
Abstract
Background
Yarrowia lipolytica is an oleaginous ascomycete yeast that stores lipids in response to limitation of nitrogen. While the enzymatic pathways responsible for neutral lipid accumulation in Y. lipolytica are well characterized, regulation of these pathways has received little attention. We therefore sought to characterize the response to nitrogen limitation at system-wide levels, including the proteome, phosphoproteome and metabolome, to better understand how this organism regulates and controls lipid metabolism and to identify targets that may be manipulated to improve lipid yield.
Results
We found that ribosome structural genes are down-regulated under nitrogen limitation, during which nitrogen containing compounds (alanine, putrescine, spermidine and urea) are depleted and sugar alcohols and TCA cycle intermediates accumulate (citrate, fumarate and malate). We identified 1219 novel phosphorylation sites in Y. lipolytica, 133 of which change in their abundance during nitrogen limitation. Regulatory proteins, including kinases and DNA binding proteins, are particularly enriched for phosphorylation. Within lipid synthesis pathways, we found that ATP-citrate lyase, acetyl-CoA carboxylase and lecithin cholesterol acyl transferase are phosphorylated during nitrogen limitation while many of the proteins involved in β-oxidation are down-regulated, suggesting that storage lipid accumulation may be regulated by phosphorylation of key enzymes. Further, we identified short DNA elements that associate specific transcription factor families with up- and down-regulated genes.
Conclusions
Integration of metabolome, proteome and phosphoproteome data identifies lipid accumulation in response to nitrogen limitation as a two-fold result of increased production of acetyl-CoA from excess citrate and decreased capacity for β-oxidation.
Similar content being viewed by others


Background
The yeast Yarrowia lipolytica has the ability to accumulate a large fraction of its mass as neutral lipids, mainly in the form of triglycerides [1]. This, along with its genetic tractability, has made it an attractive model for the production of high value lipids and biofuel precursors [2, 3]. As in other fungi, lipid accumulation in Y. lipolytica is heavily dependent on environmental conditions, particularly the relative amounts and types of carbon and nitrogen sources available [1, 4–6].
The response to nitrogen quality and quantity has been well characterized in Saccharomyces cerevisiae [7–13] and is primarily regulated by nitrogen catabolite repression, which operates in yeast and filamentous fungi [14] to allow preferential utilization of preferred nitrogen sources. When high quality nitrogen sources are available, the GATA type transcription factors Gln3p and Gat1p become phosphorylated and remain bound to Ure2p in the cytosol, whereas they localize to the nucleus and activate nitrogen utilization genes in poor nitrogen conditions [15–18]. A second set of GATA type transcription factors, Gzf3p and Dal80p, act as repressors that compete for binding sites with and are regulated by Gln3p and Gat1p [19, 20]. It is unknown whether the pathways above operate similarly in Y. lipolytica. Interestingly, work comparing the evolutionary conservation of regulatory network motifs between Y. lipolytica and other yeasts, including S. cerevisiae, identified post-transcriptional regulation as the most conserved aspect of gene regulatory networks [21]. Comparison of the transcriptional regulators found that regulation of β-oxidation and peroxisome biogenesis by FarA and amino acid biosynthesis by Gcn4 are likely to be conserved between Y. lipolytica and filamentous fungi [22].
The purpose of this study was to reveal the regulatory changes of Y. lipolytica in response to nitrogen limitation. Though transcriptional responses to nitrogen limitation have been previously characterized in Y. lipolytica [4], the regulatory mechanisms driving these responses are not well understood. We analyzed the metabolome, proteome, and phosphoproteome in nitrogen replete and limited conditions to connect intracellular metabolite pools with the expression level and phosphorylation of proteins The analysis presented is focused on changes in the expression and phosphorylation state of regulatory proteins (kinases, phosphatases, and transcription factors) to generate hypotheses regarding which of these pathways are involved in regulation of the lipid accumulation response. To our knowledge, this is the first global study of protein phosphorylation in Y. lipolytica and as such, paves the way for future work on post-translational modification and protein engineering in this organism.
Results
Nitrogen limitation results in rapid increase in lipid droplet size
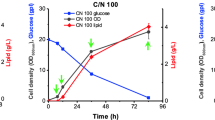
It has been shown previously that limitation of nitrogen is an excellent and well conserved method to induce lipid accumulation in oleaginous fungi [5, 23, 24]. We have compared samples of the oleaginous yeast Y. lipolytica growing in either high (C/N = 10) or low (C/N = 150) nitrogen conditions to further our understanding of the metabolic events that lead from limitation of nitrogen to lipid accumulation. We grew Y. lipolytica in high (C/N = 10) or low (C/N = 150) nitrogen minimal medium followed by measurement of dry cell weight and microscopic examination (Fig. 1). Per cell lipid droplet staining intensity increased in high and low nitrogen conditions by three hours post transfer from rich medium (p < 0.001) and differed significantly by six hours post transfer (p < 0.001) (Fig. 1b and c). Cell mass accumulation was not significantly less in the nitrogen limited cells until hour nine (p < 0.001) (Fig. 1d). We chose to further explore the response to nitrogen limitation at hour nine, where lipid accumulation was clearly proceeding and cell mass accumulation was affected due to nitrogen limitation, by analyzing intracellular and extracellular metabolites as well as the global proteome and phosphoproteome.

Experimental design for measuring the response to nitrogen limitation. a Y. lipolytica cultures grown in YPD are washed and split into YNB medium with a high (C/N = 10) or low (C/N = 150) concentration of ammonium sulfate to analyze the response to nitrogen limitation. After transfer cells were collected at regular time intervals and fixed for confocal microscopy or dried for mass measurement. We chose to collect samples at hour nine for metabolome and proteome experiments. b Fixed cells were stained with calcofluor-white (blue) to visualize the cell wall and nile-red (red) to image lipid droplets in response to nitrogen limitation. Bars indicate 5 μm. c Quantification of lipid droplet intensity. Box plot indicates the median, 10th, 25th, 75th and 90th percentiles. d Quantification of dry cell mass accumulation. Error bars represent the standard deviation of three replicates
Metabolomic, proteomic and phosphoproteomic response to nitrogen limitation
We collected and analyzed extracellular and intracellular polar metabolites from Y. lipolytica 9 h after transfer to growth medium with either a high (C/N = 10) or low (C/N = 150) concentration of nitrogen in the form of ammonium sulfate, as well as after one hour of growth from the C/N = 150 condition. 146 intracellular metabolites were quantified, 93 of which were identified. 76 extracellular metabolites were quantified and 33 of these were identified. We also analyzed the proteome from Y. lipolytica after 9 h of growth in either high (C/N = 10) or low (C/N = 150) nitrogen conditions, and identified 59,578 unique peptides in at least one biological replicate. These peptides mapped to 4926 of the Y. lipolytica protein models in at least one replicate of the experiment and to 3567 of the models in all replicates, representing 55.3 % of the annotated coding sequences [25]. We identified 2101 unique quantifiable peptides after enrichment with immobilized metal affinity chromatography (IMAC). 1219 of those peptides had at least one phosphorylated serine, threonine or tyrosine residue and mapped to a total of 599 genes, paving the way for studies of protein phosphorylation in Y. lipolytica. The 882 peptides identified after IMAC enrichment but without a phosphorylation site are skewed toward the most abundant peptides identified in the phosphoproteomics analysis, suggesting they represent contamination from highly expressed proteins (Additional file 1) – these were not considered further. All quantified metabolites and peptides are tabulated in Additional file 2. We annotated and mapped Y. lipolytica genes to metabolic pathways relevant to nitrogen limitation and lipid accumulation and overlaid the metabolome, proteome and phosphoproteome data onto this map (Fig. 2).

