
Abstract
Background
The Mediterranean mussel (Mytilus galloprovincialis) is a cosmopolitan, cultured bivalve with worldwide commercial and ecological importance. However, there is a qualitative and quantitative lack of knowledge of the molecular mechanisms involved in the physiology and immune response of this mollusc. In order to start filling this gap, we have studied the transcriptome of mantle, muscle and gills from naïve Mediterranean mussels and hemocytes exposed to distinct stimuli.
Results
A total of 393,316 million raw RNA-Seq reads were obtained and assembled into 151,320 non-redundant transcripts with an average length of 570 bp. Only 55 % of the transcripts were shared across all tissues. Hemocyte and gill transcriptomes shared 60 % of the transcripts while mantle and muscle transcriptomes were most similar, with 77 % shared transcripts. Stimulated hemocytes showed abundant defense and immune-related proteins, in particular, an extremely high amount of antimicrobial peptides. Gills expressed many transcripts assigned to both structure and recognition of non-self patterns, while in mantle many transcripts were related to reproduction and shell formation. Moreover, this tissue presented additional and interesting hematopoietic, antifungal and sensorial functions. Finally, muscle expressed many myofibril and calcium-related proteins and was found to be unexpectedly associated with defense functions. In addition, many metabolic routes related to cancer were represented.
Conclusions
Our analyses indicate that whereas the transcriptomes of these four tissues have characteristic expression profiles in agreement with their biological structures and expected functions, tissue-specific transcriptomes reveal a complex and specialized functions.
Similar content being viewed by others


Background
The Mediterranean mussel (Mytilus galloprovincialis) is a cultured bivalve species with an important commercial and ecological value worldwide [1, 2]. In contrast to other cultured bivalves such as clams and oysters, where different pathogens may result in massive mortalities and therefore, substantial economic losses [3–5], M. galloprovincialis displays an extraordinary resistance to a variety of pathogens [6]. Although molluscs lack a specific immune response, their innate response, which involves circulating hemocytes and a large variety of molecular effectors, constitutes an efficient defense mechanism [7–9]. While a wide range of molecules involved in the bivalve immune system have been described [10–13], particularly for mussels and oysters [14–18], the information is very limited compared, for example, to vertebrates.
Unfortunately, most bivalve genomic resources are not annotated or well described, with the exception of the Pacific oyster, Crassostrea gigas, whose genome has been recently published [19] or the pearl oyster, Pinctada fucata, in which genome annotation is still at the draft level [20]. Several bivalve transcriptomes are publicly available for M. galloprovincialis [21, 22], Bathymodiolus azoricus [23], Patinopecten yessoensis [24], Ruditapes philippinarum [25, 26] Corbicula fluminea [27] and Crassotrea gigas [19, 28, 29]. There are also 666 entries from the class Bivalvia deposited in the NCBI Short Read Archive (SRA) (25/03/2015). The number of available sequences for M. galloprovincialis is constantly increasing [30–33]. As an example, 23 M. galloprovincialis entries are publicly available in the SRA database, including whole-body, digestive gland and hemocytes transcriptomes, a M. galloprovincialis EST database called Mytibase [16].
In this study we show the results of the first comparative RNA-Seq analysis of gene expression in different M. galloprovincialis tissues, including gills, muscle, mantle and hemocytes. The raw data are accessible from the NCBI Short Read Archive (SRA: SRP033481). Additional files 1, 2, 3, 4 and 5 include all the transcripts obtained, together with their expression values, annotation and sequences in FASTA format.
Results and discussion
Sequence analysis and functional annotation
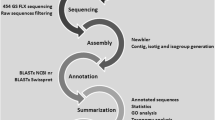
Mussel samples were processed as depicted in Fig. 1. The 7 cDNA libraries obtained (2 from stimulated hemocytes, 2 from mantle, 2 from muscle and 1 from gills) were sequenced on the Illumina HiSeq™ 2000 platform.

Flow chart summarizing the work tasks and the data processing pipeline
The sequencing and assembly statistics are summarized in Table 1. Briefly, we obtained a total of 393.3 million raw reads (with an average of 56.2 million reads per run). Of these, more than 95 % passed the quality standards and were subjected to further analyses. The filtered high-quality reads were assembled in a three-step approach with the Trinity software [34] into 1,242,475 contigs, which after clustering resulted in 479,806 unigenes. Until this point, the assembly protocol was individual for each sample, but the third and last clustering step was performed in common for the 7 samples. A total of 151,320 non-redundant unigenes (“transcripts” hereafter) were obtained, which could represent the M. galloprovincialis global transcriptome for these 4 tissues.
The length of the transcripts ranged from 200 to 17,690 bp, with an average length of 570 bp, a similar size to that obtained with Roche 454 technology in other bivalves, e.g., 582 bp in the Manila clam [26]. Furthermore, when we compared our results with SOLiD and Illumina RNA-Seq analyses conducted in oyster, we obtained larger transcripts than those reported by Gavery and Roberts [28] or Zhao et al. [29], averaging 276 bp (554 using GigasDatabase v8 as a reference for mapping) and 322 bp, respectively.
The NCBI’s nucleotide and non-redundant, SwissProt, KEGG [35] and COG [36] databases were chosen to annotate the transcripts. The percentage of transcripts annotated with an e-value threshold of 1x10e−5 was 33.7 %. The annotations and expression values are included in Additional file 1. Our annotation percentage was similar to previous transcriptome studies conducted in bivalves using 454 technology [26], with 45 % of hemocyte transcripts being annotated with an e-value threshold of 1x10e−3. Similar approaches applied in oyster using the SOLiD [28] or Illumina [29] sequencing platform resulted in an annotation success of 41 % or 16 %, respectively, while in the M. galloprovincialis digestive gland transcriptome, about half (48.1 %) of the transcripts were successfully annotated [22].
The coverage of the whole transcript for each specific tissue sample (calculated as the percentage of base pairs in a transcript covered by reads of a specific sample per transcript length) is summarized in Table 2. The mean coverage was 69.87 %, with an average of 256.55 reads being mapped to each transcript, which is lower than the values reported in the oyster gills study by Gavery and Roberts [28], in which 454 reads per transcript were mapped (376 using GigasDatabase v8 as a reference). Hemocytes were the sample with the lowest coverage, but also showed the highest number of mapped reads per transcript. If this fact is related with the specific immune function of hemocytes, as was previously reported in other bivalves [26], is something that deserves further investigation.
