
Abstract
Background
Nr2e1 (nuclear receptor subfamily 2, group e, member 1) encodes a transcription factor important in neocortex development. Previous work has shown that nuclear receptors can have hundreds of target genes, and bind more than 300 co-interacting proteins. However, recognition of the critical role of Nr2e1 in neural stem cells and neocortex development is relatively recent, thus the molecular mechanisms involved for this nuclear receptor are only beginning to be understood. Serial analysis of gene expression (SAGE), has given researchers both qualitative and quantitative information pertaining to biological processes. Thus, in this work, six LongSAGE mouse libraries were generated from laser microdissected tissue samples of dorsal VZ/SVZ (ventricular zone and subventricular zone) from the telencephalon of wild-type (Wt) and Nr2e1-null embryos at the critical development ages E13.5, E15.5, and E17.5. We then used a novel approach, implementing multiple computational methods followed by biological validation to further our understanding of Nr2e1 in neocortex development.
Results
In this work, we have generated a list of 1279 genes that are differentially expressed in response to altered Nr2e1 expression during in vivo neocortex development. We have refined this list to 64 candidate direct-targets of NR2E1. Our data suggested distinct roles for Nr2e1 during different neocortex developmental stages. Most importantly, our results suggest a possible novel pathway by which Nr2e1 regulates neurogenesis, which includes Lhx2 as one of the candidate direct-target genes, and SOX9 as a co-interactor.
Conclusions
In conclusion, we have provided new candidate interacting partners and numerous well-developed testable hypotheses for understanding the pathways by which Nr2e1 functions to regulate neocortex development.
Similar content being viewed by others

Background
The proper development of the mammalian neocortex involves a balance between cell-intrinsic developmental programs and environmental factors. In this process, neurons acting as the backbone of the neuronal circuitry are generated first. These cells arise from the dorsal telencephalon, generating cortical excitatory neurons by radial migration, and the ventral telencephalon giving rise to cortical inhibitory interneurons by tangential migration [1–5]. The neurogenic stage is followed by the integration of glial cells in the circuitry during the gliogenic stage. In mice, neurons are generated from embryonic day 12 (E12) to E18, with astrocytes appearing at around E18 [6, 7]. Ultimately, the neocortex will comprise six different radial layers with cell populations having distinct molecular identities [8].
Nr2e1 (nuclear receptor subfamily 2, group e, member 1, also known as Mtll, Tlx, Tll, and tailless) encodes a transcription factor important in the process of neocortex development [9, 10]. This complex cellular process involves a careful balance between proliferation of neural stem cells (NSC), and the proper temporal differentiation of progenitor cells (PC) (i.e. neurons versus glia). Nr2e1 is expressed along the ventricular zone (VZ) of the dorsal telencephalon during neocortex development and is crucial for NSC self-renewal and maintenance [11–14]. Absence of Nr2e1 in mouse embryos reduces the number of PC populating the VZ and subventricular zone (SVZ) during development, which results in reduced thickness of the cortical plate [9]. The reduction in PC populating the VZ is more prominent in the caudal telencephalon whereas the reduction in the SVZ is seen at all rostrocaudal levels during development. This cell-reduction ultimately results in defects in structures generated later, such as the upper cortical layers (layers II and III), the dentate gyrus, and the olfactory bulb [9, 10]. Absence of Nr2e1 in mouse embryos also results in premature neurogenesis, which contributes to the defects in the upper cortical layers [9].
