-colored curves, with the bootstrap replicates plotted in lighter hues of the corresponding colors
PSMC analysis of the Kenyan X chromosome revealed a secondary reduction in N e started 70,000 years ago to a final N e of ~2000 individuals 10,000 years ago. We are unable to infer more recent population history accurately using this method due to the limited numbers of available mutations in the short time frame [18]. Further research using historical museum Lycaon specimens would fill in this temporal gap e.g. [21]. While our reconstructions were robust to coverage variation (see ‘Demographic history reconstruction’ below), higher coverage genomes would also increase the resolution of population history reconstructions [20].
Candidate selected processes and genomic regions
Based on the domestic dog genome annotations, SnpEff 4.1 L predicted that the identified Lycaon autosomal and X-chromosomal sequence variants would cause 34,001,288 and 36,362,161 genic effects in the Kenyan and South African individuals, respectively (Additional files 1 and 2) [22]. The majority of sequence variant effects fell within introns (Kenyan: 62.604%, South African: 63.383%) and intergenic regions (Kenyan: 23.081%, South African: 23.162%). To discover candidate genes that have diverged since Lycaon’s divergence with Canis, we identified Lycaon genes that contained missense mutations and stop codon gains using SnpSift 4.1 L [23]. We identified 15,611 (15,565 genes with missense mutations and 799 with stop codon gains) Kenyan and 9793 (9440 genes containing missense mutations and 741 with stop codon gains) South African wild dog candidate divergent genes. 9506 divergent genes (9159 with missense mutations and 653 with stop codon gains) were found in both wild dogs. These divergent genes were determined by 47,059 Kenyan sequence variants (sequenced at 8.6× mean coverage) of and 27,893 South African variants (6.0× mean coverage). 25,149 variants were shared between the two Lycaon individuals. These variants were very homozygous (Kenyan: 95%, South African: 95%), which suggests that they are the result of divergence between the Lycaon and Canis clades, rather than more recent variants arising within Lycaon.
We annotated the candidate divergent genes’ functions using DAVID 6.7 with domestic dog (option “Canis lupus”) as the genomic background [24]. We found 76 and 29 enriched processes in the Kenyan and South African individuals, respectively (Additional files 3 and 4). We filtered these terms with a Benjamini-Hochberg false discovery rate of 0.05 [25]. After filtration, seven terms (‘Complement and coagulation cascades’, ‘ECM-receptor interaction’, ‘Neuroactive ligand-receptor interaction’, ‘Hematopoietic cell lineage’, ‘Lysosome’, ‘ABC transporters’, ‘Aminoacyl-tRNA biosynthesis’) were significantly enriched in the Kenyan individual. ‘Olfactory transduction’ was significant in the South African individual. The differences in significant terms may reflect population-specific selection pressures on wild dogs. Further population-level investigation is needed to determine the roles these pathways play in Lycaon evolution.
In order to identify regions of low and high diversity, we calculated the numbers of segregating SNP sites across the Lycaon autosomes in 100,000 bp non-overlapping windows using VCFtools 0.1.15 (Fig. 3) [26, 27]. By averaging over large genomic windows, we limited the effects of sequencing errors and allelic drop-out due to low sequencing coverage. We identified 768,416 segregating Lycaon SNPs (Kenyan: 398,891 SNPs, South African: 434,911 SNPs). We observed individual-specific regions of low diversity (<10 segregating SNPs/100,000 bp). The Kenyan individual had runs of low diversity (contiguous regions of low diversity at least 5 million bp long) on chromosomes 4, 6, 7, 12, 15, 21, 27, and 30, while the South African individual had runs of low diversity on chromosomes 1, 5, 8, 12, 14, 19, 27, 29, 30, 34, 36 and 38. These low-diversity regions may be the result of inbreeding and/or population-specific natural selection. Due to the long lengths of these low-diversity runs, encompassing numerous genes, we are not currently able to link low diversity levels to selection on individual genes. These results are not surprising since both populations are recently re-established, either by natural recolonization (Laikipia, Kenya) or deliberate reintroduction (KwaZulu-Natal, South Africa). Previous genetic investigations using microsatellites and mitochondrial DNA found some evidence of rare inbreeding in wild dogs from the Greater Limpopo Transfrontier Conservation Area and KwaZulu-Natal [10, 28]. However, free-ranging wild dogs strongly avoid incestuous matings [10]. For instance, at KwaZulu-Natal, Becker et al. [10] observed only one of six breeding pairs being more closely related than third-order kin. While our chromosomal diversity data do not permit us to discern between inbreeding and/or population-specific natural selection, the possibility of inbreeding is, therefore, concerning from a conservation standpoint. Additional population-level data are required to determine the causes and effects of these low-diversity regions.