We sequenced 2 samples of hemocytes, mantle and muscle, in order to understand whether a second biological replicate would effectively increase the sequencing depth (Table 2). Although this second replicate resulted in an average increase of 9.7 % transcripts for each tissue, it did not result in a significantly higher number of transcripts when all tissues were considered (Additional file 2). However, the use of pools of individuals, different tissues and biological replicates increased the reliability and robustness of the results, as previously reported [37]. For example, we achieved a transcriptome completeness of 88.71 % or 95.16 % (considering whole or partial sequence comparisons, respectively) using the CEGMA package (http://korflab.ucdavis.edu/datasets/cegma/).
Qualitative description of the M. galloprovincialis transcriptome
Using KEGG, we annotated 31,144 transcripts (20.6 %). This annotation served as a basis for analyzing not only the role of individual transcripts, but also the interaction with other genes. Figure 2 provides a representation of the global functionality of the transcripts and summarizes the 256 molecular pathways found in the transcriptomes. It was interesting that a high number of these transcripts had annotations related to the immune system, signal transduction and infectious diseases (bacterial, viral and parasitic). A possible explanation for this could be that, as a filter feeding animal, M. galloprovincialis is permanently in contact with microorganisms and with toxic/pollutant substances in their marine environment [38], and has adapted to become very resistant to these impacts [39, 40]. Another group of disease-associated pathways were those related to cancer, which ranked second among the most represented pathways, like in other studies in oysters [29]. Interestingly, although mussels and oysters shared less than 10 % of proteins with a sequence identity over 80 % (Fig. 3), the response to infectious disease and cancer were highly represented in both transcriptomes [29]. Although these cancer-related genes may have other functions, this subject obviously requires further attention specially taking into account that some bivalves are affected by a disease of the circulatory system closely resembling leukemia [41, 42].

Summary of the KEGG reference pathway results. Bars represent the percentage of the total KEGG-annotated transcripts in the transcriptomes

Comparison of the translated Mytilus galloprovincialis sequences with the Crassostrea gigas proteome downloaded from http://www.oysterdb.com/FrontDownloadAction.do?method=download
The information about the molecules that were present and absent in each pathway is available in Additional file 3. The specialization and diversification observed throughout the phylogeny of the immune system [43] suggests that the absence of some key molecules in the pathways can be an artifact. It is possible that they were not annotated or that other molecules could play a similar function.
A comparative analysis among the M. galloprovincialis transcriptomes was conducted to identify transcripts conserved in the 4 tissues and those unique to each tissue (Fig. 4). Among the total 151,320 transcripts, 54.57 % were shared by all the tissues. The most related pair of tissues, muscle and mantle, had 76.63 % transcripts in common, whereas hemocytes and gills shared only 59.56 % of the transcripts.

Venn diagram showing a comparison of the R. philippinarum tissue transcriptomes: hemocytes, mantle, muscle and gills. Numbers refer to the transcripts that belong to each group
The tissue with the fewest private transcripts was muscle, with only 769 unique transcripts (0.51 %), while gills presented the highest number of non-shared transcripts, 9178 (6.07 %). This might be due to the filter feeding behavior of bivalves, where the gills are in constant contact with the surrounding habitat and exposed to more stress factors such as microorganisms, pollutants, pH or salinity changes.
Tissue-specific transcriptome portions are presented in Table 3. A high number of lectins, C1q domain-containing proteins and fibrinogen-related proteins were detected in gills. Their direct contact with the environment could explain the high presence of these putative recognition and immune-triggering molecules [11, 12, 44]. Hemocytes, as key players in the invertebrate immune response [45], showed a high percentage of antimicrobial peptides (AMPs), such as defensin, mytilins, and myticins, as well as other immune-related proteins, such as FREPs, serine protease inhibitors, complement component C4, HSP90 and C1q domain-containing proteins. Hemocyte hematopoiesis is a poorly described process in bivalves, but some studies suggest that heart and mantle could be possible hematopoietic tissues [46, 47]. In our case, peroxidasin transcripts, an early hematopoietic differentiation marker in Drosophila [48], were found only in the mantle, reinforcing the hypothesis that mantle could be the main hematopoietic tissue of bivalves.
The lowest number of tissue-specific transcripts was observed in muscle, which could be due to its limited functional diversity (Fig. 4). In this case, the most abundant transcripts corresponded to heat shock proteins (HSP90, HSP70, HSP40, HSP20, HSP24 and GprE) and ribosome-related sequences. Interestingly, some immune-related molecules, such as myticin C, ficolin-2, C1q domain-containing protein and the scavenger receptor MARCO, were also present in the muscle. In mammals, MARCO is a pattern recognition receptor for Gram-positive and negative bacteria expressed mainly in macrophages [49] and it has not been described in invertebrates to date.
Some tissue-specific transcripts presented different variants depending on the tissue. This could mean that the gene is not tissue-exclusive but instead tissue-exclusive variants may exist. This was clearly observed for the C1q domain-containing proteins, a group of molecules that show high variability in M. galloprovincialis [12]. The C1q annotation for all the non-shared transcripts did not coincide between tissues (Additional file 4), which could suggest a high specialization of this molecule in each tissue and, possibly, specialization for non-self recognition, as might be inferred from the high abundance of the C1q sequences in gills. Without the M. galloprovincialis genome we can not know if the C1q variants are different isoforms or belong to different loci, further research is needed to clarify this issue.
Quantitative analysis between tissues: RNA-Seq
The transcriptomes were also quantitatively analyzed. We first normalized the number of reads that were mapped to each transcript into RPKM units (Reads Per Kilobase of exon model per Million mapped reads). To evaluate differentially expressed genes (d.e.g.) among tissues we used NOISeq [50], a nonparametric statistical approach that presents a low false discovery rate. The expression distribution of all the transcripts is showed in Fig. 5. As it is reflected by the red color intensity in each chart, the most similar tissues are mantle and muscle (d) while the most different are hemocytes and gills (c). The pairwise comparisons between the four tissues are summarized in Table 4. In the tissues that exhibited more transcripts in common (mantle and muscle) only 256 d.e.g. were found. In contrast, the comparison between hemocytes and gill, the tissues with the most dissimilar transcriptomes, revealed almost 2000 significantly different transcripts.