Previous work has shown that a nuclear receptor transcription factor can have hundreds of target genes [15], and the most extensively studied nuclear receptors are estimated to bind more than 300 co-interacting proteins [16, 17]. However, recognition of the critical role of nuclear receptor Nr2e1 in NSC and neocortex development is relatively recent [11, 12, 18–20], thus the molecular mechanisms involved for this nuclear receptor are only beginning to be understood. First, in forebrain development, Nr2e1 has been shown to regulate cell cycle progression via its interaction with the tumour suppressor gene Pten, and the cyclin-dependent kinase inhibitor p21 [11]. This involves a repressive mechanism mediated via the interaction of Nr2e1 with chromatin modifier proteins such as members of the histone deacetylase family (HDACs), and the demethylase protein LSD1 (KDM1A) [14, 21]. Second, the balance between NSC proliferation and differentiation has been demonstrated to be under the control of regulatory loops involving both Nr2e1, and microRNA encoding genes such as mir-9, miR-137, and let-7d [22–24]. This phenomenon includes an intricate network formed by the ability of let-7d and mir-9 to silence Nr2e1 expression by binding the 3′ UTR regions of this gene and the ability of Nr2e1 to inactivate the expression of mir-9 in a first feedback loop [22, 24]. A second loop has been reported that includes the repression of the co-interactor Lsd1 by miR-137 that can be relieved by the repression of miR-137 by Nr2e1 [23]. Finally, Nr2e1 has been shown to act as a transcriptional activator of the deacetylase gene Sirt1, which has a role in promoting neuronal differentiation [25, 26]. Thus, we hypothesize there is still much to learn about the molecular mechanisms underlying the role of NR2E1 in NSC and neocortex development. Hence, we have undertaken additional research on these mechanisms, especially focused on in vivo analyses, to inform our understanding of neocortex development.
Large-scale transcriptome-profiling experiments, using methodologies such as serial analysis of gene expression (SAGE), have given researchers the advantages of both qualitative and quantitative information pertaining to biological processes. SAGE analysis relies on sequencing and quantification of short (14 bp) cDNA fragments called tags, which are derived from messenger RNA transcripts [27]. This approach is considered an open transcriptome technology as no a priori knowledge of the transcript sequences is required [28]. For the mammalian central nervous system, SAGE profiling experiments have been used to generate knowledge on a variety of topics including; fundamental studies on brain development [29], connectivity, and aging [30–32], as well as specific neuropathologies and drug responses [33–36]. Advancement in SAGE library generation such as SAGE-lite [37], which enabled the use of extremely small quantities of tissues such as those from laser capture microdissection (LCM), and LongSAGE, which improved tag-to-gene mapping by generating longer tag fragments (21 bp) [38], made these approaches particularly appropriate to reveal in vivo molecular changes in neocortex development. One of the inherent challenges in transcriptome profiling is the effective analysis of large-scale datasets to optimize extraction of relevant biological meaning. By producing LongSAGE libraries at multiple developmental times, in the presence and absence of Nr2e1, we generated a rich dataset for comparative analysis. Additionally, we took advantage of the intrinsic nature of transcription factors, which regulate gene expression by binding to specific DNA sequences, and used it to further hone our gene list. This was partly based on the power of transcription-factor-discovery-motif algorithms that, when coupled to cross-species genome comparisons or phylogenetic footprinting, have proven successful in making reliable binding site predictions [39–42]. Returning to biology to further validate the bioinformatic studies, we of course used the literature, but most importantly, we also tested our primary new hypothesis in vitro by embryonic stem cells (ESC) differentiation and in vivo during brain development. Thus, in this work, we used a novel approach, implementing multiple computational methods to generate significant-novel-biological information regarding the molecular mechanisms underlying the role of nuclear receptor Nr2e1 in neocortex development.
Results and discussion
LongSAGE libraries generated from laser capture microdissection tissues
To identify novel-candidate-target and co-interacting genes for the nuclear receptor Nr2e1, we favoured an in vivo source of RNA in order to most accurately capture molecular events occurring during neocortex development. Thus, LongSAGE libraries were prepared using RNA purified from tissues obtained by LCM of the VZ/SVZ, of the dorsal-lateral telencephalon, from Wt and Nr2e1 frc/frc embryos. This work was undertaken at three different developmental time points (E13.5, E15.5, and E17.5), which are known to express Nr2e1 in the dissected region [9, 11, 18, 43] (Fig. 1a). These libraries were sequenced to a depth ≥100,000 tags (total number of tags per libraries, see Fig. 1b). To generate tags for analysis, we used a filtering procedure involving the DiscoverySpace 4.0 application (http://www.bcgsc.ca/platform/bioinfo/software/ds) (filtering details, see Methods) [44]. On average, ~24 % of the total tags per library were discarded in this procedure resulting in a useful tag population averaging ~83,000 tags per library, and corresponding to ~25,000 tag types per library (Fig. 1b). Singleton tags (tags counted only once) constituted ~18 % of the useful tags population per library and ~68 % of the tag type population per library (Fig. 1b). These numbers were consistent with previously published results, obtained using a similar filtering procedure [45].