Response of the lipid and nitrogen metabolic networks to nitrogen limitation. Proteome, metabolome and phosphoproteome of Y. lipolytica grown in low (C/N = 150) versus high (C/N = 10) nitrogen conditions after nine hours. Nodes represent metabolites and edges represent genes. Colors of metabolites indicate log2 fold-change. Colors of gene names indicate significantly up and down regulated proteins identified by global proteomics; up-regulated indicates higher expression in the low (C/N = 150) nitrogen condition. Underlines indicate proteins with an identified phosphorylation site. Colored arrows indicate significant changes in phosphorylation level of a protein; up indicating more phosphorylation in the low (C/N = 150) nitrogen condition. Black text indicates metabolites and genes from which proteins were not identified. Metabolic pathways and gene names are based on homology to S. cerevisiae and N. crassa
Nitrogen limitation results in nitrogenous metabolite depletion and intracellular carbon compound accumulation
We found that nitrogen containing compounds tend to be depleted in cells grown in nitrogen limited medium (Hr 9, C/N 150 : Hr 9, C/N 10; Fig. 3a), while most non-nitrogen containing carbohydrates, alcohols and acids were present at higher intracellular levels in nitrogen limited medium (Hr 9, C/N 150 : Hr 9, C/N 10; Fig. 3b). Of the amino acids we measured, only alanine was significantly depleted in nitrogen limited cells, while the other significantly depleted nitrogen containing compounds between the C/N = 10 and C/N = 150 conditions at hour nine were 3-amino,2-piperidone, putrescine, spermidine and urea (Fig. 3a). We found arabitol, fructose, nigerose, threitol and mannitol at higher extracellular levels after 9 h than one hour in the C/N = 150 condition suggesting these metabolites are released by the cells (Additional file 2). Of these only threitol was at a significantly higher extracellular concentration after nine hours at C/N = 150 versus C/N = 10 suggesting threitol production is induced by nitrogen limitation. Intracellular threitol concentration is also significantly higher in the C/N =150 condition supporting this idea (Fig. 3b). Other significantly enriched metabolites found in the nitrogen limited cells include citrate, mannitol-P, xylitol, malate, fumarate, arabitol, α-hydroxyglutarate, phenyllactate, ribonate, γ-hydroxybutyric acid, and inositol (Hr 9, C/N 150 : Hr 9, C/N 10; Fig. 3).

Intracellular metabolite pools in response to nitrogen limitation. Intracellular metabolite concentrations were measured and dry weight normalized after one and nine hours at C/N = 150 and nine hours at C/N = 10. Average fold change from three replicates each of (a) nitrogen containing metabolites and (b) non-nitrogen containing metabolites was calculated at nine hours for C/N = 150 vs. C/N = 10 (black bars) and for C/N = 150 at 9 vs. 1 h (grey bars). *significantly changing intracellular metabolites (p < 0.01). Arginine hydrolyzes to ornithine during chemical derivatization for GC-MS, thus ornithine represents a pooled value for intracellular ornithine and arginine
Proteomic response to nitrogen limitation
We tested up- and down-regulated genes for gene ontology term enrichment and found that genes associated with proteolysis are up-regulated after nine hours of nitrogen limitation while amino acid metabolism, translation and ribosome biogenesis associated genes are down-regulated along with β-oxidation (Table 1) similar to what has been found in the oleaginous yeast Rhodosporidium toruloides [26]. We also found that of 115/132 proteins annotated as being a structural component of the ribosome are significantly down-regulated (p < 0.05) while none are significantly upregulated. We tested whether inhibiting translation would result in lipid accumulation in nitrogen replete conditions by addition of cycloheximide which blocks the elongation step in eukaryotic ribosomes [27]. We found that inhibition of ribosome function resulted in lipid droplet growth but to a lesser extent than nitrogen limitation (Fig. 4). This suggests that downregulation of the structural components of the ribosome, and presumably also a decreased capacity for translation, both play a part in lipid accumulation but are not entirely responsible. We found that proteins predicted to utilize acyl-CoA’s within the peroxisome for β-oxidation are downregulated, however, we did not see a change in the number of peroxisomes during lipid accumulation (Fig. 5) and so suggest it is only their function that is changing.

Inhibition of translation induces lipid droplet growth. Y. lipolytica cultures grown for nine hours were fixed, stained with calcofluor-white (blue) to visualize the cell wall and nile-red (red) to image lipid droplets. Cells were imaged in triplicate by confocal microscopy. Representative images are shown. The area of each cell covered by lipid droplets is calculated and statistically different in all three treatments (p < 0.05). Bars indicate 5 μm. Box plot indicates the median, 10th, 25th, 75th and 90th percentiles

Number of peroxisomes does not change in response to nitrogen limitation. Y. lipolytica strain FEB64, harboring a green fluorescent protein tagged version of peroxisomal marker pex13, was grown in low (C/N = 150) and high (C/N = 10) nitrogen conditions as in Fig. 1. a At nine hours cells were collected, stained with calcofluor-white (blue), and imaged for Pex13-GFP (green). b The number of foci corresponding to peroxisomes per cell from two images from each of three replicates is not significantly different (p > 0.05)
Phosphoproteomic response to nitrogen limitation
We analyzed the 599 proteins with a phosphorylation site for gene ontology (GO) term enrichment to determine whether any particular types of proteins are prone to phosphorylation. Interestingly, this identified the process of protein phosphorylation and the functions of tyrosine and serine/threonine kinase activity as being very significantly enriched (p < 1E-10) suggesting the main function of phosphorylation in Y. lipolytica may be the regulation of kinases. We next compared the fold change values of the phosphorylated peptides with their counterparts in the global proteomics data to control for changes in protein expression and identify phosphorylation sites that change in their fractional abundance during nitrogen limitation. 53 peptides were identified as having decreased phosphorylation levels, while 80 exhibited increased phosphorylation (p < 0.05) during nitrogen limitation.
Identification of regulatory pathways associated with nitrogen limitation
We are particularly interested in identifying the regulatory pathways affected by nitrogen limitation in Y. lipolytica, and to that end we predicted 89 kinases, 45 phosphatases and 279 DNA binding proteins present in Y. lipolytica using domain analysis and by comparison with homologs in other fungi. From those, 43 proteins (11 kinases, 2 phosphatase, 30 DNA binding proteins) were found to change significantly in their abundance (Table 2), while 23 peptides were found to change in their phosphorylation state (16 kinases, 7 DNA binding proteins) (Table 3) and which define a set of nitrogen responsive regulatory factors. We found that 37 % (33/89) of the predicted kinases and 18 % (49/279) of the predicted DNA binding proteins have at least one phosphorylation site, indicating that these potentially regulatory proteins are enriched for protein phosphorylation compared to the genome as a whole (9 %; 599/6448) but that phosphatases are not enriched for phosphorylation (9 %; 4/45). The number of phosphorylation sites identified for a given protein is also greater for kinases, and to a lesser extent, DNA binding proteins (Fig. 6).

Regulatory proteins are enriched for phosphorylation. Genes were identified that are predicted to encode regulatory proteins (89 kinases, 45 phosphatases and 279 DNA binding proteins). The number of phosphorylation sites identified per protein in either of the conditions tested is quantified for each of these classes and the proteome as a whole
Identification of DNA regulatory motifs associated with nitrogen limitation
We have found genes that are up- and down-regulated by limitation of nitrogen at the protein level. We were curious whether we could predict DNA elements driving these expression changes in the absence of RNA expression data. Thus, we analyzed the promoter elements of genes that were significantly up- and down-regulated for enrichment of short motifs. While this type of analysis is more appropriate for investigation of RNA expression data and in this context does not account for post-transcriptional regulation of expression, we identified two enriched motifs in the up-regulated genes and four enriched motifs in the down regulated genes as well as a motif specifically enriched in the promoters of β-oxidation genes (Fig. 7a), which are significantly down-regulated (Fig. 2). We validated the DNA motifs identified in the up- and down-regulated genes by comparing the effect of their position relative to the transcription start site (TSS) on expression level across all genes (Fig. 7b). We found that the presence of either a CC[TG]TTAT or G[AC]TAAGC site within roughly 0.5 kb of the TSS correlates with up-regulation, while a [GA]TGAGTCA, TGAAAAA, ACCCCACA or CACGTG[AC] site correlates with down-regulation. The [AT]CCCCACA site identified in the β-oxidation specific promoters is similar to the ACCCCACA site found in the down-regulated genes and is associated with down-regulation in both cases.

Promoter elements associated with nitrogen limitation responsive genes have distance dependent effects. a Promoter regions of genes that encode up- and down-regulated proteins as well as a subset of β-oxidation specific down-regulated proteins are enriched for specific DNA motifs. b For each Y. lipolytica gene the distance to the nearest occurrence of each motif five prime of the transcription start site is determined and binned in 0.1 kb windows based on this distance. The average protein fold change values for each bin up to 1.5 kb from the transcription start site for Y. lipolytica grown in low (C/N = 150) versus high (C/N = 10) nitrogen are plotted. The grey span indicates the interquartile range. c The most strongly enriched gene ontology (GO) terms (p < 0.005) for genes with the given DNA motif present within 0.5 kb 5’ of their transcription start site
Prediction of function of DNA motifs associated with nitrogen limitation
The function of the CC[TG]TTAT motif in the up-regulated genes is unclear. The second motif, G[AC]TAAGC is clearly a binding site for GATA family transcription factors. Of the 10 predicted GATA family transcription factors in Y. lipolytica three are up-regulated (YALI0C22682g, YALI0D20482g, YALI0E16577g) and three are down-regulated (YALI0E30547g, YALI0F14267g, YALI0F17886g) during nitrogen limitation. We hypothesize that one or more of these six proteins in combination regulate genes with a G[AC]TAAGC site in their promoter region. Included in these GATA transcription factors are three proteins with similarity to Gln3p in Saccharomyces cerevisiae [28] and AreA in Aspergillus nidulans [29] both of which regulate nitrogen catabolite repression in these organisms [30, 31].
