
Abstract
Background
Pisciricketssia salmonis is the causal agent of Salmon Rickettsial Syndrome (SRS), which affects salmon species and causes severe economic losses. Selective breeding for disease resistance represents one approach for controlling SRS in farmed Atlantic salmon. Knowledge concerning the architecture of the resistance trait is needed before deciding on the most appropriate approach to enhance artificial selection for P. salmonis resistance in Atlantic salmon. The purpose of the study was to dissect the genetic variation in the resistance to this pathogen in Atlantic salmon.
Methods
2,601 Atlantic salmon smolts were experimentally challenged against P. salmonis by means of intra-peritoneal injection. These smolts were the progeny of 40 sires and 118 dams from a Chilean breeding population. Mortalities were recorded daily and the experiment ended at day 40 post-inoculation. Fish were genotyped using a 50K Affymetrix® Axiom® myDesignTM Single Nucleotide Polymorphism (SNP) Genotyping Array. A Genome Wide Association Analysis was performed on data from the challenged fish. Linear regression and logistic regression models were tested.
Results
Genome Wide Association Analysis indicated that resistance to P. salmonis is a moderately polygenic trait. There were five SNPs in chromosomes Ssa01 and Ssa17 significantly associated with the traits analysed. The proportion of the phenotypic variance explained by each marker is small, ranging from 0.007 to 0.045. Candidate genes including interleukin receptors and fucosyltransferase have been found to be physically linked with these genetic markers and may play an important role in the differential immune response against this pathogen.
Conclusions
Due to the small amount of variance explained by each significant marker we conclude that genetic resistance to this pathogen can be more efficiently improved with the implementation of genetic evaluations incorporating genotype information from a dense SNP array.
Similar content being viewed by others
Background
The control of infectious diseases is a prime concern in salmon farming due to severe economic losses, reduced animal welfare and challenges to the sustainability of the industry [1, 2]. Piscirickettsia salmonis is an intracellular bacterium, causing Salmon Rickettsial Syndrome (SRS) [3] in salmon species and is considered one of the major pathogens of the salmon farming industry. During P. salmonis outbreaks fish exhibit pale gills, abdominal swelling and haemorrhages on the base of fins [4]. Infected fish often have skin lesions [5, 6]. Internally, serosanguinous ascites and swollen kidneys, livers and spleens are common. P. salmonis is horizontally transmitted through gills or skin [7]. This pathogen has evolved over time, being insidious with each outbreak and becoming refractory to treatments [4]. Antibiotic use may inhibit the growth of the pathogen, but treatments have been unsuccessful in stopping disease outbreaks [8]. Commercial vaccines have also not proven to be as efficient as needed [4, 9, 10]. In Chile alone, P. salmonis is responsible for the 74 % of the infectious-related deaths in Atlantic salmon [11] resulting in economic losses estimated at US $100 million [12].
Disease resistance has become an increasingly important trait of interest included in the genetic programs of aquatic species [1, 13, 14]. Selective breeding for disease resistance in aquaculture species commonly includes survival information from experimental infection of full-sibs of the breeding candidates [1, 13, 14]. The identification of the most resistant families is performed using the information obtained from challenge results, which can be used to identify the genomic regions (i.e. quantitative trait loci, QTL) involved in the genetic variation of these traits [1, 13, 14]. Selection for disease resistance traits is based on estimated breeding values (EBVs) calculated using phenotypic records and pedigree. Previous studies have estimated low to moderate heritabilities (proportion of the phenotypic variance that is accounted for by the additive genetic variance), ranging between 0.11 and 0.41 for resistance to P. salmonis [15, 16] in the same commercial population of Atlantic salmon used in the present study; indicating a potential for selective breeding for P. salmonis resistance in this population.