Laser microdissected LongSAGE libraries were used to reveal the transcriptome of Wt and Nr2e1 frc/frc embryos. a The laser capture microdissection (LCM) procedure. ((a), I-IV) Embryonic day 13.5 sagittal sections stained with cresyl violet. ((a), II) The ventricular/subventricular zone (VZ/SVZ) of the dorsal lateral telencephalon cut with laser. ((a), III) The VZ/SVZ removed by LCM. ((a), IV) The VZ/SVZ of the dorsal lateral telencephalon captured by LCM for RNA extraction. LV, Lateral Ventricle; Str, striatum. Scale bars, 100 μm. b The composition of LongSAGE libraries. Column one presents the name of the library; columns two and three, the genotype and developmental stage respectively; column four, the amount of RNA used as starting material; and columns five to nine, the number of tags for each library depending on the filtering criteria used
LongSAGE libraries differential statistical analyses and gene IDs recovery
The Audic-Claverie significance test, implemented in the DiscoverySpace 4.0 application, was used to perform statistical analyses on the filtered tags [44, 46]. Tags differentially expressed between Wt and Nr2e1 frc/frclibraries at each time point (E13.5, E15.5, and E17.5), and falling within the confidence interval of 95 % (P < 0.05), according to the Audic-Claverie significance test, were retained for further analyses. The results for tags significantly increased or decreased in abundance at each time point are shown in Fig. 2a. The proportion of differentially abundant tags (either increased or decreased) varied between 15 to 25 % when compared to the combined numbers of useful tags found in the Wt and Nr2e1 frc/frc library at each time point (e.g. (Up at E13.5 “Tags (P < 0.05)” (Fig. 2a)/(Wt + Nr2e1 frc/frc at E13.5 “Total useful tags” (Fig. 1b))) × 100). LongSAGE tags were mapped to RefSeq (v52) and Ensembl (v66) databases [45]. On average, 52 % of the differentially abundant tags mapped to genes (average of (“Tags mapped to genes”/“Tags (P < 0.05)”)×100 for each library, Fig. 2a). The number of Refseq accession IDs corresponding to differentially abundant tags at the three different time points are also shown in Fig. 2a. These accession numbers, corresponding to Refseq genes, were retrieved and used in the subsequent analyses.

Comparative transcriptome investigation of Wt and Nr2e1 frc/frc embryos yielded hundreds of candidate-differentially-regulated genes. a Details of the number of differentially abundant tags (increased or decreased) and the corresponding number of genes between Wt and Nr2e1 frc/frc embryos at each time point. In the exceptional case of a gene having both a significantly up and down tag, it was counted in both categories. Column one presents the direction of change; column two, the embryonic day of tissue harvest; column three, the number of tags that had significantly different counts between Wt and Nr2e1 frc/frc embryos; column four, the number of tags having significant different counts that mapped to genes found in the Ensembl (v66) and Refseq (v52) gene collections; and column five, the number of Refseq genes mapped by the corresponding tags. b The Venn diagram presents the number of up- and down-regulated genes that were exclusive or shared at each embryonic day (E13.5, E15.5, and E17.5). Bracketed numbers correspond to the total of differentially-regulated genes between Wt and Nr2e1 frc/frc embryos at the corresponding time point
We next looked at the genes that were differentially regulated between the Wt and Nr2e1 frc/frc libraries at the E13.5, E15.5, and E17.5 time points. This resulted in a total of 1279 Refseq accession numbers, originating from a corresponding list of 1387 tag sequences (Additional file 1: Table S1 and Additional file 2: Table S2), and distributed according to the Venn diagram in Fig. 2b. Interestingly, when performing the analyses, on average 6 genes per time point corresponded to tags that were found in both the up and down regulated populations (data not shown). This suggested that the tags mapped to these genes were corresponding to alternative transcripts that were expressed in opposing directions when comparing Wt and Nr2e1 frc/frc libraries. The Venn diagram results also demonstrated that on average 54 % of the differentially-regulated genes (combined up and down) were specific for each time point: E13.5, 59 % ((383/650) × 100); E15.5, 59 % ((423/711) × 100); and E17.5, 44 % ((146/333) × 100). Furthermore, on average 17 % of the genes overlapped between at least two time points: E13.5 and E15.5, 20 % (((140 + 88)/(650 + 423 + 60)) × 100); E15.5 and E17.5, 17 % (((88 + 60)/(711 + 146 + 39)) × 100); and E13.5 and E17.5, 15 % (((88 + 39)/(650 + 146 + 60)) × 100). Finally, only 6.9 % of the genes overlapped between the three time points ((88/1279) × 100).