Segregating autosomal SNP sites across the Lycaon genomes. Chromosomes are distinguished by color and separated by black lines. The number of segregating SNPs per 100,000 bp window is plotted on the y-axis in logarithmic scale. We identified population-specific regions of low diversity in both the Kenyan (chromosomes 4, 6, 7, 12, 15, 21, 27, and 30) and South African (chromosomes 1, 5, 8, 12, 14, 19, 27, 29, 30, 34, 36 and 38) individuals. There are also highly variable regions on chromosomes 3 and 16 in both individuals, chromosome 26 in the Kenyan individual, and chromosome 19 in the South African individual
We also identified regions of high diversity (>200 segregating SNPs per 100,000 bp) on chromosomes 3 and 16 in both individuals and chromosome 19 in the South African individual (Fig. 3). The high-diversity regions comprised 0.44% of the total segregating SNPs in both individuals and included 1138 Kenyan and 970 South African segregating SNPs on chromosome 3,646 Kenyan and 704 South African segregating SNPs on chromosome 16, and 249 South African segregating SNPs on chromosome 19. Using the UCSC Genome Browser [29], we scanned the genome within 100,000 bp upstream and downstream of these high-diversity regions for known genes mapped to CanFam3.1. These included FER, FKBP3, SNRPF, and PJA2 on chromosome 3, HUS1 and PKDIL1 on chromosome 16, and RNF150 and TBC1D9 on chromosome 19. These genic regions may have undergone positive or relaxed selection since the Lycaon/Canis divergence or may represent chromosomal duplications. Future functional analyses will determine which roles (if any) these genes play in Lycaon evolution.
Positive selection on the mitochondrial genome
Each of the 13 protein-encoding mitochondrial genes from the two novel Lycaon mitochondrial genomes were aligned against the corresponding genic sequences from the domestic dog mitochondrial reference sequence and the one publically available near-complete Lycaon pictus mitochondrial genome sequence [GenBank:KT448283.1] [16, 30]. We calculated ratios of non-synonymous substitutions per non-synonymous site to synonymous substitutions per synonymous site (dN/dS) and tested for positive selection using the codon-based Z-test in MEGA6 [31]. We found evidence for positive selection on 12 of 13 genes (COX1, COX2, COX3, ATP6, ATP8, ND1, ND2, ND3, ND4, ND4L, ND5, CYTB; Additional file 5). To determine whether selection occurred primarily on the Lycaon or Canis branches, we reran these analyses excluding the domestic dog sequence. We found evidence for positive selection on all 13 Lycaon mitochondrial genes (Additional file 5).
To confirm branch specific signatures of selection, we aligned the Lycaon CYTB sequences against published canid taxa [GenBank: AY656746, EU442884, GU063864, JF342908, KF646248, KT448273–4, KU696390, KU696404, KU696408, NC_008093, NC_013445] using MAFFT 7.222 [32] as implemented in Geneious 10.0.2 (Biomatters, Ltd., Auckland, New Zealand). We then generated a phylogenetic tree with FastTree 1.0 [33]. Using the ‘codonml’ algorithm in PAML 4.8 [34], we performed pairwise comparisons on CYTB codon data and compared the likelihood of the alternate (non-fixed omega values) or null hypotheses (fixed omega values). A likelihood ratio test was calculated from ln values obtained from these comparisons to determine where evidence of selection was occurring in canids. We found significant evidence (p ≤ 0.001) of positive selection between African wild dogs and coyotes (Canis latrans) (statistic: 128.88) and between Lycaon and Cuon (statistic: 81.48). We also found branch-specific selection between the Kenyan and South African Lycaon individuals (statistic: 92.54).
The 13 candidate selected Lycaon mitochondrial genes are involved in the electron transport chain and adenosine triphosphate synthesis. This suggests natural selection on Lycaon metabolic processes e.g. [35, 36], which is likely given their unique antelope hunting strategies and diet. Moreover, these results are consistent with African wild dogs’ very high metabolic rate and hunting energy expenditure in comparison to domestic dogs [37].