The availability of molecular tools like dense panels of SNPs (Single Nucleotide Polymorphisms) in domesticated species has led to the search for genomic regions and causal mutations underlying variation in complex traits through the use of linkage and association mapping [17–19]. High-density SNP panels have recently been developed for Atlantic salmon [20, 21]. In Genome-Wide Association Studies (GWAS or association mapping) a group of individuals are phenotyped and then genotyped using a large number of SNPs, in order to detect statistical association between a marker and a trait of interest [22]. Data from GWAS experiments are commonly analyzed testing one SNP at a time using linear models [17]. Two limitations of this approach are that it does not account for epistasis effects (interaction between markers) and adjustments are needed to control the false positive rate due to the multiple testing problem
The associated markers can be used to accelerate the genetic progress by means of Marker Assisted Selection (MAS), if the total variance explained by significant markers is high [23]. MAS can use a causative mutation that has been identified in a gene or regulatory region that has a major effect over the trait; or can use SNPs that are in Linkage Disequilibrium (LD) with a QTL to increase the response to selection [17]. Information from SNP panels can be used for the evaluation of breeding candidates through Genomic Selection (GS). In GS, sibs of the selection candidates are phenotyped and genotyped, while the selection candidates are only genotyped. The sibs of the selection candidates are utilized to predict the effect of each marker, and genomic EBVs (GEBVs) are calculated for the candidates using the information of all genotyped SNPs [24]. Knowledge concerning the architecture of the resistance trait is needed before deciding on the most appropriate approach to enhance artificial selection for P. salmonis resistance in Atlantic salmon, (i.e. MAS or GS).
Here we present the first Genome Wide Association Analysis performed on Atlantic salmon to dissect the genetic architecture of resistance to P. salmonis using a 50 K SNP array in 2601 fish.
Methods
Fish material and records
The origin of the population used in the present study is a Chilean salmon strain derived from the Irish strain Fanad-Mowi (originally from Norway) [25, 26]. Fish from the Fanad-Mowi strain were introduced for farming purposes to Chile during the 1990’s through commercial agreements. In 1997, a breeding program for Atlantic salmon was started by the company AquaChile (Puerto Montt, Chile), aimed at improving economically relevant traits including growth rate, carcass quality traits and late maturation. The base population of the breeding nucleus was established mainly with fish from the Fanad-Mowi strain. More specifically, the population used in the present study corresponds to the year-class 2010, which already has four generations of selective breeding in Chilean farming conditions. Two thousand six hundred one Atlantic salmon smolts weighing on average 274.8 g (SD = 90.6 g) were experimentally challenged against P. salmonis as described previously [15]. Briefly, infection was induced through intra-peritoneal (IP) injection with 0.2 ml of a LD50 inoculum of P. salmonis. Post-injection, infected fish were distributed in three different tanks with salt water (31 ppt). These smolts were the same individuals used in a previous study [15] and were the progeny of 40 sires and 118 dams from the breeding population of Salmones Chaicas, X Region, Chile. Each individual was identified using a PIT (Passive Integrated Transponder) tag, and a similar number of fish from each of the 118 full sib families were placed in each tank. The average number of fish per family was 22, ranging from 9 to 24. In order to determine the specific response to P. salmonis infection, the absence of other pathogens is required. Fish were found to be negative for Infectious Salmon Anemia virus, Infectious Pancreatic Necrosis virus and Renibacterium salmoninarum by RT-PCR and negative for Flavobacterium spp. by culture. Mortalities were recorded daily and all survivors were anesthetized and euthanized at day 40. Final body weight was recorded at the day of death for every dead fish or at the end of the challenge for survivors. Fin samples were taken from all fish and preserved in ethanol at −80 ° C until DNA extraction. The challenge test and sampling procedures were approved by The Comité de Bioética Animal, Facultad de Ciencias Veterinarias y Pecuarias, Universidad de Chile (Certificate N° 08–2015).
Genotyping
Genomic DNA was extracted from the fin clips from all challenged fish using a commercial kit (DNeasy Blood & Tissue Kit, Qiagen), following the manufacturer’s instructions. Fish were genotyped using a 50 K Affymetrix® Axiom® myDesignTM SNP Genotyping Array designed by AquaInnovo and the University of Chile.