The functions of the motifs in the down-regulated genes are also predictable. The [GA]TGAGTCA motif is similar to sites in S. cerevisiae that are regulated by nitrogen responsive transcription factors that regulate amino acid biosynthesis including Gcn4p, Arg81p, Arg80p and Cup9p [32–35]. We did not identify significant changes for homologs of these genes in Y. lipolytica. However, the ortholog of gcn4 (YALI0E27742g) has a phosphorylation site at S219 that decreases during nitrogen limitation. We did not detect peptides specific to Gcn4 in our global proteomic data and were thus unable to calculate the fractional change for this phosphorylation site. However, in nitrogen replete conditions in S. cerevisiae, Gcn4p is rapidly turned over by a process dependent on phosphorylation of the proteins activation domain and subsequent degradation by the 26S proteosome [36]. Our results indicate a similar mechanism controlling amino acid metabolism is active in Y. lipolytica.
The [TG]GAAAAA motif resembles the ribosomal RNA processing element, bound by Stb3p in S. cerevisiae [37, 38]. The homolog in Y. lipolytica (YALI0C09232g) has a number of detected phosphopeptides, none of which change significantly. This is in contrast to studies in S. cerevisiae where nutrient limitation results in decreased phosphorylation of Stb3p by Sch9p as a mediator of ribosome biogenesis [39].
The ACCCCACA motif resembles that bound by the carbon catabolite repressor Mig1p (in S. cerevisiae) or CreA (in A. nidulans) [35, 40, 41]. We did not detect significant changes in the protein level of the Mig1 homolog in Y. lipolytica (YALI0E07942g). However, it may be differences in regulation rather than protein level that contribute to the nitrogen limitation response. Deletion of mig1 in Y. lipolytica results in lower expression of β-oxidation genes at the RNA level and increased lipid accumulation [42] Together, these results suggest Mig1 is a direct regulator of β-oxidation genes in Y. lipolytica.
The CACGTG[AC] motif resembles the E-box motif originally described as an enhancer in mammals [43] and subsequently found to be bound by a number of basic helix-loop-helix (bHLH) transcription factors in yeast including Cbf1p, Tye7p, Hac1p, Rtg3p, Ino2p, Pho4p and Ino4p [34, 35, 44]. Of the nine predicted bHLH family transcription factors in Y. lipolytica, two are up-regulated (YALI0D15334g and YALI0F11979g), one is down-regulated (YALI0F05126g) and the homolog of Rtg1p (YALI0F11979g) is significantly phosphorylated during nitrogen limitation.
Discussion
Nitrogen limitation induced lipid accumulation
Biochemical investigations into the nature of oleaginous fungi has found that lipid accumulation occurs when environmental nitrogen is depleted in the presence of excess carbon, glucose in this case. When nitrogen is limited, the cells continue to utilize glucose but will no longer divide. Excess carbon flow into the tricarboxylic acid (TCA) cycle accumulates as citrate which is transported from the mitochondria to the cytosol and utilized for de novo production of fatty acids [45]. Both decreased capacity for amino acid biosynthesis for protein production and polyamine biosynthesis, which are essential for nuclear division [46, 47], may prevent cell division. However, the ultimate reason for decreased division may depend on initial environmental conditions that determine which intracellular nitrogenous metabolite becomes limiting first. After nine hours of nitrogen limitation, we found that all three Y. lipolytica cyclins are significantly down-regulated (Additional file 2), suggesting fewer cells are in G1, S or G2 phase. Examination of the TCA cycle in an oleaginous yeast such as Y. lipolytica would suggest that αlpha-ketoglutarate would accumulate when ammonium is not available for fixation to glutamate. We found this to be the case, along with accumulation of all other detected TCA cycle intermediates (Figs. 2 and 3), highlighting that carbon intermediate build-up occurs throughout the TCA cycle and is not specific to citrate [48]. Accumulation of citrate is thought to promote lipid accumulation through the production of acetyl-CoA via ATP-citrate lyase [49]. In support of this idea, we found that Citrate Synthase, which utilizes acetyl-CoA to make citrate from oxaloacetate, and Aconitase, which isomerizes citrate to isocitrate, are down-regulated, suggesting flux through these reactions is proceeding at a lower rate (Fig. 2).
We did not find a difference in expression of any of the enzymes involved in de novo lipid production from citrate. Rather, ATP-Citrate Lyase and Acetyl-CoA Carboxylase are phosphorylated (Fig. 1, Additional file 2), suggesting that if their activity is changing due to rapid accumulation of lipids, it is due to posttranslational modification of the enzymes. Overexpression of ATP-Citrate Lyase [50] and Acetyl-CoA Carboxylase [51, 52] have yielded positive results when applied to lipid accumulation in Y. lipolytica. However, if the activity or localization of these enzymes is regulated by phosphorylation, then it may be possible to achieve better results using strains where the phosphorylated residue(s) have been mutated to mimic constitutively phosphorylated or unphosphorylated enzymes. Work in mammals has identified phosphorylation of ATP-citrate lyase by insulin responsive pathways as a regulator of fatty acid biosynthesis in the liver [53, 54], and global analysis of protein phosphorylation has identified numerous sites on Acetyl-CoA Carboxylase that are subject to modification in S. cerevisiae [55], some of which have been mutated in recent attempts to enhance lipogenesis in S. cerevisiae [56, 57]. An additional general driver of lipid synthesis in oleaginous organisms is thought to be Malic Enzyme which oxidizes malate to pyruvate and CO2 and reduces NADP+ to NADPH - the production of NADPH as a reducing agent for fatty acid synthesis being the key aspect of this reaction [58]. In agreement, we found the gene for Malic Enzyme to be upregulated in Y. lipolytica during lipid accumulation (Fig. 2).
While the enzymes for synthesis of acyl-CoA from acetyl-CoA appear to be controlled by phosphorylation, the production of storage lipids is a different story. De novo production of triglycerides can proceed by repeated acylation of glycerol, which becomes the backbone. However, production of glycerol-3P via either phosphorylation of glycerol or reduction of DHAP is unlikely as both enzymes are down regulated during nitrogen limitation. Instead, the expression of Slc1 and Ale1, which make phosphatidic acid, and Are1 and Are2, which acylate sterols to steryl-esters, increases during nitrogen limitation, suggesting that most of the acyl-CoA produced is being transferred to sterols and 1-acyl-sn-glycerol-3P. This suggests that, for production of neutral lipids rather than sterol esters, it may be useful to engineer strains with lower expression of Are1/2.
Nitrogen assimilation
Of particular interest is the effect of limitation of extracellular nitrogen on intracellular nitrogen carriers and intracellular sensing and signaling in response to depleted metabolites. Nitrogen assimilation occurs via either (1) NADP dependent Glutamate Dehydrogenase (Gdh1), which utilizes NADP+ to convert αlpha-ketoglutarate and NH3 to glutamate, or (2) Glutamine Synthetase (Gln1/2), which produces glutamine from glutamate and NH3 with energy from ATP. During nitrogen limitation we found both Gln1 and Gln2 to be strongly up-regulated, while Gdh1 was somewhat down-regulated suggesting route (2) is used for nitrogen assimilation during nitrogen starvation, as has been reported previously in other organisms [59–61]. Glutamate Synthase (Glt1) which drives the production of two molecules of glutamate from glutamine and αlpha-ketoglutarate, thus completing the cycle of formation of glutamate from αlpha-ketoglutarate and NH3, is down-regulated, suggesting the cells are limited for glutamine rather than glutamate and are free to make it using ATP when glucose is available in excess.
Of the amino acids we detected, only alanine is significantly depleted in nitrogen limited cells while the other significantly depleted nitrogen containing compounds we measured are 3-amino, 2-piperidone, putrescine, spermidine and urea. Urea production occurs via Arginase mediated degradation of arginine to ornithine and urea [62] or as a biproduct of purine catabolism [63]. The decrease in urea confirms that ammonium is not available in excess in the C/N = 150 condition as expected. The physiological relevance of 3-amino,2-piperidone in fungi is unclear; however, it is found at elevated levels in Homo sapiens with a defect in SLC25A15, which transports ornithine across the mitochondrial membrane [64, 65], suggesting it may be involved in urea cycling. The polyamines putrescine and spermidine are produced from ornithine via the activity of Ornithine Decarboxylase (spe1) and Spermidine Synthase (spe3) [66] and their production is regulated at the level of spe1 in Neurospora crassa [67]. Putrescine and spermidine decrease during nitrogen limitation, as does intracellular ornithine to a less significant degree; these decreases occur concurrently with down-regulation of spe1 and spe2 (Figs. 2 and 3). This result is unexpected when compared to studies in N. crassa where polyamine starvation results in high spe1 activity [68] but may reflect repression due to low ornithine availability during nitrogen limitation.