NOISeq log2-transformed expression charts. Red indicates the differentially expressed genes (d.e.g.) with a p-value < 0.01. a. Hemocytes and Mantle; b. Hemocytes and Muscle; c. Hemocytes and Gills; d. Mantle and Muscle; e. Mantle and Gills; f. Muscle and Gills
The heatmap provided in Fig. 6 illustrates, as an example, the quantitative expression of 5 among the top-expressed genes in each tissue, showing also the high reproducibility of the two biological replicates.

Heatmap of a selection of 5 of the most highly expressed genes by tissue, which shows the expression level of all the biological replicates used in this study. The scale bar is a non-linear representation of the normalized expression: Saturated green: no expression, 0 %. Black: 2 % of maximum expression. Saturated red: maximum expression, 100 %
Table 5 shows the 25 most highly expressed genes in each tissue compared to the other 3 transcriptomes. The top d.e.g. in hemocytes were immune-related, including AMPs, such as myticin A, mytilin B, mytilin 6 and 7 or defensin 2b; pore-forming molecules, such as apextrin and MAC/perforin; lectins (C-type, nacre protein, macrophage mannose receptor) and many other genes that are directly or indirectly related to the immune system, such as ADAMTS16, a metalloprotease required for remodeling the basement membrane during cell migration [51] (Table 5). This list also included C1q, a serine protease inhibitor that modulates host-pathogen interactions [52] and HSP70, ISG12 and IAP, which play important roles in apoptosis and immunity [53–55]. The expression fold change was relatively high, varying approximately between 200 and 2000. Nevertheless, we have to consider that hemocytes were stimulated with different treatments whereas the other tissues were sampled from unstimulated mussels.
In mantle the most highly expressed protein was usherin, showing a fold change of 5008 (Table 5). This protein is involved in visual and auditory transduction in mammals [56]. Other bivalves, such as scallops, possess ‘eyes’ at the mantle edge that influence their relationship with the environment [57]. The presence of this highly expressed gene in the mantle suggests that it might play a sensory role, in addition to its shell-forming and reproduction functions, which are also represented by genes such as vitellogenin 6, which is a precursor to egg-yolk proteins during embryonic development [58], or fibronectin 3, which is involved in shell formation in bivalves but also in mammal spermatogenesis [59, 60]. Interestingly, 3 of the 25 most highly expressed genes in mantle were related to antifungal functions or chitin metabolism: the PIF protein, mytimycin and chitinase 3, which showed fold changes of over 1000 compared with the other tissues. The shell of bivalves is a substrate for epibiotic communities, including fungi. Some fungi possess the ability to penetrate into the internal organs of animals and cause mycoses if the host-pathogen relationship is altered [61]. Therefore, the shell and the mantle could represent the first antifungal barrier, which would explain the presence of these d.e.g.
The muscle showed many typical myofibril molecules presenting fold changes of over 400, such as myosin light and heavy chain; paramyosin, which is typical of invertebrates; obscurin, which is involved in myofibrillogenesis [62]; calcium-related proteins, such as calmodulin or calpain, which are linked to muscle remodeling and contraction [63, 64]; and angiogenesis- and migration-related genes, such as angiopoietin-4 [65] and viral response molecules (e.g., TRIM56) [66], which exhibited expression increases of 465 and 776 fold, respectively (Table 5). These results suggest other possible functions of muscle in bivalves, as mentioned above.
The expression profile observed in gills confirmed previous studies showing that collagen is a major compound of this tissue [67]. Collagen was represented at more than 1000 fold the levels found in the other tissues, and showed higher levels than other extracellular matrix-related genes, such as dermatopontin, ST14 and TLL1. Dermatopontin accelerates and stabilizes collagen fibril formation, but this protein also presents other functions that are closely related to immune defense, such as cell adhesion via integrin binding, enhancing Transforming Growth Factor β1 activity or inhibiting cell proliferation [68]. ST14 degrades the extracellular matrix [69] and TLL1 processes procollagen C-propeptides [70]. However, as previously noted, the gills showed a significant expression of some immune-related molecules, such as the PIF protein, perlucin, LPS binding protein, big defensin or apextrin, which displayed fold changes ranging from 1243, in the case of apextrin, to 2837, in the case of PIF.
Enrichment analyses to compare qualitative and quantitative results
Gene Ontology (GO) terms were assigned to the non-redundant transcripts. A total of 18,899 (12.5 %) transcripts matched at least one GO term, which is twice as much as what was obtained in other reported Illumina transcriptomes (6 % GO annotation) [29]. This GO information was used to identify overrepresented biological processes in each transcriptome and in each group of d.e.g. by tissue. The results are summarized in Fig. 7 for the different transcriptomes and in Fig. 8 for the d.e.g. Figure 8 also shows overrepresented cellular components and molecular functions of hemocytes, mantle and muscle transcriptomes. First, it is important to note the large differences in enriched terms when the whole transcriptomes are compared with those of the d.e.g., which do not present a single term in common. The complete transcriptomes appeared to show more general functions, such as metabolism, transport or transcription, whereas the differentially expressed transcriptomes presented more detailed terms and functions, such as defense or regulation of specific signaling processes.

Classification of the complete transcriptomes by tissue type after a Blast2GO Enrichment Analysis. Only overrepresented biological process GO terms are shown

Classification of the d.e.g. by tissue after a Blast2GO Enrichment Analysis. Only overrepresented GO terms are shown and for the gills, only the biological process enriched GO terms are shown
Mussel hemocytes exhibited the most divergent transcriptome due to the large number of significantly enriched processes found (Fig. 7). These processes included immune-related functions, regulation of the apoptotic process, cellular response to chemical stimulus, intracellular protein kinase cascade and hemopoiesis. Other functions regarding cell proliferation (regulation of cell cycle, DNA-dependent DNA replication, DNA repair) or migration (cell junction assembly) were also found to be overrepresented. In contrast, the hemocyte d.e.g. (Fig. 8) showed a quite different profile, with a high representation of categories related to the immune response, such as innate immune response (12 % of d.e.g.), defense response to bacterium (21 %), regulation of immune defense to virus (12 %) or defense response to fungus (7 %). Figure 8 provides other interesting results as well; for example, almost 50 % of the hemocyte d.e.g. had functions involved in the response to stimulus, and over 20 % of them had ontologies for extracellular proteins and functions related to receptor binding.