50 K genotyping array
The markers included in this array were chosen from a 200 K SNP array previously developed and validated in six commercial populations (five with European Origin and one with North American origin) and two wild populations (one from Europe and another one from North America) [21]. Affymetrix software SNPolisher [27] was used to retain 159,099 Poly-high-resolution (SNPs that form distinct clusters of good resolution and have at least two occurrences of minor allele) [27] and No- minor-homozygote (SNPs forming two distinct clusters and with no occurrences of minor homozygous genotypes) [27] SNPs. SNPs were further filtered based on Mendelian error in genotypes of 14 trios (sire, dam and offspring) and one sire-offspring pair from one commercial population. 137,712 SNPs with less than 1 Mendelian error were selected. SNPs were further selected on the basis of minor allele frequency (MAF) in five commercial populations of European origin. We gave preference to SNPs with higher MAF in the commercial populations of European origin because the target populations which will be used for further implementation of genomic selection and Genome Wide Association Studies have European origin. For this purpose four related European populations were grouped together as Group A and a fifth population was called Group B. SNPs were selected if they met one of the following two criteria: i) MAF > =0.05 in Group A and MAF > =0.1 in Group B or ii) MAF > =0.05 in Group B and MAF > =0.1 in Group A. From 112,241 SNPs that passed the above criteria, 55,591 SNPs were selected so that they are as evenly distributed along the genome as possible. This was done by selecting SNPs from windows of equal size across various linkage groups of the genome. When selecting SNPs from windows, higher preference was given to SNPs with lower Mendelian error. Pearson correlation was used to estimate LD among the SNP pairs belonging to same linkage group. Only one member of each SNP pair with R-square > =0.99 was retained. For each pair higher preference was given to SNPs near the end of the linkage group or having lower Mendelian error. From the remaining 53,998 candidates, 50,000 SNPs were selected by removing a SNP that had a distance of less than 10Kb from its neighbor, so markers will be distributed equally across the genome.
Genotypes were obtained for our samples following the Best Practice Analysis Workflow from Affymetrix [28] and selecting the Poly-high-resolution and the No-minor-homozygote SNPs from SNPolisher [27]. Quality control was performed on the genotypes to filter based on Hardy Weinberg equilibrium (p < 1 × 10−10), Minor Allele Frequency (>0.001) and call rate for SNPs and samples (>0.95). For the quality control the R statistical software and the GENAbel library [29, 30] were used.
Trait definition and genome wide association analysis
The resistance phenotypes were defined as the time to death (TD), measured in days with values ranging from 1 to 40, depending on the day the fish died; and as a binary survival (BS), scored as 1 if the fish died during the 40-day challenge and 0 if the fish survived until the end of the trial. Fish weight at the end of the experiment and tank designation were also recorded to be included as covariate and factor within the model, respectively.
Linear regression and logistic regression were used to identify association between SNPs and resistance to P. salmonis, for the TD and BS traits, respectively, using the mlreg function implemented in GenABEL [29, 30]. Logistic regression was used to account for the binary nature of the BS trait. A genomic kinship matrix was calculated from SNP data using the gkin function. The inbreeding coefficient (F) was estimated using PLINK [31] to determine the extent of inbreeding in our samples. Heritability values were also estimated from genotype data, using a linear mixed model implemented on polygenic function [29, 30, 32–34]. The general formula used for the linear regression model is as follows:
Where Y i is the phenotypic record (TD); β 0 the intercept; β 1 the effect of each SNP; β 2 is the effect of each tank (Tk); β 3 is the effect of the weight (W) and e i the random residual. The general formula for the logistic regression model for BS is:
Where π i is the probability of the random variable being one, β 0 the intercept; β 1 the effect of each SNP; β 2 is the effect of each tank (Tk) and β 3 is the effect of the weight (W).
The -log10 (p-value) for each SNP across the genome was plotted to summarize the GWAS results. The significance threshold was determined using False Discovery Rate correction [35]. This correction was performed as follows: (1) we ordered p values p 1 ≤ p 2 ≤ … ≤ p k where k is the number of markers tested. (2) Starting with the largest p value, we found the first individual p value (p i ) that satisfied: p i ≤ i/k *0.05, where i = ith observation. (3) The p i that satisfied the condition above became the critical value for the experiment.