Pelage genes
We extracted CDS corresponding to 11 genes involved in canid coat color (agouti signaling peptide [ASIP], β-defensin 103 [DEFB103A], melanocortin 1 receptor [MC1R], melanophilin [MLPH], microphthalmia-associated transcription factor [MITF], premelanosome protein [PMEL], tyrosinase-related protein 1 [TYRP1]) and type (fibroblast growth factor 5 [FGF5], keratin 71 [KRT71], R-spondin 2 [RSPO2]) [38–46] using Geneious 9.0.4. In cases where there were multiple isoforms or CDS annotations (DEFB103A, FGF5, MITF, PMEL), we chose the longest variant alignable to the domestic dog CDS reference sequence to maximize detection power. To detect positively selected genes, we identified non-synonymous and synonymous SNPs compared to the domestic dog sequence and calculated the non-synonymous/synonymous (N/S) ratio (Additional file 6).
ASIP and PMEL had elevated N/S ratios suggestive of positive selection (5.00 and 9.00 respectively). Lycaon PMEL also had a stop codon gain at amino acid 341, suggesting selection at this locus. Additionally, we found a threonine insertion at amino acid 371 in the Lycaon MLPH gene and a six amino acid deletion corresponding to domestic dog amino acids 186–191 in the Lycaon MITF gene. To further characterize these four candidate genes, we compared the Lycaon coding sequences against all publically available canid sequences using BLAST+ 2.5.0 [47]. None of the Lycaon ASIP, PMEL, MITF, and MLPH CDS haplotypes have been identified in other canids previously. Lycaon ASIP shares 99% nucleotide identity, but only 96% amino acid identity, with domestic dogs. Wild dog PMEL shares both 99% nucleotide identity and amino acid identity with domestic dogs. Excluding the six amino acid deletion, Lycaon MITF haplotypes had >99% nucleotide and amino acid identity with domestic dogs. Lycaon MLPH haplotypes had 98% nucleotide identity and 97–98% amino acid identity (excluding the amino acid insertion) to domestic dogs.
These four genes are strong candidates to explain the characteristic Lycaon pelage. Mutations in ASIP alter relative production of eumelanin and pheomelanin, resulting in lighter and darker hair colors, in numerous species including domestic dogs [41]. Variants in PMEL cause merle patterning in domestic dogs [40]. MITF is associated with white-spotting phenotypes in domestic dogs and causes coat color variants in laboratory mice (Mus musculus). MITF variants are also associated with deafness, small eye size, and poor bone resorption in mice [46]. Nevertheless, further laboratory assays (such as transgenic experiments) are needed to confirm that these identified variants are functional and to determine their phenotypic effects. Furthermore, our data do not permit us to distinguish between positive selection on the Lycaon and Canis branches (e.g. coat variation associated with dog domestication and breed development).
Conclusions
We provide two genome sequences of Lycaon pictus, representing two individuals from highly divergent ecological regions (Laikipia County, Kenya and KwaZulu-Natal Province, South Africa). We identified over a million polymorphic Lycaon SNPs, useful for further population-level analyses. Analyses of these genomes showed that extant Lycaon populations have endured at least two population contractions within the last 1,000,000 years. We identified chromosomal regions of high and low diversity and over 15,000 candidate divergent genes. Furthermore, Lycaon mitochondrial genomes have undergone positive selection, suggestive of selection for metabolic processes. Finally, we identified four candidate genes (ASIP, MITF, MLPH, PMEL) that may be involved in Lycaon pelage patterns.
Methods
Samples
We sequenced two Lycaon pictus individuals sampled during previous studies: a female from Laikipia County, Kenya [2, 9] and a male from Hluhluwe-Imfolozi Park, KwaZulu-Natal Province, South Africa [10, 48, 49]. The Kenyan individual was sampled under an Institutional Animal Care and Use Committee (IACUC) protocol approved by the University of California, Davis (10813) [9], while the South African individual was sampled under IACUC protocols approved by the Smithsonian National Zoological Park (08-21) and Humboldt State University (06/07.W.209.A) [10].