The mantle transcriptome showed some remarkable enriched processes (Fig. 7), such as the evolutionarily conserved Wnt receptor signaling pathway, which plays a key role in development, including stem cell proliferation and cancer [71]. This finding is also congruent with the mantle being the hematopoietic tissue in mussels. Moreover, the differentially expressed transcriptome of the mantle confirmed the functional similarity between the mantle and muscle, as it included three GO terms related to muscle contraction: myofibril, myosin filament and calmodulin binding (Fig. 8). The results of the muscle d.e.g. enrichment analysis fully coincide with the mantle analysis, adding more terms related to contraction, such as motor activity, sarcomere, actin binding or calcium binding (Fig. 8). The complete muscle transcriptome, as well as the transcriptomes of the other tissues, presented general processes such as metabolism or transcription, but also two specific processes: response to DNA damage stimulus and hemopoietic or lymphoid organ development, including differentiation of resident and migratory cell types (Fig. 7). The GO terms related to immune response may be due to the normal presence of some hemocytes in the muscle.
The enriched functions of the gill transcriptome showed a similar, but reduced profile compared with that from hemocytes (Fig. 7). The gill transcriptome was not as closely related to immunity as it was to signaling (intracellular protein kinase cascade) and cell proliferation (cell cycle phase, chromosome organization and cytokinesis), which were most likely overrepresented due to the direct contact of this tissue with the environment, as such contact could lead to a regular renewal of the tissue. The enrichment analysis of the gill d.e.g. (Fig. 8) produced the highest number of results among all the analyzed data, with 37 GO biological process categories being overrepresented. All of these processes can be grouped into three main categories: calcium homeostasis, coagulation and defense, which are intimately related to each other. The identified coagulation processes (blood coagulation, platelet activation and degranulation) could also be included in the defense group because coagulation triggers the complement cascade [72] and is critical in immune defense, as well as the production of toxic radicals such as nitric oxide (NO) (represented with the categories nitric oxide metabolic process and regulation of nitric oxide synthase activity), which has been shown to occur in the gills. NO production is known to be up-modulated in bivalves stimulated with bacteria and parasites [73, 74]. Calcium homeostasis processes were clearly represented in the gill d.e.g. (Fig. 8), such as the detection and response to calcium ions, regulation of the release of sequestered calcium and activation of phospholipase C activity. In addition to their role in gas exchange, gills exhibit osmoregulatory, ion transport and homeostasis functions in crustaceans and fish [75, 76]; however, these functions have not been studied in bivalves. The cells involved in these processes in fish are ionocytes, a mitochondria-rich cell (MRC) type. In bivalves, three types of MRCs are present in the gills [67]. These factors suggest that there is calcium homeostasis activity in mussel gills.
Conclusions
We have shown the value of whole-transcriptome analysis generated via RNA-Seq for accurate quantification of gene expression. Using almost 400 million reads, we described the transcriptome and expression profiles of M. galloprovincialis tissues and the generated data has enriched the genomic resources available for this organism.
This study represents the first RNA-Seq approach applied in bivalves to describe and analyze tissue-specific transcriptomes. We identified a high number of transcripts related to the immune system, signal transduction and infectious diseases that highlight immune functions in all the tissues studied, probably as a result of mussel’s open circulatory system. Another group of disease-associated pathways were those related to cancer, which ranked second among the most represented pathways. Moreover, we also found specific and unexpected functions in specific tissues: mussel hemocytes showed the greatest number of antimicrobial and defense proteins; mantle appeared to exhibit a more specific antifungal function and even to be a firm candidate of the hematopoietic tissue; gills presented a large number of putative recognition molecules; and muscle expressed stress- and defense-related proteins.
Our results shed light into the transcriptomics and physiology of the Mediterranean mussel. This species has a great economical and ecological importance, it has been extensively used as pollution sentinel and the present findings related to immunity, hematopoiesis and cancer confirm that M. galloprovincialis is a very interesting candidate to be the model species for bivalves and even molluscs. The mussel genome project, that will come soon, will further support this candidature.
Methods
Tissue sampling, in vitro stimulation of hemocytes and RNA isolation
M. galloprovincialis mussels were obtained from a commercial shellfish farm (Vigo, Galicia, Spain) after depuration. The animals were maintained in open-circuit filtered sea water tanks at 15 °C with aeration and were fed daily with Phaeodactylum tricornutum and Isochrysis galbana until 2 days before sampling. Prior to the experiments, the mussels were acclimatized to aquarium conditions for one week.
The mantle, muscle and gill tissues from 5 mussels were sampled, pooled and conserved in 1 ml of TRIzol (Invitrogen). All samplings were performed as 2 biological replicates from all the tissues, except for the gills (which included only 1 biological replicate).
For hemolymph collection, approximately 50 mussels were notched in the shell and hemolymph (1–3 ml) was withdrawn from the adductor muscle of each mussel with a 0.5-mm-diameter (25G) disposable needle. The hemolymph was pooled and distributed in 6-well plates, with 7 ml per well, in a total of 9 wells, one for each treatment. The hemocytes were allowed to settle to the base of the wells for 30 min at 15 °C in the dark. Then, the hemocytes were stimulated for 3 h at 15 °C with 50 μg/ml polyinosinic:polycytidylic acid (Poly I:C), peptidoglycans (PG), zymosan, Vibrio anguillarum DNA (CpG), lipopolysaccharide (LPS), lipoteichoic acid (LTA), 100 ng/ml flagellin and 1 x 106 CFU/ml of heat-inactivated Vibrio anguillarum (one stimulus per well). The last group of hemocytes remained unstimulated. All the stimuli were purchased from SIGMA, except for CpG and V. anguillarum, which were produced in our laboratory. This procedure was performed twice to obtain 2 biological replicates. Hemolymph was centrifuged at 4 °C at 3000 g for 10 min and the pellet was resuspended in 500 μl of TRIzol (Invitrogen).