The proportion of the heritability explained by each significant marker was obtained by comparing the heritability estimated with the polygenic function and the heritability estimated with the inclusion of the significant SNP genotype as factor [36]. The proportion of the phenotypic variance explained by each marker for both traits was estimated multiplying the heritability by the proportion of heritability explained by each marker. The approximate effect of each marker was obtained from the linear regression analysis performed in GenABEL [29, 30]. The level of linkage disequilibrium between significant markers was determined by calculating r 2 statistics using the r2fast function implemented in GenABEL [29, 30, 37]. For r 2 calculations we used all genotyped samples which passed the QC.
Results and discussion
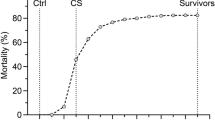
The average mortality per family at the end of the test was 38.4 % (SD = 17.1, minimum = 8.3 %, maximum = 73.7 %) (Fig. 1). We observed considerable variation in the phenotypic survival across families: some families showed more resistance with a low mortality (as low as 8 %), while other families had high mortality (up to 73 %).

Cumulative mortality by family after Piscirickettsia salmonis experimental infection. The average mortality after 40 days of experimental infection against P. salmonis is plotted for each of the 118 full-sib families of Atlantic salmon included in the present study
Final body weight was recorded as a covariate in the statistical models as previously described [15, 16]. Average final body weight was 326.7 g (SD = 137 g; CV = 42 %). Fish grew during the trial, and the increased SD and CV reflects a higher level of dispersion of final body weight compared with body weight measured at the beginning of the trial (SD = 90.6; CV = 33 %). This is likely due to varying days of death among animals, as some fish died early in the trial (having less time to grow) and some survived the entire trial period.
Forty-eight thousand eight hundred eighty-six SNPs and 2391 samples passed all quality control criteria. Average F was 0.017 (SD = 0.008). Thus, the inbreeding level in this population is considered low and will have a small impact on the power of the association analysis. Furthermore, we accounted for population structure within the sample aimed at decreasing the chances of occurrence of type I and type II errors in the association analysis. The heritability estimated using the genomic information was 0.19 for TD and 0.20 for BS (p values < 0.00001) [30], which is consistent with the results obtained in previously reported estimations using pedigree information [15, 16]. In these previous studies when the binary survival (0/1) was analysed using a threshold model, a heritability of 0.24 ± 0.04 was calculated [15]. When a linear model was used to analyse the day of death, a heritability of 0.18 ± 0.03 was estimated [15, 16].
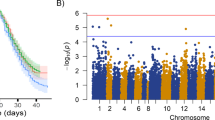
The p values obtained from the GWAS indicate evidence of significant associations on Atlantic salmon chromosomes Ssa01 and Ssa17 (Figs. 2 and 3).

GWAS for Piscirickettsia salmonis resistance measured as time to death: Red line indicates False Discovery Rate significance threshold

GWAS for Piscirickettsia salmonis resistance measured as binary survival: Red line indicates False Discovery Rate significance threshold
Results were consistent between models after multiple testing corrections (Table 1). The linear model performed slightly better in terms of detecting significant markers associated with TD than the logistic model for BS. A linear model using TD appears to be a better fit than a logistic model using BS; which leads to more efficient identification of important SNPs. Similarly, a slightly higher accuracy of EBVs was obtained when fitting a linear model to analyse day of death when compared to a threshold model to analyse overall test period survival (binary) for P. salmonis resistance in Atlantic salmon [15].
The logistic regression model revealed two significant SNP associated with BS located in one genomic region of chromosome Ssa01 (AQI_UCh-93346943 and AQI_Uch-93451692). In the linear regression model, five significant SNPs associated with TD were identified, the same two identified by the logistic regression and three additional SNPs. One marker is located in the same genomic region of the afore-mentioned SNPs (AQI_UCh-93323028) and two are located in different regions of chromosomes Ssa01 and Ssa17 (AQI_UCh-93268476 and AQI_UCh-93384759). Two of these markers are significant for both trait definitions and models used. Markers that were significant only for TD were suggestive for BS. The resistance alleles are the minor alleles, with frequencies ranging between 0.24 and 0.31 (Table 2), indicating that there is still potential to increase their frequencies in the breeding population.