Laboratory methods
DNA was extracted from blood samples using Qiagen blood and tissue kits (Qiagen, Valencia, CA, USA) and sheared to ~350 bp using a Q800R sonicator (Qsonica, LLC, Newtown, CT, USA). Double-indexed Illumina libraries were built from the sheared DNA using the KAPA Library Preparation Kit – Illumina (Kapa Biosystems, Wilmington, MA, USA) with purification steps performed using carboxyl paramagnetic beads [50]. Library quality was ensured via fluorometric analysis using Qubit® dsDNA HS assays (Life Technologies, Carlsbad, CA, USA), quantitative PCR using the KAPA Library Quantification Kit – Illumina/Universal (Kapa Biosystems, Wilmington, MA, USA), and analysis on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) high-sensitivity DNA chip. Libraries were pooled equimolarly and 2 × 250 bp paired-end sequenced on a HiSeq 2500 lane (Illumina, Inc., San Diego, CA, USA).
Sequence quality control
Read pairs were demultiplexed using the BaseSpace® pipeline (Illumina, Inc., San Diego, CA, USA). 75,651,396 and 63,766,266 read pairs were generated for the Kenyan and South African Lycaon samples respectively. Raw reads were trimmed and adapter artifacts were removed using Trimmomatic 0.33 (options ILLUMINACLIP: NexteraPE-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36) [51]. Library sequence quality was confirmed using FastQC 0.11.2 [52].
Mitochondrial genome assembly
The quality-controlled reads were aligned against the circularized domestic dog reference mitochondrial genome [GenBank:NC_002008.4] [16] using Geneious 8.1.6 (medium-low sensitivity, 5 alignment iterations, minimum mapping quality 30). The aligned reads were merged using FLASH 1.2.11 (option –M 250) [53]. PCR duplicates were removed from the merged reads using CD-HIT-DUP 0.5 [54]. The deduplicated reads were then realigned against the dog reference mitochondrial genome in Geneious 8.1.6 (medium sensitivity alignment, 10 alignment iterations, minimum mapping quality 30) to generate the final sequences.
Autosomal assembly
The non-mitochondrial reads were merged using FLASH 1.2.11 (option –M 250) [51]. The merged, unmerged, and unpaired reads were then concatenated and treated as single-end sequences for downstream processing. The concatenated reads were aligned against the autosomes of the domestic dog genome build CanFam3.1 [15] using the ‘mem’ algorithm in BWA 0.7.12 [55, 56]. Aligned reads below mapping quality 30 and PCR duplicates were removed using the ‘view’ (option –q 30) and ‘rmdup’ (option –s) commands in SAMtools 1.3 [57]. Sequence variants (minimum quality 20) were identified using the SAMtools 1.3 ‘mpileup’ command (option –C50) and BCFtools 1.3 ‘call’ command (option –m) pipeline [57, 58]. Genome completeness was evaluated using BUSCO 1.1b1 and the ‘Vertebrates’ gene set (Additional files 7, 8 and 9) [59]. Genome assemblies were very complete: compared to the 1663 ‘complete’ BUSCO autosomal orthologs found in the domestic dog, 1614 (97%) of the Kenyan and 1665 (100.1%) of the South African individual’s orthologs were complete. Similarly, 747 ‘fragmented’ BUSCO autosomal orthologs were found in the domestic dog, compared to 688 (92.1%) for the Kenyan individual and 707 (94.6%) for the South African individual.
Allosomal assembly
The Kenyan Lycaon individual was a female, while the South African individual was a male. To reconstruct the allosomal sequences, we aligned the unmapped, concatenated nuclear reads against either the CanFam3.1 X chromosome assembly (Kenyan individual) [15] or both the CanFam3.1 X chromosome and domestic dog MSY chromosome assemblies (South African individual) [17] using the ‘mem’ algorithm in BWA 0.7.12 [55, 56]. Aligned reads below mapping quality 30 and PCR duplicates were removed using the ‘view’ (option –q 30) and ‘rmdup’ (option –s) commands in SAMtools 1.3 [57]. Sequence variants (minimum quality 20) were identified using the SAMtools 1.3 ‘mpileup’ command (option –C50) and BCFtools 1.3 ‘call’ command (option –m) pipeline [57, 58]. The mapped MSY reads were then realigned against the reference sequence [GenBank:KP081776.1] using Geneious 8.1.7 (medium sensitivity alignment, five alignment iterations). Y coding region sequences were extracted based on the domestic dog MSY annotations, and consensus sequences were generated using Geneious 8.1.7 (options Highest Quality and Total). We excluded non-coding regions from analysis due to the Y chromosome’s large number of repetitive elements, which complicates accurate alignment [17]. We identified 87 Y SNPs between the African wild dog and the domestic dog, of which 32 were silent mutations, 53 produced amino acid substitutions, and two caused gene truncations (Additional file 10).