From this step onwards the methodology used was the same for all the tissues. Total RNA isolation was conducted following the manufacturer’s protocol using the RNeasy Mini kit (Qiagen) for RNA purification after DNase I treatment. Next, the concentration and purity of the RNA were measured using a NanoDrop ND1000 spectrophotometer. Finally, RNA integrity was tested on an Agilent 2100 Bioanalyzer (Agilent Technologies) to produce cDNA libraries for Illumina sequencing.
cDNA production and Illumina sequencing
The mRNA-Seq sample preparation kit from Illumina was used according to the manufacturer’s instructions. Briefly, eukaryotic mRNA was extracted from total RNA using oligo (dT) magnetic beads and cleaved into short fragments using fragmentation buffer. A cDNA library compatible with the Illumina NGS technology was then prepared from the fragmented mRNA via reverse transcription, second-strand synthesis and ligation of specific adapters (paired-ends) after cDNA purification using the QIAquick PCR Purification Kit (Qiagen). The amount of cDNA in each library was quantified through spectrofluorometric analysis using the Qbit system. Next-generation sequencing was performed using Illumina HiSeq™ 2000 technology at the Beijing Genomics Institute (BGI-HongKong Co., Ltd., Tai Po, Hong Kong).
Bioinformatics workflow
Assembly and functional annotation
The image data output from the sequencing apparatus was transformed via base calling into raw data and stored in FASTQ format. The raw data were cleaned with filter_fq software to discard low-quality reads, reads with regions with greater than 5 % unknown bases or reads with adapters.
De novo transcriptome assembly was conducted with the short reads assembly program Trinity [34, 77] (minimal contig_length: 100; group_pairs distance: 250; minimal kmer_cov: 2). Trinity first combined overlapping reads to form contigs with at least a 100-bp length and a minimum of 2 reads to be assembled. Then, the contigs were assembled again to obtain longer sequences that could not be further extended, which are unigenes. During this process and before obtaining the final unigenes, the reads were mapped against the contigs to confirm the assembly procedure. When multiple samples from the same species are sequenced (biological replicates or different tissues), unigenes from each sample can be applied together to perform another assembly step. This process detects sequence splicing and redundancy to acquire the longest sequences and group them into clusters. Each cluster is formed by several unigenes with more than 70 % similarity. To simplify the terminology employed in this study, all the non-redundant sequences will be called “transcripts”, regardless of whether they are unique unigenes or belong to a cluster. The completeness of the mussel transcriptome was confirmed with the CEGMA package (http://korflab.ucdavis.edu/datasets/cegma/).
A total of 151,320 transcripts were obtained following this protocol. This number represents all the detectable variability in the mRNAs from the four studied tissues, including splicing variants, non-overlapping fragments of the same mRNA, UTRs or mRNAs in different splicing stages.
The transcripts were first annotated using BLASTx and BLASTn (with an e-value threshold of 10e−5) against the NCBI nr, Swiss-Prot, KEGG and COG protein databases and the NCBI nt nucleotide database. The annotation step provided the identity of the transcript with the species harboring the matching sequence, which is useful for detecting possible contaminants in our samples. Using the KEGG database information, the metabolic pathways and functions of the annotated transcripts could be obtained and presented.
The oyster proteome was downloaded from http://www.oysterdb.com/FrontDownloadAction.do?method=download and compared with the translated mussel transcripts.
RNA-Seq with NOISeq: Quantitative analysis between tissues
RNA-Seq compares the number of reads that align to a specific transcript in different samples or cDNA libraries. The calculation of expression uses the RPKM (Reads Per Kilobase of exon model per Million mapped reads) normalization, while accounting for the length of the transcript that they belong to, its number of base pairs and the total number of reads in the transcriptome [78]. This normalization can eliminate the influence of different gene lengths and sequencing levels on the calculation of the gene expression. Therefore, the calculated gene expression can be directly used for comparison of the differences in gene expression between tissues in pairwise comparisons. The chosen method for evaluating the d.e.g. between tissues was NOISeq (http://bioinfo.cipf.es/noiseq) [50]. NOISeq is a nonparametric statistical approach that creates an empirical distribution of count changes that are adapted to the available data. This method has been proven to be the most effective in controlling the false discovery rate. The p-value threshold used to detect d.e.g. was 0.01.
To present the quantitative results and to facilitate their visualization, the pairwise comparisons (three per tissue) were fused, calculating the average of the three fold change values of the transcripts with the same annotation. Only one table/figure per tissue is presented, rather than all the possible comparisons.
The heatmap shown in Fig. 6 was designed with the software TMeV [79]. The normalized values (RPKM) for each gene by tissue and biological replicate were used to represent their expression in a green/0 – red/100 scale, with green representing the lower expression values and red the higher expression values.
GO classification and enrichment analysis
The nr annotation was used to obtain the GO term assignments of the transcripts with the Blast2GO program [80]. Then, enrichment analyses were conducted with the total information from all the tissues, including the reference set and each tissue and expression analysis test set. Next, Fisher’s exact test was run with default values (a two-tailed test that removes double IDs, with a false discovery rate (FDR) cut-off of 0.01). The Blast2GO option to show only the most specific terms (0.01 FDR cut-off) was used once. To reduce the dimensions of Fig. 8, the enrichment analyses of the expression results were combined according to the tissue types. Thus, only one graph per tissue is represented, instead of all the possible comparisons. Non-redundant categories were aggregated. For the coincident categories, the average of the percent representation was calculated.