It is interesting to note that three of the identified markers are located in a ~660Kb region (contig Jcf1000860129) in chromosome Ssa01. This region contains several predicted genes [38] and some of them could play an important role in the resistance against P. salmonis (Table 3). AQI_UCh-93451692 is located in an intronic region of Alpha-(1,3)-Fucosyltransferase, which is involved in neutrophil and T cell recruitment and lymphocyte trafficking [39–41]. The SNP marker located on chromosome Ssa17 is close to a Metalloprotease, described as up-regulated in Atlantic salmon infected with P. salmonis [42]. SNP AQI_UCh-93384759 is located in an intronic region of a predicted gene corresponding to Tenascin, which has been described to generate an effective immune response to bacterial lipopolysaccharide [43]. Other genes like interleukin receptors are located in the genomic regions identified in the GWAS and could be involved in differential susceptibility to P. salmonis infection. Some of these genes have been previously identified in gene expression assays of diseased fish [42], and thus these results support the evidence that they may be involved in Atlantic salmon’s response to P. salmonis. Further assays like differential expression or sequencing of selected genes and regions in susceptible and resistance fish could lead to a better understanding of the biological response to this pathogen.
The proportion of the heritability explained by each identified marker ranged between 0.09 and 0.18 for TD and 0.03 and 0.24 for BS (Table 4). The proportion of phenotypic variance explained by each marker was small, ranging from 0.007 to 0.045 for BS and from 0.017 to 0.035 for TD.
Markers located in the same contig (Jcf1000860129) exhibit high level of LD, ranging between 0.53 and 0.84 (Table 5). The LD between markers in this contig and the marker located in the other contig (Ccf000000073) in chromosome Ssa01 is also relatively high (0.37-0.65), which is consistent with the physical proximity of both contigs. The marker located on chromosome Ssa17 showed low LD with other markers. The closer the markers are located, the higher the value of r2 is to be expected [17]. Due to the high amount of LD present between the markers identified, their effect is likely to be shared.
Many genes usually affect quantitative traits and consequently the benefit of conducting MAS depends on the proportion of the variance explained by each QTL [44]. We found five markers significantly associated with resistance to P. salmonis, but the proportion of phenotypic variance and heritability explained are relatively small. In a typical GWAS analysis, markers need to surpass a specific threshold to be considered significant. In the case of traits that are affected by many genes of small effect, their effect can be underestimated [45]. On the other hand, polygenic approaches like genomic selection take into account the nature of such traits and include the information of all marker genotypes. Although there is no major effect QTL, the marker information can still be used for breeding programs [46] to predict the GEBVs of selection candidates [44] through GS. GS takes into account all markers to estimate the GEBVs, without the need to surpass a significance threshold for association with a particular trait [44]. The sib-test design can be used for MAS [47] or GS, where the association between markers and phenotypes is estimated in the sibs of the candidates, and the candidates are selected on breeding values that result from summing the estimates of the effects of their marker alleles [48]. Different approaches can be used to determine the marker effects to estimate the breeding values of the candidates. The GS methods facilitates computation of individual breeding values for all genotyped animals and do not require any prior knowledge of the underlying QTL [49]. Taking the relatively small proportion of the genetic variance explained by the significant markers into account, the results presented in this study suggest that a GS approach will be the most appropriate way of incorporating molecular information to assist artificial selection for P. salmonis resistance in Atlantic salmon.
Conclusions
This is the first study designed to dissect the genetic basis for the resistance for P. salmonis using a dense SNP array in Atlantic salmon. Resistance to P. salmonis can be described as a moderate polygenic trait, as there are likely several loci involved but each with a small effect. Five SNPs could be identified as significantly associated with P. salmonis resistance. These markers explained a relatively small proportion of the variance for the trait.