Kinase signaling in response to nitrogen limitation
Categorically, we observed the highest number of phosphorylation sites on kinases. In total we identified 16 kinases that change significantly in their phosphorylation state and thus can begin to construct a network of phosphorylation dependent signaling cascades in Y. lipolytica. We compared the significantly changing phosphorylation sites with the wealth of phosphorylation data available for S. cerevisiae through Phophogrid [69] but found most (10/16) occur in regions of the proteins without significant homology between S. cerevisiae and Y. lipolytica. We can say little about the function of the sites we observed that change in their phosphorylation state in the absence of clear homology with other organisms. However, global studies on protein-protein interactions and functional studies using phosphosite mutants will aid our effort to construct a complete regulatory network for Yarrowia. Of the six phosphorylated sites that are in conserved regions between S. cerevisiae and Y. lipolytica, four have not been observed to be phosphorylated and two have been observed as phosphorylated on a homologous site in S. cerevisiae. These two phosphorylation sites are present in homologs of Rim11p (YALI0D20966g) and Yak1p (YALI0C16665g) and in both cases are required for, or enhance, the kinase activity of the enzyme. Phosphorylation of Tyr-181 in YALI0D20966g is a conserved site of modification that regulates the activity of the glycogen synthase kinase 3 enzymes Rim11p (at Tyr-199 in S. cerevisiae) and GSK-3B (at Try-216 in H. sapiens). In both yeast and humans phosphorylation is associated with an increase in the activity of the kinase [70, 71], suggesting that in Y. lipolytica this kinase is activated during nitrogen limitation. Phosphorylation of Tyr-583 of YALI0C16665g is homologous with Tyr-530 phosphorylation in S. cerevisiae Yak1p, which is autophosphorylated, resulting in kinase activation [72]. Dephosphorylation of YALI0C16665g suggests the activity of a Yak1-like pathway is decreased during nitrogen limitation.
Regulatory response to nitrogen limitation
The effect of limiting nitrogen on the transcriptome has been previously characterized in Y. lipolytica and results in changes in transcripts related to nitrogen metabolism along with repression of protein production [4]. However, the regulatory factors driving these changes have not been previously investigated in Y. lipolytica. Our results show that the response to nitrogen limitation involves changes in the intracellular metabolite pools that affect the expression and activity of genes through specific transcription factor binding domains and kinase signaling. We identified short DNA elements that associate GATA family transcription factors with up-regulated genes and leucine zipper family transcription factors with down-regulated genes (Fig. 7). GATA family transcription factors primarily control amino acid biosynthesis in S. cerevisiae [12]. This function appears to be conserved in Y. lipolytica as genes with a G[AC]TAAGC motif within 500 base pairs 5’ of their transcription start site are enriched for amino acid biosynthetic processes (Fig. 7). Amino acid biosynthetic GO terms were also identified for upregulated genes with a [GA]TGAGTCA site in their promoter that is similar to that bound by Gcn4p [32, 73], which activates genes in response to amino acid starvation [33]. In the nitrogen limited cells, genes with the [GA]TGAGTCA motif tend to be down- rather than up-regulated suggesting the nitrogen replete cells represent conditions more akin to amino acid starvation or that this motif represents regulation by a novel pathway in Y. lipolytica.
A motif similar to that bound by the carbon catabolite repressor creA, ACCCCACA, is associated with genes that are down-regulated during nitrogen limitation. This is quite interesting as the sole carbon source for the experiment is glucose. During nitrogen limitation, the intracellular concentration of glucose and glucose-6P is higher which may explain greater repression of genes with the ACCCCACA motif in their promoter. Depictions of carbon catabolite repression in S. cerevisiae often emphasize repression of genes involved in utilization of alternative carbon sources [12]. In Y. lipolytica the alternative carbon source genes are those associated with fatty acid metabolism and peroxisomal β-oxidation which makes sense in light of the fact that Yarrowia is often isolated from lipid rich substrates [74–76] and whose metabolism is geared toward utilization of fats [77].
Conclusion
In summary, we have used a variety of techniques to characterize the response to nitrogen limitation in the oleaginous yeast Y. lipolytica. In our growth conditions we observed complex changes in intra- and extracellular metabolite levels during batch culture and correlate these with microscopically observed cellular features and changes in protein expression and phosphorylation state. Substantial effort has been applied in the past few years to understanding the nature of and engineering oleaginous yeast [4, 6, 52, 78, 79], but to date only limited work has been done to characterize lipid accumulation from a regulatory perspective. Here we provide a comprehensive dataset describing nitrogen limitation induced lipid accumulation in Y. lipolytica that will enable more detailed experiments to understand the oleaginous nature of Yarrowia and provide genetic targets for future metabolic engineering efforts.
Methods
Yeast strains, cultivation and sample collection
Wild-type Y. lipolytica strain W29 (ATCC20460™) (American Type Tissue Culture; Manassas, VA) was used for metabolome and proteome experiments. Strains were maintained in YPD medium (1 % yeast extract, 1 % peptone, 2 % glucose) at 28 °C. Frozen stocks were maintained at −80 °C in 25 % glycerol. For experiments, Y. lipolytica was pregrown overnight in YPD broth in baffled 250 mL shake flasks at 28 °C and 200 rpm. The cells were pelleted and washed twice with 1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate (BD; Franklin Lakes, NJ) and inoculated into 50 mL YNB10 (1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate, 25 g/L glucose, 5.5 g/L ammonium sulfate) or YNB150 (1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate, 25 g/L glucose, 0.366 g/L ammonium sulfate) to OD600 = 5 in baffled 250 mL shake flasks at 28 °C and 200 rpm in triplicate. The concentration of ammonium sulfate affects the pH of the medium. YNB10 is pH 5.4 while YNB150 is pH 4.9. Cycloheximide was added to 100 μg/mL when appropriate. For protein and intracellular metabolite measurements, 10 mL samples were collected by vacuum filtration on 0.45 μm nylon Whatman filters (GE Healthcare; Little Chalfont, UK) and washed with 5 mL 1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate. The filters were transferred to Eppendorf tubes, weighed and then flash frozen in liquid nitrogen. For extracellular metabolites, cells were pelleted at 22,000 × g for 1 min and 1 mL of supernatant was collected and frozen in liquid nitrogen. For microscopy, 1 mL samples were collected and fixed in 4 % formaldehyde (Ted Pella; Redding, CA) for 30 min, washed 3 times with PBS (8 g/L NaCl, 2.56 g/L Na2HPO4 · 7H2O, 0.2 g/L KCL, 0.2 g/L KH2PO4) and then stored at 4 °C. For dry weight, 1.5 mL samples were washed once with H20 and their weight measured after drying the cells in a speed vac.
Strain construction
PCR used Q5 high fidelity DNA polymerase (New England Biolabs; Ipswich, MA). PCR products were purified using the GeneJET gel extraction and DNA cleanup micro kit (Thermo Scientific; Rockford, IL). Transformations were performed using a quick version of the lithium acetate transformation procedure [80]. Transformants were selected on YNB10 agar and verified by PCR and microscopy. The pex13 ortholog (YALI0C05775g) was -GFP tagged using a split marker strategy. The 5’ and 3’ sequences flanking the end of pex13 and the entire leu2 gene were amplified from W29 gDNA with primers OEB172 (GGCTACGGAGGCATGAACT) and OEB173 (CCTCCGCCTCCGCCTCCGCCGCCTCCGCCATTAACGGGAACGGCACTG), OEB174 (TGCTATACGAAGTTATGGATCCGAGCTCGACACATACTGGTGCGACTGC) and OEB175 (TCGTTTTCGTCCTTCACCTC), and OEB170 (CGTAACCCTGATCGTACCTTGATGTCGACCCGTTGCTATCTCCACAC) and OEB171 (CGAGCTCGGATCCATAACTTCGTATAGCAGTGCAGTCGCCAGCTTAAA) respectively while gfp was amplified with primers OKP31 (GGCGGAGGCGGCGGAGGCGGAGGCGGAGG) and OEB169 (CGACATCAAGGTACGATCAGGGTTACGGATATCTTACTTGTAAAGCTC) from plasmid pYL4. These were then assembled into pex13-gfp:leu2 split marker cassettes using overlapping PCRs and transformed into strain FKP355, derived from the Po1g [81] genetic background in which ku70 was replaced with hph (Bredeweg EL, Pomraning KR, Dai Z Baker SE, in prep).