Abbreviations
- ACC Nº:
-
Accession number
- ADAMTS16:
-
A disintegrin and metalloproteinase with thrombospondin motifs 16
- AMPs:
-
Antimicrobial peptides
- BLAST:
-
Basic local alignment search tool
- bp:
-
Base pairs
- C1q:
-
C1q domain-containing proteins
- CEGMA:
-
Core Eukaryotic Genes Mapping Approach
- COG:
-
Clusters of orthologous groups of proteins
- d.e.g.:
-
Differentially expressed genes
- EST:
-
Expressed sequence tags
- FC:
-
Fold change
- FDR:
-
False discovery rate
- FREPs:
-
Fibrinogen-related proteins
- GO:
-
Gene Ontology
- GrpE:
-
GroP-like gene E
- HSP:
-
Heat shock protein
- IAP:
-
Inhibitor of apoptosis protein
- ID:
-
Identity
- IFI27:
-
Interferon alpha-inducible protein 27-like protein
- ISG12:
-
Interferon-stimulated gene 12 protein
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LPS:
-
Lipopolysaccharide
- LTA:
-
Lipoteichoic acid
- M. galloprovincialis :
-
Mytilus galloprovincialis
- MAC:
-
Membrane attack complex
- MARCO:
-
Macrophage receptor with collagenous structure
- MRC:
-
Mitochondria-rich cell
- NCBI:
-
National Center for Biotechnology Information
- NGS:
-
Next generation sequencing
- NO:
-
Nitric oxide
- PG:
-
Peptidoglycans
- Poly I:C:
-
Polyinosinic:polycytidylic acid
- RNA-Seq:
-
RNA sequencing
- RPKM:
-
Reads Per Kilobase of exon model per Million mapped reads
- SRA:
-
Short read archive
- ST14:
-
Suppressor of tumorigenicity 14 protein
- TLL1:
-
Tolloid like-1 precursor
- TMeV:
-
TIGR Multiexperiment Viewer
- TRIM56:
-
Tripartite motif-containing protein 56
- V. anguillarum :
-
Vibrio anguillarum
References
Figueras A. Biología y cultivo de mejillón (Mytilus galloprovincialis) en Galicia. Madrid: Consejo Superior de Investigaciones Científicas; 2007.
Gosling E. Bivalve Molluscs: Biology, Ecology and Culture. New York: John Wiley & Sons; 2008.
Gestal C, Roch P, Renault T, Pallavicini A, Paillard C, Novoa B, et al. Study of diseases and the immune system of bivalves using molecular biology and genomics. Rev Fish Sci. 2008;16:131–54.
Paillard C, Leroux F, Borrego JJ. Bacterial disease in marine bivalves, Review of recent studies. Trends and evolution. Aquat Living Resour. 2004;17:477–98.
Villalba A, Reece KS, Ordás MC, Casas SM, Figueras A. Perkinsosis in molluscs: A review. Aquat Living Resour. 2004;17:411–32.
Figueras A. Cultured Aquatic Species Information Programme. Mytilus galloprovincialis. In: FAO Fisheries and Aquaculture Department. 2004. http://www.fao.org/fishery/culturedspecies/Mytilus_galloprovincialis/en. Accessed 10 February 2015.
Canesi L, Gallo G, Gavioli M, Pruzzo C. Bacteria–hemocyte interactions and phagocytosis in bivalves. Microsc Res Technol. 2002;57:469–76.
Olafsen JA. Role of lectins (C-reactive protein) in defense of marine bivalves against bacteria. Adv Exp Med Biol. 1995;371A:343–8.
Ordás MC, Novoa B, Figueras A. Modulation of the chemiluminescence response of Mediterranean mussel (Mytilus galloprovincialis) haemocytes. Fish Shellfish Immunol. 2000;10:611–22.
Pallavicini A, Costa MM, Gestal C, Dreos R, Figueras A, Venier P, et al. Sequence variability of myticins identified in haemocytes from mussels suggests ancient host-pathogen interactions. Dev Comp Immunol. 2008;32:213–26.
Romero A, Dios S, Poisa-Beiro L, Costa MM, Posada D, Figueras A, et al. Individual sequence variability and functional activities of fibrinogen-related proteins (FREPs) in the Mediterranean mussel (Mytilus galloprovincialis) suggest ancient and complex immune recognition models in invertebrates. Dev Comp Immunol. 2011;35:334–44.
Gerdol M, Manfrin C, De Moro G, Figueras A, Novoa B, Venier P, et al. The C1q domain containing proteins of the Mediterranean mussel Mytilus galloprovincialis: a widespread and diverse family of immune-related molecules. Dev Comp Immunol. 2011;35:635–43.
Criscitiello MF, de Figueiredo P. Fifty shades of immune defense. PLoS Pathog. 2013;9:e1003110.
Fleury E, Huvet A, Lelong C, de Lorgeril J, Boulo V, Gueguen Y, et al. Generation and analysis of a 29,745 unique Expressed Sequence Tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database, the GigasDatabase. BMC Genomics. 2009;10:341.
Gueguen Y, Cadoret JP, Flament D, Barreau-Roumiguière C, Girardot AL, Garnier J, et al. Immune gene discovery by expressed sequence tags generated from hemocytes of the bacteria-challenged oyster, Crassostrea gigas. Gene. 2003;303:139–45.
Venier P, De Pittà C, Bernante F, Varotto L, De Nardi B, Bovo G, et al. MytiBase: a knowledgebase of mussel (M. galloprovincialis) transcribed sequences. BMC Genomics. 2009;10:72.
Wang A, Wang Y, Gu Z, Li S, Shi Y, Guo X. Development of expressed sequence tags from the Pearl Oyster, Pinctada martensii dunker. Mar Biotechnol. 2011;13:275–83.
Tanguy A, Bierne N, Saavedra C, Pina B, Bachère E, Kube M, et al. Increasing genomic information in bivalves through new EST collections in four species, development of new genetic markers for environmental studies and genome evolution. Gene. 2008;408:27–36.
Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012;490:49–54.
Takeuchi T, Kawashima T, Koyanagi R, Gyoja F, Tanaka M, Ikuta T, et al. Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Res. 2012;19:117–30.
Rosani U, Varotto L, Rossi A, Roch P, Novoa B, Figueras A, et al. Massively parallel amplicon sequencing reveals isotype-specific variability of antimicrobial peptide transcripts in Mytilus galloprovincialis. PLoS One. 2011;6:e26680.
Gerdol M, De Moro G, Manfrin C, Milandri A, Riccardi E, Beran A, et al. RNA sequencing and de novo assembly of the digestive gland transcriptome in Mytilus galloprovincialis fed with toxinogenic and non-toxic strains of Alexandrium minutum. BMC Res Notes. 2014;7:722.