Availability of supporting data
SNP data from the 50 K Array used in this study have been deposited on SalmonDB database [38] [http://salmondb.cmm.uchile.cl/download/Array-Aquainnovo-UChile/]. Sequences flanking significant markers can be found as Additional file 1: Table S1.
Abbreviations
- GWAS:
-
Genome Wide Association Study
- LD:
-
Linkage disequilibrium
- MAS:
-
Marker assisted selection
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
- SRS:
-
Salmon Rickettsial syndrome
- GS:
-
Genomic selection
References
Yáñez JM, Houston RD, Newman S. Genetics and genomics of disease resistance in salmonid species. Front Genet. 2014;5:1–13.
Bishop SC, Woolliams JA. Genomics and disease resistance studies in livestock. Livest Sci. 2014;166:190–8.
Fryer JL, Hedrick RP. Piscirickettsia salmonis: a Gram-negative intracellular bacterial pathogen of fish. J Fish Dis. 2003;26:251–62.
Rozas M, Enriquez R. Piscirickettsiosis and Piscirickettsia salmonis in fish: a review. J Fish Dis. 2014;37:163–88.
Cvitanich J, Garate O, Smith CE. Etiological agent in a Chilean coho disease isolated and confirmed by Koch’s postulates. FHS/AFS Newsletter. 1990;18:1–2.
Fryer JL, Lannan C, Garces L, Larenas J, Smith P. Isolation of a rickettsiales-like organism from diseased coho salmon (Oncorhynchus kisutch) in Chile. Fish Pathol. 1990;25:107–14.
Smith PA, Pizarro P, Ojeda P, Contreras J, Oyanedel S, Larenas J. Routes of entry of Piscirickettsia salmonis in rainbow trout Oncorhynchus mykiss. Dis Aquat Organ. 1999;37:165–72.
Cabello FC. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol. 2006;8:1137–44.
Leal J, Woywood D. Piscirickettsiosis en Chile: avances y perspectivas para su control. Salmociencia. 2007;2:34–42.
Marshall S, Conejeros P, Zahr M, Olivares J, Gomez F, Cataldo P, et al. Immunological characterization of a bacterial protein isolated from salmonid fish naturally infected with Piscirickettsia salmonis. Vaccine. 2007;25:2095–102.
Sernapesca. Informe sanitario de salmonicultura en centros marinos 2014. http://www.sernapesca.cl/index.php?option=com_remository&Itemid=246&func=fileinfo&id=11083. Accessed date 15/5/2015.
Cabezas M. Fármacos naturales en el cultivo de Salmonídeos: una alternativa en el control de enfermedades. Salmociencia. 2006;1:27–33.
Ødegård J, Baranski M, Gjerde B, Gjedrem T. Methodology for genetic evaluation of disease resistance in aquaculture species: challenges and future prospects. Aquac Res. 2011;42:103–14.
Yáñez JM, Martínez V. Genetic factors involved in resistance to infectious diseases in salmonids and their application in breeding programmes. Arch Med Vet. 2010;42:1–13.
Yáñez JM, Bangera R, Lhorente JP, Oyarzún M, Neira R. Quantitative genetic variation of resistance against Piscirickettsia salmonis in Atlantic salmon (Salmo salar). Aquaculture. 2013;414–415:155–9.
Yáñez JM, Lhorente JP, Bassini LN, Oyarzún M, Neira R, Newman S. Genetic co-variation between resistance against both Caligus rogercresseyi and Piscirickettsia salmonis, and body weight in Atlantic salmon (Salmo salar). Aquaculture. 2014;433:295–8.
Goddard M, Hayes M. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet. 2009;10:381–91.
Andersson L, Georges M. Domestic-animal genomics: deciphering the genetics of complex traits. Nat Rev Genet. 2004;5:202–12.
Dekkers JCM. Application of genomic tools to animal breeding. Curr Genomics. 2012;13:207–12.