LongSAGE expression results suggested distinct roles for Nr2e1 in different stages of neocortex development
To understand the role of Nr2e1 in gene expression during neocortex development, we performed hierarchical clustering on the tag ratio values corresponding to each of the 1279 Refseq accession numbers of differentially-regulated genes (Fig. 3, Additional file 1: Table S1 and Additional file 2: Table S2). Tag sequences and corresponding tag counts of the 1279 Refseq accession numbers were retrieved for each LongSAGE library using the DiscoverySpace 4.0 application. Fold changes from tags statistically differentially abundant, at least at one time point between the Wt and Nr2e1 frc/frc libraries, were calculated as previously described [45], and hierarchical clustering was performed using the Gene Cluster software as described in Methods [47]. The clustering results were visualized in a heat-map display using Java TreeView (Fig. 3a) [48]. Spearman-rank-ordering correlation was additionally performed on the fold changes dataset at each time point as described in Methods. The results demonstrated that at the E13.5 and E15.5 time-points, differential tag ratios correlated positively (Spearman R = 0.28, P < 0.001). In contrast, comparing E13.5 and E17.5, as well as E15.5 and E17.5 yielded negative correlation values (E13.5 vs. E17.5, Spearman R = −0.24, P < 0.001; E15.5 vs. E17.5, Spearman R = −0.23, P < 0.001) (Fig. 3b). This demonstrated that the differential-tag ratio found between the E13.5, and E15.5 libraries were more similar than the one observed in the E17.5 library. This also suggested that Nr2e1 expression has a more similar effect on genes in early and mid-stages of neurogenesis, than during the switch from neurogenesis to gliogenesis occurring around E17.5. These results correlated with previously published observations, demonstrating a progression of the Nr2e1-null phenotype during neocortex development, with a greater effect between E13 and E15 [9].

Hierarchical clustering suggested a stage-specific role for Nr2e1 in neocortex development. a Tag numbers for each of the corresponding 1279 genes were retrieved from DiscoverySpace and hierarchical-clustering was performed prior to visualization in a heat-map; displaying significant-differential-tag ratios from Wt vs. Nr2e1 frc/frc libraries. The relative expression was calculated based on the tag ratios from Wt vs. Nr2e1 frc/frc libraries and corrected to account for library sizes; (observed tag counts/total useful tags) X 100,000. Tags having a count value of “0” (no expressed tags) were adjusted to a value of “1” for fold change calculations only. Green, down; red, up; black, no difference; grey, no expressed tags. b Embryonic-stage-specific differences in expression profiles of the 1279 differently abundant genes in neocortex development of Wt vs. Nr2e1 frc/frc embryos was demonstrated using Spearman rank correlation. Spearman rank correlation was performed on gene lists from pairs of embryonic stages using STATISTICA. Results revealed a significant positive correlation between the lists corresponding to early and mid-neurogenic stages (E13.5 vs. E15.5); whereas significant negative correlations were obtained with the tag ratios corresponding to the early and mid-neurogenic stages versus the early gliogenic stages (E13.5 vs. E17.5, and E15.5 vs. E17.5). Column one presents the time-point comparisons; column two, the valid number of genes included in the analysis; columns three and four, the corresponding R and P values obtained by Spearman rank correlation
Bioinformatics analyses for the prediction of Nr2e1 direct targets
Considering that Nr2e1 encodes for a transcription factor, and that transcription factors regulate transcription by binding the promoter regions of their target genes; we hypothesized that a list of genes, differentially regulated between Wt and Nr2e1 frc/frc, would comprise genes containing Nr2e1 binding sites within their promoter regions. Thus, interrogation of our pooled list of 1279 Refseq accession numbers was undertaken using three different software tools; the ORCA toolkit (tk) to perform the initial orthologous-sequence alignment and phylogenetic footprinting [49], a customized version of oPOSSUM for prediction and storage of transcription factor binding sites (TFBSs) (http://www.cisreg.ca/oPOSSUM/) [39, 40], and a DAVID GO term analysis to evaluate if the resulting genes were found in biological processes relevant to Nr2e1 (http://david.abcc.ncifcrf.gov/summary.jsp) [50, 51]. The “modified” version of the oPOSSUM database used a position-weight matrix (PWM) that we designed based on the nine sequences available from the literature, which were known to be bound by NR2E1 (Additional file 3: Table S3) [21, 22, 52–56]. The resulting matrix and logo are depicted in Fig. 4a.