Demographic history reconstruction
Autosomal population history parameters were reconstructed using PSMC r62 (options –N25 –t15–r5 –p “64*1”, minimum quality 20) and tested with 100 bootstrap replicates [18]. We calculated the autosomal mutation rate for each Lycaon individual using the total number of identified autosomal sequence variants (13,985,381 and 15,132,667 for the Kenyan and South African individuals respectively), an estimated autosomal genome size of 2.3 Gbp, an estimated generation time of 5 years/generation [7] and an estimated divergence time from the Canis/Cuon clade of 2.74 million years ago (95% highest posterior density: 2.15–3.38 million years ago) [30]. We estimated the Kenyan and South African Lycaon mutation rates as 5.5 × 10−9 mutations/site/generation (range: 5.0–7.1 × 10−9 mutations/site/generation) and 6.0 × 10−9 mutations/site/generation (range: 4.9–7.7 × 10−9 mutations/site/generation) respectively. Final PSMC demographic reconstructions were scaled based on an estimated generation time of 5 years/generation [7] and a mutation rate of 5.8 × 10−9 mutations/site/generation. While variation of the reconstruction scaling within the extremes of the estimated mutation rates (4.9–7.7 × 10−9 mutations/site/generation) varied estimates of N e and timing of population history events, overall demographic history patterns remained similar.
The Kenyan X-chromosomal history was reconstructed separately from the autosomal history. We calculated the X-chromosomal mutation rate for the Kenyan individual using the total number of observed Kenyan X-chromosomal variants (634,216), the same divergence and generation times as for the autosomal analyses and a chromosome size of 124 Mbp. We estimated the Kenyan X-chromosomal mutation rate as 4.7 × 10−9 mutations/site/generation (range: 3.8–5.9 × 10−9 mutations/site/generation). We did not estimate the X-chromosomal mutation rate for the South African male due to his hemizygosity. X-chromosomal PSMC demographic reconstruction and scaling parameters were the same as for the autosomal analyses except that the results were scaled with a mutation rate of 4.7 × 10−9 mutations/site/generation. Variation of the mutation rate scaling again did not affect inference of demographic history.
To test the effects of coverage on our demographic reconstructions, we repeated the PSMC analyses under medium depth stringency settings (minimum sequencing depth 2, maximum sequencing depth 12) recommended by PSMC’s authors and high depth stringency settings (minimum sequencing depth 10) recommended in [20]. We recovered nearly identical demographic reconstructions under the medium depth stringency settings (Additional file 11). Under the high depth stringency settings, we were unable to resolve the Kenyan X-chromosomal reconstruction due to missing data. Nevertheless, our autosomal reconstructions under the high depth stringency recovered very similar demographic histories for the last 1,000,000 years, except that N e estimates were larger (particularly in the South African individual) (Additional file 12). Increased N e estimates are expected since heterozygotes are more likely to be observed in the higher coverage regions [20]. Therefore, we conclude that our reconstructed demographic patterns were robust to coverage variation. Interestingly, under the high depth stringency settings, we also reconstructed two additional contraction and expansion cycles between 10,000,000 and 600,000 years ago in the South African individual’s population history (Additional file 12). These results require further verification with additional genome sequences.
Abbreviations
- bp:
-
Base pairs
- CDS:
-
Coding sequence
- dN/dS :
-
Non-synonymous substitutions per non-synonymous site to synonymous substitutions per synonymous site
- IACUC:
-
Institutional Animal Care and Use Committee
- MSY:
-
Male-specific Y chromosome
- N/S:
-
Non-synonymous substitutions to synonymous substitutions
- N e :
-
Effective population size
- SNP:
-
Single nucleotide polymorphism
References
Woodroffe R, Sillero-Zubiri C. Lycaon pictus. The IUCN Red List of Threatened Species. 2012;2012:e.T12436A16711116.
Marsden CD, Woodroffe R, Mills MGL, McNutt W, Creel S, Groom R, et al. Spatial and temporal patterns of neutral and adaptive genetic variation in the endangered African wild dog (Lycaon pictus). Mol Ecol. 2012;21:1379–93.
Woodroffe R, Ginsberg JR. Introduction. In: Woodroffe R, Ginsberg J, Macdonald D, editors. The African wild dog. Gland and Cambridge: IUCN; 1998. p. 1–6.