Houston R, Taggart J, Cézard T, Bekaert M, Lowe N, Downing A, et al. Development and validation of a high density SNP genotyping array for Atlantic salmon (Salmo salar). BMC Genomics. 2014;15:9. doi:10.1186/1471-2164-15-90.
Yáñez JM, Naswa S, López ME, Bassini L, Cabrejos ME, Gilbey J, Bernatchez L, Norris A, Soto C, Eisenhart J, Simpson B, Neira R, Lhorente JP, Schnable P, Newman S, Mileham A, Deeb N. Development of a 200 K SNP array for Atlantic Salmon: exploiting across continents genetic variation. In: Proceedings of the 10th World Congress on Genetics Applied to Livestock Production. Vancouver; 2014.
Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. doi:10.1371/journal.pcbi.1002822.
Hayes B, Goddard M. Genome-wide association and genomic selection in animal breeding. Genome. 2010;53:876–83.
Villanueva B, Fernández J, García-Cortés LA, Varona L, Daetwyler HD, Toro MA. Accuracy of genome-wide evaluation for disease resistance in aquaculture breeding programs. J Anim Sci. 2011;89:3433–42.
Solar II. Use and exchange of salmonid genetic resources relevant for food and aquaculture. Rev Aquac. 2009;1:174–96.
Norris A. Microsatellite genetic variation between and within farmed and wild Atlantic salmon (Salmo salar) populations. Aquaculture. 1999;180:247–64.
SNPolisher User Guide (Version 1.4) Affymetrix, Inc. October 14, 2013.
Axiom® Genotyping Solution Data Analysis User Guide, Affymetrix, Inc. http://www.affymetrix.com/support/downloads/manuals/axiom_genotyping_solution_analysis_guide.pdf . Accessed date 15/3/2015.
Aulchenko YS, Ripke S, Isaacs A, van Dujin CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;10:1294–6.
GenAbel Project Developers. GenABEL Tutorial. 2015. DOI 10.5281/zenodo.19738
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–75.
Teyssèdre S, Elsen JM, Ricard A. Statistical distributions of test statistics used for quantitative trait association mapping in structured populations. Genet Sel Evol. 2012;44:32.
Wei W, Hemani G, Hicks AA, Vitart V, Cabrera-Cardenas C, Navarro P, et al. Characterisation of genome-wide association epistasis signals for serum uric acid in human population isolates. PLoS ONE. 2011;6:e23836. doi:10.1371/journal.pone.0023836.
Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;1:577–85.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300.
Korte A, Farlow A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods. 2013;9:29.
Hao K, Di X, Cawley S. LdCompare: rapid computation of single- and multiple-marker r2 and genetic coverage. Bioinformatics. 2006;23:252–4.
Di Genova A, Aravena A, Zapata L, González M, Maass A, Iturra P. SalmonDB: a bioinformatics resource for Salmo salar and Oncorhynchus mykiss. Database. 2011. doi:10.1093/database/bar050.
Maly P, Thall AD, Petryniak B, Rogers CE, Smith PL, Marks RM, et al. The (1,3) fucosyltransferase FucT-VII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. 1996;86:643–53.
Smith PL, Gersten KM, Petryniak B, Kelly RJ, Rogers C, Natsuka Y, et al. Expression of the a(1,3)fucosyltransferase Fuc-TVII in lymphoid aggregate high endothelial venules correlates with expression of L-selectin ligands. J Biol Chem. 1996;271:8250–9.
Parisi MG, Benenati G, Cammarata M. Sea bass Dicentrarchus labrax (L.) bacterial infection and confinement stress acts on F-type lectin (DlFBL) serum modulation. J Fish Dis. 2014. doi:10.1111/jfd.12309.
Rise ML, Jones SRM, Brown GD, von Schalburg KR, Davidson WS, Koop BF. Microarray analyses identify molecular biomarkers of Atlantic salmon macrophage and hematopoietic kidney response to Piscirickettsia salmonis infection. Physiol Genomics. 2004;20:21–35.