Novel implementation of three computational methods to generate a focused list of biologically-relevant Nr2e1-candidate-target genes. a Data mining from the literature allowed generation of a position-weight matrix representing the binding properties of NR2E1. The matrix and resulting logo are presented. b Flow chart describing the novel implementation of three computational methods to generate a focused list of biologically-relevant Nr2e1-candidate-target genes. DiscoverySpace was used to generate a compiled list of 1279 Refseq accession numbers, corresponding to genes differentially regulated between Wt and Nr2e1 frc/frc embryos. A customized oPOSSUM database of predicted conserved TFBS was created by first aligning all orthologous human-mouse genes using ORCAtk. During this alignment, 304 of the 1279 differentially regulated genes were excluded due to a lack of ortholog information or poor ORCAtk alignment quality. Then the remaining 975 genes with conserved promoter regions were scanned with the NR2E1 matrix (as well as all the vertebrate matrices from the JASPAR CORE collection of transcription factor binding site profiles). Of the 975 scanned genes, 770 contained conserved NR2E1 binding sites. These 770 Refseq accession numbers were submitted to a gene ontology (GO) term analysis using DAVID. From the DAVID analysis, 479 Refseq accession numbers were found to be enriched in GO term categories related to biological processes
The results from these sequential analyses are summarized in the flowchart of Fig. 4b. ORCAtk orthologous sequence alignments between human and mouse for each gene was initially performed; resulting in the exclusion of 304 Refseq accession numbers due to poor conservation between human and mouse within the promoter sequences of these genes. This resulted in 975 Refseq accession numbers that were used in the modified oPOSSUM database. Of these 975 accession numbers, 770 (79 %) were found to have predicted binding sites for NR2E1 within their promoter regions (Fig. 4b) (Additional file 2: Table S2). [57, 58] These 770 accession numbers were further studied in a GO term enrichment analysis using the DAVID service. The 770 Refseq accession numbers were first converted to DAVID IDs using the DAVID knowledgebase, and then compared to the DAVID mouse-background list of genes [50, 59]. The enrichment results were visualized using the functional annotation module based on the relevance for each enriched gene to “biological process” with an initial P value < 0.1, using the modified Fisher exact test (EASE score) [51, 60, 61]. In this process, 291 Refseq accession numbers were discarded as they were not enriched in our list compared to the mouse background (Fig. 4b). The remaining 479 Refseq accession numbers were interrogated based on their “biological process” terms. Only terms with a P value < 0.05 after multiple test correction, using the Bonferroni approach [59, 62], were considered interesting for further investigation. Table 1 shows the list of GO terms passing this criterion, Additional file 2: Table S2 list the differentially expressed genes within these GO terms. As expected, numerous terms related to cell cycle regulation were found after performing the GO term enrichment analysis on the 770 Refseq list. However, terms related to cell cycle regulation were also found in a similar analysis, using the initial 1279 Refseq list. This suggested that genes involved in cell cycle regulation were differentially expressed in our LongSAGE results when comparing Wt to Nr2e1 frc/frc, but were not enriched for the presence of NR2E1 binding sites within the promoter regions. In contrast, the term “nervous system development” (P < 0.01), with 64 differentially-regulated genes, was found to be enriched only after performing the analysis on the 770 Refseq list, again, suggesting the presence of NR2E1 binding sites within the promoter regions of genes enriched for this term. Interestingly, similar results were also obtained using a different GO term enrichment software; “GOstats” yielded significant results for the “nervous system development” term (P < 0.001) [63]. Thus, we subsequently used the “nervous system development” gene list from the “DAVID analysis” for further investigations.