Bettencourt R, Pinheiro M, Egas C, Gomes P, Afonso M, Shank T, et al. High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics. 2010;11:559.
Hou R, Bao Z, Wang S, Su H, Li Y, Du H, et al. Transcriptome sequencing and de novo analysis for Yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PLoS One. 2011;6:e21560.
Milan M, Coppe A, Reinhardt R, Cancela LM, Leite RB, Saavedra C, et al. Transcriptome sequencing and microarray development for the Manila clam, Ruditapes philippinarum: genomic tools for environmental monitoring. BMC Genomics. 2011;12:234.
Moreira R, Balseiro P, Planas JV, Fuste B, Beltran S, Novoa B, et al. Transcriptomics of in vitro immune-stimulated hemocytes from the Manila clam Ruditapes philippinarum using high-throughput sequencing. PLoS One. 2012;7:e35009.
Chen H, Zha J, Liang X, Bu J, Wang M, Wang Z. Sequencing and De Novo Assembly of the Asian Clam (Corbicula fluminea) Transcriptome Using the Illumina GAIIx Method. PLoS One. 2013;8:e79516.
Gavery MR, Roberts SB. Characterizing short read sequencing for gene discovery and RNA-Seq analysis in Crassostrea gigas. Comp Biochem Physiol Part D Genomics Proteomics. 2012;7:94–9.
Zhao X, Yu H, Kong L, Li Q. Transcriptomic responses to salinity stress in the Pacific oyster Crassostrea gigas. PLoS One. 2012;7:e46244.
Apeti DA, Lauenstein GG, Christensen JD, Kimbrough K, Johnson WE, Kennedy M, et al. A historical assessment of coastal contamination in Birch Harbor, Maine based on the analysis of mussels collected in the 1940s and the Mussel Watch Program. Mar Pollut Bull. 2010;60:732–42.
Montes MO, Hanna SK, Lenihan HS, Keller AA. Uptake, accumulation, and biotransformation of metal oxide nanoparticles by a marine suspension-feeder. J Hazard Mater. 2012;225–6:139–45.
Venier P, Varotto L, Rosani U, Millino C, Celegato B, Bernante F, et al. Insights into the innate immunity of the Mediterranean mussel Mytilus galloprovincialis. BMC Genomics. 2011;12:69.
Balseiro P, Falcó A, Romero A, Dios S, Martínez-López A, Figueras A, et al. Mytilus galloprovincialis myticin C: a chemotactic molecule with antiviral activity and immunoregulatory properties. PLoS One. 2011;6:e23140.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4.
Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6.
St Laurent G, Shtokalo D, Tackett MR, Yang Z, Vyatkin Y, Milos PM, et al. On the importance of small changes in RNA expression. Methods. 2013;63:18–24.
Gonzalez-Rey M, Lau TC, Gomes T, Maria VL, Bebianno MJ, Wu R. Comparison of metal accumulation between ‘Artificial Mussel’ and natural mussels (Mytilus galloprovincialis) in marine environments. Mar Pollut Bull. 2011;63:149–53.
Romero A, Costa M, Forn-Cuni G, Balseiro P, Chamorro R, Dios S, et al. Occurrence, seasonality and infectivity of Vibrio strains in natural populations of mussels Mytilus galloprovincialis. Dis Aquat Organ. 2014;108:149–63.
Kurelec B, Pivčević B. Evidence for a multixenobiotic resistance mechanism in the mussel Mytilus galloprovincialis. Aquat Toxicol. 1991;19:291–301.
Carella F, Figueras A, Novoa B, De Vico G. Comparative cytomorphological features and PCNA expression pattern in Haemic Neoplasia from mediterranean mussels (Mytilus galloprovincialis) and Galician common cockles (Cerastoderma edule). Dis Aquat Organ. 2013;105:81–7.
Arriagada G, Metzger MJ, Muttray AF, Sherry J, Reinisch C, Street C, et al. Activation of transcription and retrotransposition of a novel retroelement, Steamer, in neoplastic hemocytes of the mollusk Mya arenaria. Proc Natl Acad Sci U S A. 2014;111:14175–80.
Boon E, Faure MF, Bierne N. The flow of antimicrobial peptide genes through a genetic barrier between Mytilus edulis and M. galloprovincialis. J Mol Evol. 2009;68:461–74.
Wang N, Lee YH, Lee J. Recombinant perlucin nucleates the growth of calcium carbonate crystals: molecular cloning and characterization of perlucin from disk abalone, Haliotis discus discus. Comp Biochem Physiol B Biochem Mol Biol. 2008;149:354–61.
Bayne CJ, Sminia T, Van der Knaap WPW. Immunological memory: status of molluscan studies. In: Manning MJ, editor. Phylogeny of immunological memory. Developments in immunology, vol. 10. Amsterdam: Elsevier; 1980. p. 57–64.
Bachère E, Gueguen Y, Gonzalez M, de Lorgeril J, Garnier J, Romestand B. Insights into the anti-microbial defense of marine invertebrates: the penaeid shrimps and the oyster Crassostrea gigas. Immunol Rev. 2004;198:149–68.
Balseiro P, Moreira R, Chamorro R, Figueras A, Novoa B. Immune responses during the larval stages of Mytilus galloprovincialis: metamorphosis alters immunocompetence, body shape and behavior. Dev Comp Immunol. 2013;35:438–47.
Grigorian M, Mandal L, Hartenstein V. Hematopoiesis at the onset of metamorphosis: terminal differentiation and dissociation of the Drosophila lymph gland. Dev Genes Evol. 2011;221:121–31.
Ojala JR, Pikkarainen T, Tuuttila A, Sandalova T, Tryggvason K. Crystal structure of the cysteine-rich domain of scavenger receptor MARCO reveals the presence of a basic and an acidic cluster that both contribute to ligand recognition. J Biol Chem. 2007;282:16654–66.
Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-Seq: a matter of depth. Genome Res. 2011;21:2213–23.
Surridge AK, Rodgers UR, Swingler TE, Davidson RK, Kevorkian L, Norton R, et al. Characterization and regulation of ADAMTS-16. Matrix Biol. 2009;28:416–24.