Confocal microscopy
Fixed cells were stained in PBS with nile red and calcofluor-white and visualized without additional washing using a Zeiss LSM710 confocal laser-scanning microscope (Carl Zeiss MicroImaging GmbH, Munchen, Germany) and Plan-Apochromate 63x/1.4 Oil or Plan-Apochromate 100x/1.4 Oil objective as described previously [82, 83]. All images were processed using imageJ [84] and Cell profiler [85].
Metabolomics sample preparation and analysis
Both extracellular and intracellular metabolites were analyzed in this study. For extracellular metabolite measurements, 20 μL of spent medium was dried in vacuo and kept in −80 °C before chemical derivatization. For intracellular metabolite measurements, frozen cells on membranes (see above) were transferred to a fresh microcentrifuge tube and 100 μL of nanopure water was added prior to vortexing. Metabolites were extracted with addition of 400 μL of chloroform/methanol mixture (2:1, v/v) and vigorous vortexing. After centrifugation (15,000 × g, 4 °C, 5 min), the aqueous layer was transferred to glass vials and dried in a vacuum concentrator. Chemical derivatization of extracted metabolites and subsequent GC-MS analysis were performed as reported previously [82, 86]. Briefly, dried metabolites were chemically modified with two derivatizations including methoxyamination and trimethylsilylation. The derivatized samples were analyzed by GC-MS, and the resulting data processed with MetaboliteDetector [87]. Retention times were calculated based on a mixture of fatty acid methyl esters.
Protein isolation and enzymatic digestion
To increase the coverage of both the phosphoproteome and global proteome, two methods of protein isolation were performed – a standard protocol based on solubilization of proteins in buffer containing 8 M urea (protocol 1), and a protocol based on precipitation of protein using chloroform/methanol (as described above for metabolite extraction; protocol 2), followed by solubilization using the first protocol. Briefly, for protocol 1, a lysis buffer was added to each sample containing 8 M urea, 75 mM NaCL in 100 mM NH4HCO3 pH 7.8, and 10 mM NaF, 1 % cocktail 2 (Sigma, P 5726) and 1 % cocktail 3 (Sigma, P 0044) as phosphatase inhibitors. For protocol 2, a modified lysis buffer was added to the samples, which contained 1 % inhibitor cocktail 2 (Sigma, P 5726) and 1 % cocktail 3 (Sigma, P 0044) in 25 mM NH4HCO3. All samples were bead beat in a Bullet Blender (Next Advance, Averill Park, NY) at speed 8 for 3 min at 4 °C, and the lysate was spun into a falcon tube at 2000xg for 10 min at 4 °C. 0.5 mL of each sample was transferred into a fresh, Eppendorf tube and a BCA assay (Thermo Scientific, Rockford, IL) was performed to determine the protein concentration. A chloroform/methanol extraction was performed on six samples to precipitate protein. Briefly, −20 °C chloroform:methanol mix (prepared 2:1 (v/v)) was added to the samples in a 5:1 ratio over sample volume and vigorously vortexed. The samples were then placed on ice for 5 min and then vortexed for 10 s followed by centrifugation at 10,000xg for 10 min at 4 °C. The precipitated protein was removed and washed with 1 mL of ice cold methanol. The samples were centrifuged at 10,000xg for 10 min to pellet the protein and the methanol decanted off. The remaining protein was placed in a fume hood to dry. Lysis buffer (from protocol 1) was then added to the protein pellets. 10 mM DTT was added to all samples, followed by sonication and incubation at 60 °C for 30 min with constant shaking at 800 rpm. Samples were then diluted 8-fold for preparation for digestion with 100 mM NH4HCO3, 1 mM CaCl2, and sequencing-grade modified porcine trypsin (Promega, Madison, WI) was added to at a 1:50 (w/w) trypsin-to-protein ratio, followed by incubation for 3 h at 37 °C. Digested samples were desalted using a 4-probe positive pressure Gilson GX-274 ASPEC™ system (Gilson Inc., Middleton, WI) with Discovery C18 100 mg/1 mL solid phase extraction tubes (Supelco, St. Louis, MO), using the following protocol: 3 mL of methanol was added for conditioning followed by 2 mL of 0.1 % TFA in H2O. The samples were then loaded onto each column followed by 4 mL of 95:5: H2O:ACN, 0.1 % TFA. Samples were eluted with 1 mL 80:20 ACN:H2O, 0.1 % TFA. The samples were concentrated down to ~30 μL using a Speed Vac, and a final BCA was performed to determine the peptide concentration.
Isobaric chemical labeling of peptides
The samples were measured and vialed to contain 100 μg each, and the volumes were brought up using 0.5 M TEAB (or dried down to) 15 μl in a low-protein binding 1.5 mL centrifuge tube. The pH of each sample was measured and brought to over pH 8 using 1 M TEAB. Vials of 4-plex iTRAQ reagent (AB Sciex, Framingham, MA) were brought to room temperature. The reagents were pulse spun to ensure the contents were collected at the bottom and 60 μl of isoproponal was added to each reagent vial. The reagents were thoroughly vortexed, spun down and added to the appropriate sample. Each sample was vortexed and spun down to incubate at room temperature for 2 h at which time 100 μL of nanopure water was added to hydrolyze the sample, followed by incubation for an additional 30 min. The samples were partially dried down in a speed vac to remove the organic solvent and then pooled together to obtain three samples each containing all 4 of the iTRAQ labels and C18 cleaned again, as described above, followed by another BCA assay to determine the final peptide mass for HPLC fractionation.
Fractionation of labeled peptides
All samples were diluted to a volume of 900 μL with 10 mM ammonium formate buffer (pH 10.0), and resolved on a reversed-phase HPLC column (XBridge C18, 250x4.6 mm, 5 μM; Waters, Milford, MA) with a guard column (4.6x20 mm) of the same material (Waters). Samples were fractionated at 0.5 mL/min using an Agilent 1100 series HPLC system (Agilent Technologies, Santa Clara, CA) with mobile phases (A) 10 mM Ammonium Formate, pH 10.0 and (B) 10 mM Ammonium Formate, pH 10.0/acetonitrile (10:90, v/v). The gradient was adjusted from at 100 % A to 95 % A over the first 10 min, 95 % A to 65 % A over min 10 to 70, 65 % A to 30 % A over min 70 to 85, maintained at 30 % A over min 85 to 95, re-equilibrated with 100 % A over min 95 to 105, and held at 100 % A until min 120. Fractions were collected every 1.25 min (96 fractions over the entire gradient) in a 96-well plate. The plate was partially dried in a speed vac and then every 24th fraction was combined for a total of 24 samples (each with n = 4 fractions pooled), using 50 % acetonitrile in water to transfer fractions. The fractions were then completely in vacuo and 25 μL of 25 mM ammonium bicarbonate was added to each. The samples were then divided in half, and one half was placed into autosampler vials and stored at at −20 °C until LC-MS/MS analysis. The remaining half were further pooled into 4 fractions for phosphopeptide enrichment using immobilized metal-ion (Fe3+) affinity chromatography [88], as previously described [89, 90].
LC-MS/MS analysis of peptides
A Waters Nano Acquity LC (Waters, Milford, MA) LC system was coupled to a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific, San Jose, CA) and used for the proteomics analyses. Orbitrap spectra (AGC 1 × 106) were collected from 300 to 1800 m/z at a resolution of 60 k HMS, profile mode, followed by data dependent Orbitrap HCD top 10 MS/MS, centroid mode, at a resolution of 7500. Tandem mass spectra were acquired with a normalized collision energy set to 45. The heated capillary temperature and spray voltage were 325 °C and 2.2 kV, respectively. Charge state screening was enabled to reject singly charged ions and the default charge state was set to five. The mass spectrometer was outfitted with a custom electrospray ionization (ESI) interface, using a custom made, chemically etched electrospray emitter,as previously described [91].
Data were acquired for 100 min after a 25 min delay from when the LC gradient started. Residual salts were first removed using a SPE column prepared in-house by slurry packing 3.6-μm Aeris Widepore C18 particles (Phenomenex, Torrance, CA) into a 5 cm × 360 μm o.d. × 150 μm i.d fused silica capillary (Polymicro Technologies Inc., Phoenix, AZ) using a 1-cm sol–gel frit on each end for media retention [92]. The reversed-phase column was prepared in-house by slurry packing 3.0-μm Jupiter C18 particles (Phenomenex, Torrance, CA) into a 70-cm × 360 μm o.d. × 75 μm i.d fused silica capillary (Polymicro Technologies Inc., Phoenix, AZ) using a 1-cm sol–gel frit for media retention. Mobile phases consisted of 0.1 % formic acid in water (A) and 0.1 % formic acid in acetonitrile (B) operated at a flow rate of 300 nL/min. The 5 μL injection volume was focused onto the analytical column and then eluted with a gradient profile as follows (min:%B); 0:0.1, 2:8, 20:12, 75:30, 97:45, 100:95 and held for 10 min before going back to starting conditions. The column was equilibrated for 52 min before the next sample was loaded onto the column.