Hartstone-Rose A, Werdelin L, De Ruiter DJ, Berger LR, Churchill SE. The Plio-Pleistocene ancestor of wild dogs, Lycaon sekowei n. sp. J Paleont. 2010;84:299–308.
Girman DJ, Mills MGL, Geffen E, Wayne RK. A molecular genetic analysis of social structure, dispersal, and interpack relationships of the African wild dog (Lycaon pictus). Behav Ecol Sociobiol. 1997;40:187–98.
Girman DJ, Kat PW, Mills MGL, Ginsberg JR, Borner M, Wilson V, et al. Molecular genetic and morphological analyses of the African wild dog (Lycaon pictus). J Hered. 1993;84:450–9.
Girman DJ, Vilà C, Geffen E, Creel S, Mills MGL, McNutt JW, et al. Patterns of population subdivision, gene flow and genetic variability in the African wild dog (Lycaon pictus). Mol Ecol. 2001;10:1703–23.
Marsden CD, Mable BK, Woodroffe R, Rasmussen GSA, Cleaveland S, McNutt JW, et al. Highly endangered African wild dogs (Lycaon pictus) lack variation at the major histocompatibility complex. J Hered. 2009;100 Suppl 1:S54–65.
Woodroffe R. Demography of a recovering African wild dog (Lycaon pictus) population. J Mammal. 2011;92:305–15.
Becker PA, Miller PS, Szykman Gunther M, Somers MJ, Wildt DE, Maldonado JE. Inbreeding avoidance influences the viability of reintroduced populations of African wild dogs (Lycaon pictus). PLoS ONE. 2012;7, e37181.
Gusset M, Slotow R, Somers MJ. Divided we fail: the importance of social integration for the re-introduction of endangered African wild dogs (Lycaon pictus). J Zool. 2006;270:502–11.
Davies-Mostert HT, Mills MGL, Macdonald DW. A critical assessment of South Africa’s managed metapopulation recovery strategy for African wild dogs. In: Hayward MW, Somers MJ, editors. Reintroduction of top-order predators. London: Wiley-Blackwell; 2009.
Lindsey PA, Alexander R, Du Toit JT, Mills MGL. The cost efficiency of wild dog conservation in South Africa. Conserv Biol. 2005;19:1205–14.
Woodroffe R, Prager KC, Munson L, Conrad PA, Dubovi EJ, Mazet JA. Contact with domestic dogs increases pathogen exposure in endangered African wild dogs (Lycaon pictus). PLoS ONE. 2012;7, e30099.
Hoeppner MP, Lundquist A, Pirun M, Meadows JRS, Zamani N, Johnson J, et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS ONE. 2014;9, e91172.
Kim KS, Lee SE, Joeng HW, Ha JH. The complete nucleotide sequence of the domestic dog (Canis familaris) mitochondrial genome. Mol Phylogenet Evol. 1998;10:210–20.
Li G, Davis BW, Raudsepp T, Pearks Wilkerson AJ, Mason VC, Ferguson-Smith M, et al. Comparative analysis of mammalian Y chromosomes illuminates ancestral structure and lineage-specific evolution. Genome Res. 2013;23:1486–95.
Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–6.
Xue Y, Prado-Martinez J, Sudmant PH, Narasimhan V, Ayub Q, Szpak M, Frandsen P, et al. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science. 2015;348:242–5.
Nadachowska-Brzyska K, Burri R, Smeds L, Ellegren H. PSMC analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol Ecol. 2016;25:1058–72.
Librado P, Der Sarkssian C, Ermini L, Schubert M, Jónsson H, Albrechtsen A, et al. Tracking the origins of Yakutian horses and the genetic basis for their fast adaptation to subarctic environments. Proc Natl Acad Sci U S A. 2015;112:E6889–97.
Cingolani P, Platts A, Coon M, Nguyen T, Wang L, Land SJ, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly. 2012;6:80–92.
Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program. SnpSift Front Genet. 2012;3:35.
Huang DW, Sherman BT, Lempick RA. Systematic and integrative analysis of large gene list using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57:289–300.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Callicrate T, Dikow R, Thomas JW, Mullikin JC, Jarvis ED, Fleischer RC, et al. Genomic resources for the endangered Hawaiian honeycreepers. BMC Genomics. 2014;15:1098.