Xue Q, Itoh N, Schey KL, Cooper RK, La Peyre JF. Evidence indicating the existence of a novel family of serine protease inhibitors that may be involved in marine invertebrate immunity. Fish Shellfish Immunol. 2009;27:250–9.
Parcellier A, Gurbuxani S, Schmitt E, Solary E, Garrido C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem Biophys Res Commun. 2003;304:505–12.
Rosebeck S, Leaman DW. Mitochondrial localization and pro-apoptotic effects of the interferon-inducible protein ISG12a. Apoptosis. 2008;13:562–72.
Silke J, Meier P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol. 2013;5(2):a008730.
Yang J, Liu X, Zhao Y, Adamian M, Pawlyk B, Sun X, et al. Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 2010;6:e1000955.
Speiser DI, Loew ER, Johnsen S. Spectral sensitivity of the concave mirror eyes of scallops: potential influences of habitat, self-screening and longitudinal chromatic aberration. J Exp Biol. 2011;214:422–31.
Nakamura A, Yasuda K, Adachi H, Sakurai Y, Ishii N, Goto S. Vitellogenin-6 is a major carbonylated protein in aged nematode, Caenorhabditis elegans. Biochem Biophys Res Commun. 1999;264:580–3.
Joubert C, Piquemal D, Marie B, Manchon L, Pierrat F, Zanella-Cléon I, et al. Transcriptome and proteome analysis of Pinctada margaritifera calcifying mantle and shell:focus on biomineralization. BMC Genomics. 2010;11:613.
Obholz KL, Akopyan A, Waymire KG, MacGregor GR. FNDC3A is required for adhesion between spermatids and Sertoli cells. Dev Biol. 2006;298:498–513.
Morley NJ. Interactive effects of infectious diseases and pollution in aquatic molluscs. Aquat Toxicol. 2010;96:27–36.
Young P, Ehler E, Gautel M. Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol. 2001;154:123–36.
Tavi P, Westerblad H. The role of in vivo Ca2+ signals acting on Ca2+-calmodulin-dependent proteins for skeletal muscle plasticity. J Physiol. 2011;589:5021–31.
Murphy RM. Calpains, skeletal muscle function and exercise. Clin Exp Pharmacol Physiol. 2010;37:385–91.
Lee HJ, Cho CH, Hwang SJ, Choi HH, Kim KT, Ahn SY, et al. Biological characterization of angiopoietin-3 and angiopoietin-4. FASEB J. 2004;18:1200–8.
Shen Y, Li NL, Wang J, Liu B, Lester S, Li K. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J Biol Chem. 2012;287:36404–13.
de Oliveira David JA, Salaroli RB, Fontanetti CS. Fine structure of Mytella falcata (Bivalvia) gill filaments. Micron. 2008;39:329–36.
Okamoto O, Fujiwara S. Dermatopontin, a novel player in the biology of the extracellular matrix. Connect Tissue Res. 2006;47:177–89.
Ge W, Hu H, Ding K, Sun L, Zheng S. Protein interaction analysis of ST14 domains and their point and deletion mutants. J Biol Chem. 2006;281:7406–12.
Berry R, Jowitt TA, Garrigue-Antar L, Kadler KE, Baldock C. Structural and functional evidence for a substrate exclusion mechanism in mammalian tolloid like-1 (TLL-1) proteinase. FEBS Lett. 2010;584:657–61.
Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–79.
Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50.
Costa MM, Prado-Alvarez M, Gestal C, Li H, Roch P, Novoa B, et al. Functional and molecular immune response of Mediterranean mussel (Mytilus galloprovincialis) haemocytes against pathogen-associated molecular patterns and bacteria. Fish Shellfish Immunol. 2009;26:515–23.
Villamil L, Gomez-Leon J, Gomez-Chiarri M. Role of nitric oxide in the defenses of Crassostrea virginica to experimental infection with the protozoan parasite Perkinsus marinus. Dev Comp Immunol. 2007;31:968–77.
Freire CA, Onken H, McNamara JC. A structure-function analysis of ion transport in crustacean gills and excretory organs. Comp Biochem Physiol A Mol Integr Physiol. 2008;151:272–304.
Conte FP. Origin and differentiation of ionocytes in gill epithelium of teleost fish. Int Rev Cell Mol Biol. 2012;299:1–25.
Yang Y, Smith SA. Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genomics. 2013;14:328.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8.
Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, et al. TM4 microarray software suite. Methods Enzymol. 2006;411:134–93.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO, a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Acknowledgments
This work has been funded by the projects BIVALIFE, FP7-KBBE-2010-4/266157 and 10 PXIB 402 096 PR from Xunta de Galicia. RM wishes to acknowledge the Spanish MICINN for her FPI Spanish research grant (BES-2009-029765) and PP gratefully acknowledges the Spanish Ministerio de Educación for her FPU fellowship (AP2010-2408). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BN, AF, DP and CC conceived and designed the experiments. RM, PP prepared the samples. RM and BN analyzed the data. RM wrote the paper. BN, AF, DP, CC and PP provided corrections to the manuscript. All authors read and approved the manuscript.
Additional files
Additional file 1:
Table listing Mytilus galloprovincialis transcripts, including the sequence, length, RPKM, description, accession number of the description (Hit ACC) and e-value obtained in each database used for annotation and the GO terms ascribed to each sequence. (XLSX 38412 kb)
Additional file 2:
Figure representing the number of transcripts obtained after the assembly of increasing number of mapped reads. (XLSX 743 kb)
Additional file 3:
Pathways found in the annotated portion of the transcriptomes. (ZIP 4950 kb)
Additional file 4:
Tables and figures showing the non-shared C1q variants found in the hemocytes, mantle, muscle and gills. (XLSX 17 kb)
Additional file 5:
Mytilus galloprovincialis transcriptome in FASTA format. (ZIP 25884 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Moreira, R., Pereiro, P., Canchaya, C. et al. RNA-Seq in Mytilus galloprovincialis: comparative transcriptomics and expression profiles among different tissues. BMC Genomics 16, 728 (2015). https://doi.org/10.1186/s12864-015-1817-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1817-5