Proteomics data processing
Raw tandem mass spectra from the mass spectrometer were processed using DtaRefinery [93] coupled to MSGFPlus [94] and searched against a collection of Y. lipolytica proteins obtained from Uniprot. Briefly, a parent mass tolerance of +/− 20 ppm, dynamically phosphorylated (+79.966331 Da) serine, threonine or tyrosine, static iTRAQ 4-plex (+144.102066 Da) modification of lysine and peptide N-terminus, with partial tryptic cleavages were considered. An in-house analysis pipeline was used to combine the peptide identifications with their related iTRAQ 4-plex reporter ion abundances derived from MASIC [95]. Briefly, the pipeline imports the peptide identification results, filters the peptide results to 1 % FDR (using MSGFPlus’ reported Q-value < = 0.01, which is derived using the standard decoy approach [96]), sums reporter ion intensities per peptide across multiple strong cation exchange fraction within a given sample, and outputs the peptide sequence with associated reporter ion intensities for each fractionated samples. Proteins associated with each peptide are reported separately. For this dataset the occurrence of peptide sequences occurring in more than one protein were rare, but in those cases where redundancy did occur all proteins associated with a peptide were reported. For quantitative comparison purposes, only the first protein reference associated with a peptide was used for further protein level analyses.
Bioinformatic analysis
ITRAQ reporter ion intensities for peptides were summed per protein. Each replicate was then mean centered and log2 transformed using Inferno [97], the results of which are presented in Additional file 1. Positive fold-change values represent metabolites and proteins up-regulated in C/N = 150 condition when compared with C/N = 10. Phosphorylation levels were calculated for individual phosphorylated peptides and normalized to their protein abundance level. Thus changes in phosphorylation state represent the fraction of the protein present that is phosphorylated. Y. lipolytica gene models were annotated using Blast2GO [98] and orthologs of Y. lipolytica genes were identified in Saccharomyces cerevisiae and Neurospora crassa by reciprocal BlastP analysis. For Blast2GO analysis, proteins changing in concentration with p < 0.001 following a two tailed t-test were considered as being up- or down-regulated during nitrogen limitation. For simplicity, genes were assigned the symbol of their ortholog in S. cerevisiae from SGD [99] when appropriate. Up- and down-regulated proteins were tested for enrichment of gene ontology terms using Fisher’s exact test. Fold-change quantities were mapped to the yli organism KEGG maps [100] to investigate trends identified by Blast2GO. Transcription start sites (TSS) were annotated by mapping high-throughput RNA-seq data, kindly provided by Jens Nielsen’s group prior to publication (Kerkhoven E et al., in prep), to the Y. lipolytica reference genome [25] using Bowtie [101] and Samtools [102]. The TSS for each gene model is defined as the point 5’ of the start codon when the read count diminishes to the background level. Promoter regions were defined as the 500 bp upstream of the TSS for identification of enriched motifs using DREME [103] and as the region upstream of the transcription start site to the next transcript for motif location analysis using custom Perl scripts. TOMTOM [104] was used to identify proteins potentially binding the enriched motifs.
Availability of supporting data
All supporting data are included as additional supplementary files.
Abbreviations
- bHLH:
-
basic helix-loop-helix
- GO:
-
gene ontology
- IMAC:
-
immobilized metal affinity chromatography
- TCA:
-
tricarboxylic acid
- TSS:
-
transcription start site
References
Athenstaedt K, Jolivet P, Boulard C, Zivy M, Negroni L, Nicaud JM, et al. Lipid particle composition of the yeast Yarrowia lipolytica depends on the carbon source. Proteomics. 2006;6(5):1450–9. doi:10.1002/pmic.200500339.
Beopoulos A, Chardot T, Nicaud JM. Yarrowia lipolytica: A model and a tool to understand the mechanisms implicated in lipid accumulation. Biochimie. 2009;91(6):692–6. doi:10.1016/j.biochi.2009.02.004.
Ageitos JM, Vallejo JA, Veiga-Crespo P, Villa TG. Oily yeasts as oleaginous cell factories. Appl Microbiol Biotechnol. 2011;90(4):1219–27. doi:10.1007/s00253-011-3200-z.
Morin N, Cescut J, Beopoulos A, Lelandais G, Le Berre V, Uribelarrea JL, et al. Transcriptomic Analyses during the Transition from Biomass Production to Lipid Accumulation in the Oleaginous Yeast Yarrowia lipolytica. Plos One. 2011;6(11):e27966. 10.1371/journal.pone.0027966.
Sitepu IR, Sestric R, Ignatia L, Levin D, German JB, Gillies LA, et al. Manipulation of culture conditions alters lipid content and fatty acid profiles of a wide variety of known and new oleaginous yeast species. Bioresour Technol. 2013;144:360–9. doi:10.1016/j.biortech.2013.06.047.
Blazeck J, Hill A, Liu LQ, Knight R, Miller J, Pan A, et al. Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nat Commun. 2014;5:3131. doi:10.1038/Ncomms4131.
Cooper TG. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol Rev. 2002;26(3):223–38.
Gagiano M, Bauer FF, Pretorius IS. The sensing of nutritional status and the relationship to filamentous growth in Saccharomyces cerevisiae. Fems Yeast Res. 2002;2(4):433–70.
Magasanik B, Kaiser CA. Nitrogen regulation in Saccharomyces cerevisiae. Gene. 2002;290(1–2):1–18.
Wong KH, Hynes MJ, Davis MA. Recent advances in nitrogen regulation: a comparison between Saccharomyces cerevisiae and filamentous fungi. Eukaryot Cell. 2008;7(6):917–25. doi:10.1128/EC.00076-08.
Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces responds to nutrients. Annu Rev Genet. 2008;42:27–81. doi:10.1146/annurev.genet.41.110306.130206.
Conrad M, Schothorst J, Kankipati HN, Van Zeebroeck G, Rubio-Texeira M, Thevelein JM. Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev. 2014;38(2):254–99. doi:10.1111/1574-6976.12065.
Rodkaer SV, Faergeman NJ. Glucose- and nitrogen sensing and regulatory mechanisms in Saccharomyces cerevisiae. Fems Yeast Res. 2014;14(5):683–96. doi:10.1111/1567-1364.12157.
Marzluf GA. Genetic regulation of nitrogen metabolism in the fungi. Microbiol Mol Biol Rev. 1997;61(1):17–32.
Kulkarni AA, Abul-Hamd AT, Rai R, El Berry H, Cooper TG. Gln3p nuclear localization and interaction with Ure2p in Saccharomyces cerevisiae. J Biol Chem. 2001;276(34):32136–44. doi:10.1074/jbc.M104580200.
Cox KH, Rai R, Distler M, Daugherty JR, Coffman JA, Cooper TG. Saccharomyces cerevisiae GATA sequences function as TATA elements during nitrogen catabolite repression and when Gln3p is excluded from the nucleus by overproduction of Ure2p. J Biol Chem. 2000;275(23):17611–8. doi:10.1074/jbc.M001648200.
Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402(6762):689–92. doi:10.1038/45287.
Cunningham TS, Andhare R, Cooper TG. Nitrogen catabolite repression of DAL80 expression depends on the relative levels of Gat1p and Ure2p production in Saccharomyces cerevisiae. J Biol Chem. 2000;275(19):14408–14.
Georis I, Feller A, Vierendeels F, Dubois E. The yeast GATA factor Gat1 occupies a central position in nitrogen catabolite repression-sensitive gene activation. Mol Cell Biol. 2009;29(13):3803–15. doi:10.1128/MCB.00399-09.
Coffman JA, Rai R, Loprete DM, Cunningham T, Svetlov V, Cooper TG. Cross regulation of four GATA factors that control nitrogen catabolic gene expression in Saccharomyces cerevisiae. J Bacteriol. 1997;179(11):3416–29.
Mazurie A, Bottani S, Vergassola M. An evolutionary and functional assessment of regulatory network motifs. Genome Biol. 2005;6(4):R35. doi:10.1186/gb-2005-6-4-r35.
Lavoie H, Hogues H, Whiteway M. Rearrangements of the transcriptional regulatory networks of metabolic pathways in fungi. Curr Opin Microbiol. 2009;12(6):655–63. doi:10.1016/j.mib.2009.09.015.
Ratledge C. Fatty acid biosynthesis in microorganisms being used for Single Cell Oil production. Biochimie. 2004;86(11):807–15. doi:10.1016/j.biochi.2004.09.017.
Garay LA, Boundy-Mills KL, German JB. Accumulation of high-value lipids in single-cell microorganisms: a mechanistic approach and future perspectives. J Agric Food Chem. 2014;62(13):2709–27. doi:10.1021/jf4042134.
Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, et al. Genome evolution in yeasts. Nature. 2004;430(6995):35–44. doi:10.1038/nature02579.
Zhu Z, Zhang S, Liu H, Shen H, Lin X, Yang F, et al. A multi-omic map of the lipid-producing yeast Rhodosporidium toruloides. Nat Commun. 2012;3:1112. doi:10.1038/ncomms2112.
Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513(7519):517–22. doi:10.1038/nature13737.
Blinder D, Magasanik B. Recognition of nitrogen-responsive upstream activation sequences of Saccharomyces cerevisiae by the product of the GLN3 gene. J Bacteriol. 1995;177(14):4190–3.
Ravagnani A, Gorfinkiel L, Langdon T, Diallinas G, Adjadj E, Demais S, et al. Subtle hydrophobic interactions between the seventh residue of the zinc finger loop and the first base of an HGATAR sequence determine promoter-specific recognition by the Aspergillus nidulans GATA factor AreA. EMBO J. 1997;16(13):3974–86. doi:10.1093/emboj/16.13.3974.
Courchesne WE, Magasanik B. Regulation of nitrogen assimilation in Saccharomyces cerevisiae: roles of the URE2 and GLN3 genes. J Bacteriol. 1988;170(2):708–13.
Kudla B, Caddick MX, Langdon T, Martinez-Rossi NM, Bennett CF, Sibley S, et al. The regulatory gene areA mediating nitrogen metabolite repression in Aspergillus nidulans. Mutations affecting specificity of gene activation alter a loop residue of a putative zinc finger. EMBO J. 1990;9(5):1355–64.
Arndt K, Fink GR. GCN4 protein, a positive transcription factor in yeast, binds general control promoters at all 5' TGACTC 3' sequences. Proc Natl Acad Sci USA. 1986;83(22):8516–20.
Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, et al. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol. 2001;21(13):4347–68. doi:10.1128/MCB.21.13.4347-4368.2001.
MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi:10.1186/1471-2105-7-113.
Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, et al. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell. 2008;32(6):878–87. doi:10.1016/j.molcel.2008.11.020.
Meimoun A, Holtzman T, Weissman Z, McBride HJ, Stillman DJ, Fink GR, et al. Degradation of the transcription factor Gcn4 requires the kinase Pho85 and the SCF(CDC4) ubiquitin-ligase complex. Mol Biol Cell. 2000;11(3):915–27.
Liko D, Slattery MG, Heideman W. Stb3 binds to ribosomal RNA processing element motifs that control transcriptional responses to growth in Saccharomyces cerevisiae. J Biol Chem. 2007;282(36):26623–8. doi:10.1074/jbc.M704762200.
Liko D, Conway MK, Grunwald DS, Heideman W. Stb3 plays a role in the glucose-induced transition from quiescence to growth in Saccharomyces cerevisiae. Genetics. 2010;185(3):797–810. doi:10.1534/genetics.110.116665.
Huber A, French SL, Tekotte H, Yerlikaya S, Stahl M, Perepelkina MP, et al. Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. EMBO J. 2011;30(15):3052–64. doi:10.1038/emboj.2011.221.
Lundin M, Nehlin JO, Ronne H. Importance of a flanking AT-rich region in target site recognition by the GC box-binding zinc finger protein MIG1. Mol Cell Biol. 1994;14(3):1979–85.
Cubero B, Scazzocchio C. Two different, adjacent and divergent zinc finger binding sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J. 1994;13(2):407–15.
Wang ZP, Xu HM, Wang GY, Chi Z, Chi ZM. Disruption of the MIG1 gene enhances lipid biosynthesis in the oleaginous yeast Yarrowia lipolytica ACA-DC 50109. Biochim Biophys Acta. 2013;1831(4):675–82. doi:10.1016/j.bbalip.2012.12.010.
Ephrussi A, Church GM, Tonegawa S, Gilbert W. B lineage--specific interactions of an immunoglobulin enhancer with cellular factors in vivo. Science. 1985;227(4683):134–40.
Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009;37(Database issue):D77–82. doi:10.1093/nar/gkn660.
Ratledge C. Regulation of lipid accumulation in oleaginous micro-organisms. Biochem Soc Trans. 2002;30(Pt 6):1047–50. doi:10.1042/.
Pomraning KR, Smith KM, Freitag M. Bulk segregant analysis followed by high-throughput sequencing reveals the Neurospora cell cycle gene, ndc-1, to be allelic with the gene for ornithine decarboxylase, spe-1. Eukaryot Cell. 2011;10(6):724–33. doi:10.1128/EC.00016-11.
Serna L, Stadler D. Nuclear division cycle in germinating conidia of Neurospora crassa. J Bacteriol. 1978;136(1):341–51.
Evans CT, Ratledge C. Biochemical activities during lipid accumulation in Candida curvata. Lipids. 1983;18(9):630–5.
Ratledge C, Wynn JP. The biochemistry and molecular biology of lipid accumulation in oleaginous microorganisms. Adv Appl Microbiol. 2002;51:1–51.
Zhang H, Zhang L, Chen H, Chen YQ, Chen W, Song Y, et al. Enhanced lipid accumulation in the yeast Yarrowia lipolytica by over-expression of ATP:citrate lyase from Mus musculus. J Biotechnol. 2014;192(Pt A):78–84. doi:10.1016/j.jbiotec.2014.10.004.
Qiao K, Imam Abidi SH, Liu H, Zhang H, Chakraborty S, Watson N, et al. Engineering lipid overproduction in the oleaginous yeast Yarrowia lipolytica. Metab Eng. 2015;29:56–65. doi:10.1016/j.ymben.2015.02.005.
Tai M, Stephanopoulos G. Engineering the push and pull of lipid biosynthesis in oleaginous yeast Yarrowia lipolytica for biofuel production. Metab Eng. 2013;15:1–9. doi:10.1016/j.ymben.2012.08.007.
Wagner PD, Vu ND. Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J Biol Chem. 1995;270(37):21758–64.
Ramakrishna S, Benjamin WB. Rapid purification of enzymes of fatty acid biosynthesis from rat adipose tissue. Prep Biochem. 1983;13(5):475–88.
Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, et al. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods. 2013;10(7):676–82. doi:10.1038/nmeth.2519.
Shi S, Chen Y, Siewers V, Nielsen J. Improving production of malonyl coenzyme A-derived metabolites by abolishing Snf1-dependent regulation of Acc1. mBio. 2014;5(3):e01130–14. doi:10.1128/mBio.01130-14.
Choi JW, Da Silva NA. Improving polyketide and fatty acid synthesis by engineering of the yeast acetyl-CoA carboxylase. J Biotechnol. 2014;187:56–9. doi:10.1016/j.jbiotec.2014.07.430.
Wynn JP, Kendrick A, Hamid AA, Ratledge C. Malic enzyme: a lipogenic enzyme in fungi. Biochem Soc Trans. 1997;25(4):S669.
Shapiro BM, Stadtman ER. The regulation of glutamine synthesis in microorganisms. Annu Rev Microbiol. 1970;24:501–24. doi:10.1146/annurev.mi.24.100170.002441.
Hummelt G, Mora J. NADH-dependent glutamate synthase and nitrogen metabolism in Neurospora crassa. Biochem Biophys Res Commun. 1980;92(1):127–33.
Dunn-Coleman NS, Robey EA, Tomsett AB, Garrett RH. Glutamate synthase levels in Neurospora crassa mutants altered with respect to nitrogen metabolism. Mol Cell Biol. 1981;1(2):158–64.
Middelhoven WJ. The Pathway of Arginine Breakdown in Saccharomyces Cerevisiae. Biochim Biophys Acta. 1964;93:650–2.
Magasanik B et al. Regulation of Nitrogen Utilization. In: Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1992. p. 283–317.
Oberholzer VG, Briddon A. 3-Amino-2-piperidone in the urine of patients with hyperornithinemia. Clin Chim Acta. 1978;87(3):411–5.
Camacho JA, Obie C, Biery B, Goodman BK, Hu CA, Almashanu S, et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet. 1999;22(2):151–8. doi:10.1038/9658.
Pitkin J, Davis RH. The genetics of polyamine synthesis in Neurospora crassa. Arch Biochem Biophys. 1990;278(2):386–91.
Pitkin J, Perriere M, Kanehl A, Ristow JL, Davis RH. Polyamine metabolism and growth of neurospora strains lacking Cis-acting control sites in the ornithine decarboxylase gene. Arch Biochem Biophys. 1994;315(1):153–60. doi:10.1006/abbi.1994.1484.
Davis RH, Krasner GN, DiGangi JJ, Ristow JL. Distinct roles of putrescine and spermidine in the regulation of ornithine decarboxylase in Neurospora crassa. Proc Natl Acad Sci U S A. 1985;82(12):4105–9.