Tensen L, Groom RJ, Van Belkom J, Davies-Mostert HT, Marnewick K, Van Vuuren BJ. Genetic diversity and spatial genetic structure of African wild dogs (Lycaon pictus) in the Greater Limpopo transfrontier conservation area. Conserv Genet. 2016;17:785. doi:10.1007/s10592-016-0821-x.
Kent WJ, Sugnet WC, Furey TS, Rosin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006.
Koepfli K-P, Pollinger J, Godinho R, Robinson J, Lea A, Hendricks S, et al. Genome-wide evidence reveals that African and Eurasian jackals are distinct species. Curr Biol. 2015;25:2158–65.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30:2725–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Price MN, Dehal PS, Arkin AP. FastTree computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50.
Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24:1586–91.
Finch TM, Zhao N, Korkin D, Frederick KH, Eggert LS. Evidence of positive selection in mitochondrial complexes I and V of the African Elephant. PLoS ONE. 2014;9, e92587.
Melo-Ferreira J, Vilela J, Fonseca MM, Da Fonseca RR, Boursot P, Alves PC. The elusive nature of adaptive mitochondrial DNA evolution of an Arctic lineage prone to frequent introgression. Genome Biol Evol. 2014;6:886–96.
Gorman ML, Mills MG, Raath JP, Speakman JR. High hunting costs make African wild dogs vulnerable to kleptoparasitism by hyaenas. Nature. 1998;391:479–81.
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, et al. Coat variation in the domestic dog is governed by variants in three genes. Science. 2009;326:150–3.
Candille SI, Kaelin CB, Cattanach BM, Yu B, Thompson DA, Nix MA, et al. A β-defensin mutation causes black coat color in domestic dogs. Science. 2007;318:1418–23.
Clark LA, Wahl JM, Rees CA, Murphy KE. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc Natl Acad Sci U S A. 2006;103:1376–81.
Kerns JA, Newton J, Berryere TG, Rubin EM, Cheng J-F, Schmutz SM, et al. Characterization of the dog Agouti gene and a nonagouti mutation in German Shepherd dogs. Mamm Genome. 2004;15:798–808.
Drögemüller C, Philipp U, Haase B, Günzel-Apel A-R, Leeb T. A noncoding melanophilin gene (MLPH) SNP at the splice donor of exon I represents a candidate causal mutation for coat color dilution in dogs. J Heredity. 2007;98:468–73.
Newton JM, Wilkie AL, He L, Jordan SA, Metallinos DL, Holmes NG, et al. Melanocortin 1 receptor variation in the domestic dog. Mamm Genome. 2000;11:24–30.
Philipp U, Hamann H, Mecklenburg L, Nishino S, Mignot E, Günzel-Apel A-R, et al. Polymorphisms within the canine MLPH gene are associated with dilute coat color in dogs. BMC Genet. 2005;16:34.
Schmutz SM, Berryere TG, Goldfinch AD. TYRP1 and MC1R genotypes and their effects on coat color in dogs. Mamm Genome. 2002;13:380–7.
Schmutz SM, Berryere TG, Dreger DL. MITF and white spotting in dogs: a population study. J Heredity. 2009;100:S66–74.
Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–14.
Spiering PA, Szykman Gunther M, Wildt DE, Somers MJ, Maldonado JE. Sampling error in non-invasive genetic analyses of an endangered social carnivore. Conserv Genet. 2009;10:2005–7.
Spiering PA, Somers MJ, Maldonado JE, Wildt DE, Szykman GM. Reproductive sharing and proximate factors mediating cooperative breeding in the African wild dog (Lycaon pictus). Behav Ecol Sociobiol. 2010;64:583–92.
Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012;22:939–46.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Andrews S. FastQC: a quality control tool for high throughput sequence data. Version 0.11.2. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 11 Dec 2014.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93.
Simão FA, Waterhouse RM, Ioannadis P, Kriventseva EV, Zdobnov M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
Acknowledgements
We thank Penny Becker (United States Fish and Wildlife Service), Michael Somers (University of Pretoria) and David Wildt (Smithsonian Conservation Biology Institute) for facilitating collection of the South African sample. We are grateful for the assistance of Adam Ferguson (Mpala Research Centre) in the production of the Lycaon range map. We thank the members of the Center for Conservation Genomics, Smithsonian Conservation Biology Institute for helpful advice on this manuscript.
Funding
The Morris Animal Foundation (D14ZO-308) and the National Geographic Society (8846–10) supported this research.