Piccinini AM, Midwood KS. Endogenous control of immunity against infection: Tenascin-C regulates TLR4-Mediated inflammation via MicroRNA-155. Cell Reports. 2012;2:914–26.
Meuwissen THE, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–29.
Stringer S, Wray NR, Kahn RS, Derks EM. Underestimated effect size in GWAS: fundamental limitations of single SNP analysis for dichotomous phenotypes. Plos One. 2011;6:e27964.
Goddard M, Hayes BJ. Genomic selection. J Anim Breed Genet. 2007;124:323–30.
Habier D, Fernando RL, Dekkers JCM. The impact of genetic relationship information on genome-assisted breeding values. Genetics. 2007;177:2389–97.
Sonesson AK, Meuwissen THE. Testing strategies for genomic selection in aquaculture breeding programs. Genet Sel Evol. 2009;41:37.
Ødegård J, Moen T, Santi N, Korsvoll SA, Kjøglum S, Meuwissen THE. Genomic prediction in an admixed population of Atlantic salmon (Salmo salar). Front Genetics. 2014;5:1.
Ghilardi N, Hongo J, Yi S, Gurney A, de Sauvage FJ. A novel type I cytokine receptor is expressed on monocytes, signals proliferation, and activates STAT-3 and STAT-5. J Biol Chem. 2002;277:16831–6.
Dreuw A, Radtke S, Pflanz S, Lippok B, Heinrich PC, Hermanns HM. Characterization of the signaling capacities of the novel gp130-like cytokine receptor. J Biol Chem. 2004;279:36112–20.
NCBI. Gene ID: 3572. [http://www.ncbi.nlm.nih.gov/gene/?term=3572#summary]. Accessed date 15/6/2015.
Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78:2791–808.
Katzenback BA, Katakura F, Belosevic M: Regulation of Teleost Macrophage and Neutrophil Cell Development by Growth Factors and Transcription Factors. http://dx.doi.org/10.5772/53589. Accessed date 15/6/2015.
Morrison D. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012;4:a011254.
Baud V, Liu Z-G, Bennet B, Suzuki N, Xia Y, Karin M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1998;13:1297–308.
Blackwell K, Zhang L, Thomas GS, Sun S, Nakano H, Habelhah H. TRAF2 phosphorylation modulates tumor necrosis factor alpha-induced gene expression and cell resistance to apoptosis. Mol Cell Biol. 2008;29:303–14.
Acknowledgements
KC & MEL want to acknowledge the National Commission of Scientific and Technologic Research (CONICYT) for the funding through the National PhD funding program. KC wants to acknowledge the Government of Canada for the funding through the Canada-Chile Leadership Exchange Scholarship. LB wish to thank CAPES scholarship for PhD studies, Government of Brazil. This project was funded by the project CORFO Innova-Chile (11IEI-12843) and U-Inicia Grant, from the Vicerrectoría de Investigación y Desarrollo, Universidad de Chile. This work has been conceived on the frame of the grant FONDEF NEWTON-PICARTE (IT14I10100), funded by CONICYT (Government of Chile) and the Newton Fund - The British Council (Government of United Kingdom). The authors thank René Figueroa for his help in developing informatics tools.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KC performed GWAS analysis, discussion and wrote the initial version of the manuscript. JPL contributed with study design. MEL contributed in the data analysis. LB managed samples and performed DNA extractions. SN and ND contributed with the filtering of the 200 K Array. ADG and AM contributed with prediction and functional annotation of S. salar genes, markers anchoring and variant effects. WD contributed with discussion. JMY conceived and designed the study, supervised the work of KC and contributed to the analysis, discussion and writing. All authors have reviewed and approved the manuscript.
Additional file
Additional file 1: Table S1.
Sequences flanking significant markers identified in GWAS. (DOCX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Correa, K., Lhorente, J.P., López, M.E. et al. Genome-wide association analysis reveals loci associated with resistance against Piscirickettsia salmonis in two Atlantic salmon (Salmo salar L.) chromosomes. BMC Genomics 16, 854 (2015). https://doi.org/10.1186/s12864-015-2038-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-2038-7