Sadowski I, Breitkreutz BJ, Stark C, Su TC, Dahabieh M, Raithatha S et al. The PhosphoGRID Saccharomyces cerevisiae protein phosphorylation site database: version 2.0 update. Database : the journal of biological databases and curation. 2013;2013:bat026. doi:10.1093/database/bat026.
Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32(4–5):577–95. doi:10.1007/s11064-006-9128-5.
Zhan XL, Hong Y, Zhu T, Mitchell AP, Deschenes RJ, Guan KL. Essential functions of protein tyrosine phosphatases PTP2 and PTP3 and RIM11 tyrosine phosphorylation in Saccharomyces cerevisiae meiosis and sporulation. Mol Biol Cell. 2000;11(2):663–76.
Kassis S, Melhuish T, Annan RS, Chen SL, Lee JC, Livi GP, et al. Saccharomyces cerevisiae Yak1p protein kinase autophosphorylates on tyrosine residues and phosphorylates myelin basic protein on a C-terminal serine residue. Biochem J. 2000;348(Pt 2):263–72.
Oliphant AR, Brandl CJ, Struhl K. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol Cell Biol. 1989;9(7):2944–9.
Hassanshahian M, Tebyanian H, Cappello S. Isolation and characterization of two crude oil-degrading yeast strains, Yarrowia lipolytica PG-20 and PG-32, from the Persian Gulf. Mar Pollut Bull. 2012;64(7):1386–91. doi:10.1016/j.marpolbul.2012.04.020.
Viljoen BC, Greyling T. Yeasts associated with Cheddar and Gouda making. Int J Food Microbiol. 1995;28(1):79–88.
Zinjarde SS, Pant AA. Hydrocarbon degraders from tropical marine environments. Mar Pollut Bull. 2002;44(2):118–21.
Barth G, Gaillardin C. Physiology and genetics of the dimorphic fungus Yarrowia lipolytica. FEMS Microbiol Rev. 1997;19(4):219–37.
Beopoulos A, Mrozova Z, Thevenieau F, Le Dall MT, Hapala I, Papanikolaou S, et al. Control of lipid accumulation in the yeast Yarrowia lipolytica. Appl Environ Microbiol. 2008;74(24):7779–89. doi:10.1128/AEM.01412-08.
Papanikolaou S, Chevalot I, Komaitis M, Marc I, Aggelis G. Single cell oil production by Yarrowia lipolytica growing on an industrial derivative of animal fat in batch cultures. Appl Microbiol Biot. 2002;58(3):308–12. doi:10.1007/s00253-001-0897-0.
Gietz RD and Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96.
Madzak C, Treton B, Blanchin-Roland S. Strong hybrid promoters and integrative expression/secretion vectors for quasi-constitutive expression of heterologous proteins in the yeast Yarrowia lipolytica. J Mol Microbiol Biotechnol. 2000;2(2):207–16.
Pomraning KR, Wei S, Karagiosis SA, Kim YM, Dohnalkova AC, Arey BW, et al. Comprehensive Metabolomic, Lipidomic and Microscopic Profiling of Yarrowia lipolytica during Lipid Accumulation Identifies Targets for Increased Lipogenesis. Plos One. 2015;10(4):e0123188. doi:10.1371/journal.pone.0123188.
Wolinski H, Kohlwein SD. Microscopic analysis of lipid droplet metabolism and dynamics in yeast. Methods Mol Biol. 2008;457:151–63.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5.
Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, et al. Improved structure, function and compatibility for Cell Profiler: modular high-throughput image analysis software. Bioinformatics. 2011;27(8):1179–80. doi:10.1093/bioinformatics/btr095.
Kim YM, Schmidt BJ, Kidwai AS, Jones MB, Kaiser BLD, Brewer HM, et al. Salmonella modulates metabolism during growth under conditions that induce expression of virulence genes. Mol Biosyst. 2013;9(6):1522–34. doi:10.1039/C3mb25598k.
Hiller K, Hangebrauk J, Jager C, Spura J, Schreiber K, Schomburg D. MetaboliteDetector: comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal Chem. 2009;81(9):3429–39. doi:10.1021/ac802689c.
Ficarro SB, Adelmant G, Tomar MN, Zhang Y, Cheng VJ, Marto JA. Magnetic bead processor for rapid evaluation and optimization of parameters for phosphopeptide enrichment. Anal Chem. 2009;81(11):4566–75. doi:10.1021/ac9004452.
Yang F, Waters KM, Webb-Robertson BJ, Sowa MB, von Neubeck C, Aldrich JT, et al. Quantitative phosphoproteomics identifies filaggrin and other targets of ionizing radiation in a human skin model. Exp Dermatol. 2012;21(5):352–7. doi:10.1111/j.1600-0625.2012.01470.x.
Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13(7):1690–704. doi:10.1074/mcp.M113.036392.
Kelly RT, Page JS, Luo Q, Moore RJ, Orton DJ, Tang K, et al. Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal Chem. 2006;78(22):7796–801. doi:10.1021/ac061133r.
Maiolica A, Borsotti D, Rappsilber J. Self-made frits for nanoscale columns in proteomics. Proteomics. 2005;5(15):3847–50. doi:10.1002/pmic.200402010.
Petyuk VA, Mayampurath AM, Monroe ME, Polpitiya AD, Purvine SO, Anderson GA, et al. DtaRefinery, a software tool for elimination of systematic errors from parent ion mass measurements in tandem mass spectra data sets. Mol Cell Proteomics. 2010;9(3):486–96. doi:10.1074/mcp.M900217-MCP200.
Kim S, Pevzner PA. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun. 2014;5:5277. doi:10.1038/ncomms6277.
Monroe ME, Shaw JL, Daly DS, Adkins JN, Smith RD. MASIC: a software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput Biol Chem. 2008;32(3):215–7. doi:10.1016/j.compbiolchem.2008.02.006.
Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi:10.1007/978-1-60761-444-9_5.
Polpitiya AD, Qian WJ, Jaitly N, Petyuk VA, Adkins JN, Camp 2nd DG, et al. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics. 2008;24(13):1556–8. doi:10.1093/bioinformatics/btn217.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6. doi:10.1093/bioinformatics/bti610.
Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40(Database issue):D700–5. doi:10.1093/nar/gkr1029.
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):D199–205. doi:10.1093/nar/gkt1076.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi:10.1186/gb-2009-10-3-r25.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi:10.1093/bioinformatics/btp352.
Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27(12):1653–9. doi:10.1093/bioinformatics/btr261.
Tanaka E, Bailey T, Grant CE, Noble WS, Keich U. Improved similarity scores for comparing motifs. Bioinformatics. 2011;27(12):1603–9. doi:10.1093/bioinformatics/btr257.
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy (DOE), Office of Science, Office of Biological and Environmental Research (OBER), Genomic Science program, under Award Number DE- SC0008744. Support was also provided by a William Wiley postdoctoral fellowship. Phosphoproteomics analyses were supported by the OBER-funded Pan-omics program at Pacific Northwest National Laboratory (PNNL). Part of this research was performed at the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the U.S. DOE OBER, and located at PNNL. PNNL is a multiprogram national laboratory operated by Battelle for the DOE under Contract DE-AC05-76RLO 1830.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KRP, YMK, TOM and SEB conceived and designed the experiments. KRP, YMK, ELB, CDN and RKC performed the experiments. KRP, YMK, ELB and SOP analyzed the data. KRP, YMK, ELB, CDN, RKC, SOP, DH, TOM and SEB contributed reagents/materials/analysis tools. KRP, YMK, CDN, TOM and SEB wrote the paper. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Supplementary Figures. Includes supplemental Figure S1, principal component analysis of intracellular and extracellular metabolite datasets; supplemental Figure S2, mean centered proteome quantification; supplemental Figure S3, global protein abundance level of phosphorylated and non-phosphorylated peptides identified after metal affinity chromatography. (PDF 334 kb)
Additional file 2:
Metabolite, global peptide, phosphopeptide, global proteome, phosphoproteome, and phosphosite quantification. Tables contain dry-weight normalized metabolite relative quantities, mean-centered and log2 transformed global peptide quantities with and without methanol/chloroform utilization for extraction, mean-centered and log2 transformed phosphopeptide quantities with and without methanol/chloroform utilization for extraction, mean-centered and log2 transformed global protein quantities with and without methanol/chloroform utilization for extraction, mean-centered and log2 transformed phosphoproteins quantities with and without methanol/chloroform utilization for extraction, and global protein abundance normalized log2 fold change quantities for phosphopeptides. (XLSX 10435 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pomraning, K.R., Kim, YM., Nicora, C.D. et al. Multi-omics analysis reveals regulators of the response to nitrogen limitation in Yarrowia lipolytica . BMC Genomics 17, 138 (2016). https://doi.org/10.1186/s12864-016-2471-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-2471-2