Availability of data and materials
The data sets supporting the results of this article are available in the Sequence Read Archive and GenBank repositories, (BioProject PRJNA304992; http://www.ncbi.nlm.nih.gov/bioproject/PRJNA304992; GenBank LPRA00000000 and LPRB00000000; http://www.ncbi.nlm.nih.gov/nuccore/LPRA00000000 and http://www.ncbi.nlm.nih.gov/nuccore/LPRB00000000).
Authors’ contributions
MGC performed the molecular genetic assays, carried out bioinformatics analyses, and wrote the manuscript. KHM and RFC conceived the study. MTRH and LDP performed bioinformatics analyses and contributed to the manuscript. RW, MSG, and JEM provided the Lycaon samples. KHM, RFC, HSY, RW and JEM participated in the design of the study. All authors reviewed and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The Lycaon pictus individuals were sampled under IACUC protocols approved by the University of California, Davis (10813), the Smithsonian National Zoological Park (08–21), and Humboldt State University (06/07.W.209.A). Ezemvelo KZN Wildlife, the Kenya Wildlife Service, the National Museums of Kenya, the Kenya National Council for Science and Technology, and Mpala Research Centre gave permission and relevant permits to conduct this research and provided logistical support. All experiments were conducted in compliance with the Convention on the Trade in Endangered Species of Wild Fauna and Flora and the International Union for the Conservation of Nature Policy Statement on Research Involving Species at Risk of Extinction.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Genic effects of SnpEff-annotated autosomal and X-chromosomal variants for the Kenyan Lycaon individual. (HTML 1596 kb)
Additional file 2:
Genic effects of SnpEff-annotated autosomal and X-chromosomal variants for the South African Lycaon individual. (HTML 1535 kb)
Additional file 3:
DAVID-annotated enriched functional processes for the Kenyan Lycaon individual. For each functional term, the total gene count, the associated genes, and the Benjamini-Hochberg false discovery rate value are given. (XLSX 23 kb)
Additional file 4:
DAVID-annotated enriched functional processes for the South African Lycaon individual. For each functional term, the total gene count, the associated genes, and the Benjamini-Hochberg false discovery rate value are given. (XLSX 16 kb)
Additional file 5:
Positive selection test results on the Lycaon mitochondrial genome. Z statistics are included in parentheses after the probability for each of the three competing models (Neutrality, Positive Selection, Purifying Selection). Results for the tests excluding the domestic dog sequence are denoted by ‘Lycaon’ in the column heading. (XLSX 48 kb)
Additional file 6:
Positive selection scan results on candidate Lycaon pelage genes. Genes were aligned to the reference CDS provided in the table. Gene coordinates are given with reference to the domestic dog genome [GenBank:CanFam3.1]. The total numbers of fixed and polymorphic synonymous and non-synonymous SNPs and the N/S ratios are provided. Also listed are non-SNP variants identified during the scan. (XLSX 41 kb)
Additional file 7:
Autosomal BUSCO orthologs identified in the CanFam3.1 assembly. Scaffolds are identified by GenBank accession. (XLSX 182 kb)
Additional file 8:
Autosomal BUSCO orthologs identified in the Kenyan Lycaon pictus genome. Scaffolds are identified by GenBank accession. (XLSX 166 kb)
Additional file 9:
Autosomal BUSCO orthologs identified in the South African Lycaon pictus genome. Scaffolds are identified by GenBank accession. (XLSX 170 kb)
Additional file 10:
Genic effects of Lycaon MSY coding sequence variants. (XLSX 55 kb)
Additional file 11:
Reconstruction of the Lycaon individuals’ autosomal and X-chromosomal demographic history using the pairwise sequentially Markovian coalescent with medium coverage stringency. Initial results are plotted using dark-colored curves, with the bootstrap replicates plotted in lighter hues of the corresponding colors. (EPS 252 kb)
Additional file 12:
Reconstruction of the Lycaon individuals’ autosomal and X-chromosomal demographic history using the pairwise sequentially Markovian coalescent with high coverage stringency. Initial results are plotted using dark-colored curves, with the bootstrap replicates plotted in lighter hues of the corresponding colors. (EPS 189 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Campana, M.G., Parker, L.D., Hawkins, M.T.R. et al. Genome sequence, population history, and pelage genetics of the endangered African wild dog (Lycaon pictus). BMC Genomics 17, 1013 (2016). https://doi.org/10.1186/s12864-016-3368-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-3368-9