Differential expression results validated by literature
We used the tag ratio values of the 64 differentially expressed genes found in the “nervous system development” GO term category to perform hierarchical clustering (Fig. 5a, Additional file 2: Table S2). We used the same hierarchical procedure as the one described for the 1279 genes list. Similarly to previously obtained results, the E13.5 and E15.5 time-points, differential tag ratios correlated positively (Spearman R = 0.41, P < 0.001), and the E13.5 and E17.5 yielded a negative correlation value (Spearman R = −0.34, P < 0.001). However, no significance was observed when comparing the E15.5 and E17.5 time points (Fig. 5b). This suggested that the differential-tag ratio found between the E13.5, and E15.5 libraries were more similar than the one observed in the E17.5 library; highlighting again the possibility of distinct roles for Nr2e1 in the neurogenic versus early gliogenic stages of neocortex development.

Hierarchical clustering suggested a stage-specific role for Nr2e1 in nervous system development. a Tag numbers for each of the 64 genes were retrieved from DiscoverySpace and hierarchical-clustering was performed prior to visualization in a heat-map; displaying significant-differential-tag ratios from Wt vs. Nr2e1 frc/frc libraries. The relative expression was calculated based on the tag ratios from Wt vs. Nr2e1 frc/frc libraries and corrected to account for library sizes; (observed tag counts/total useful tags) X 100,000. Tags having a count value of “0” (no expressed tags) were adjusted to a value of “1” for fold change calculations only. Green, down; red, up; black, no difference; grey, no expressed tags; bold, key genes in this analysis. b Embryonic-stage-specific differences in expression profiles of “nervous system development” genes in neocortex development of Wt vs. Nr2e1 frc/frc embryos was demonstrated using Spearman rank correlation. Spearman rank correlation was performed on gene lists from pairs of embryonic stages using STATISTICA. Results revealed a significant positive correlation between the lists corresponding to early and mid-neurogenic stages (E13.5 vs. E15.5); whereas a significant negative correlation was obtained with the tag ratios corresponding to the early neurogenic and early gliogenic stages (E13.5 vs. E17.5). No significance was obtained between the mid-neurogenic and early gliogenic stages (E15.5 vs. E17.5). Column one presents the time-point comparisons; column two, the valid number of genes included in the analysis; columns three and four, the corresponding R and P values obtained by Spearman rank correlation
As expected, all the Nr2e1 frc/frc libraries showed no tags for Nr2e1. Interestingly, even the Wt libraries, despite being obtained by LCM for a focused region of Nr2e1 expression, showed low abundance of Nr2e1 tags (E13.5, 4; E15.5, 2; and E17.5, 0). Thus, only E13.5 reached significant differential expression between Wt and Nr2e1 frc/frc (−4.5 fold, P < 0.05). As expected, the number of tags mapping to Nr2e1 in the Wt libraries showed a declining trend (Wt E13.5 vs. Wt E17.5, P = 0.06). This is in agreement with published and publicly-available expression results [18] (Allen Mouse Brain Atlas, http://www.brain-map.org/); where Nr2e1 expression has been observed as early as E8, peaks at E13, sharply decreases until E18, and is barely detectable in new-born brains [18]. Hence, at the time point of lowest Nr2e1 expression (E17.5) the LongSAGE approach was insufficiently sensitive to detect this latter gene transcript. Interestingly, our bioinformatics enrichment analysis included Nr2e1 in the list of genes with predicted NR2E1 binding sites within their promoter regions, adding support to previous observations proposing a self-regulating mechanism for Nr2e1 [22, 64].
When analysing large-scale transcriptome-profiling datasets, the overall level of expression is an important factor influencing the outcome of statistical significance. In our LongSAGE libraries, Pten and P21 (Cdkn1a), two direct targets of Nr2e1 [11, 14, 21], were expressed at low levels in the VZ/SVZ (total number of tags, Pten: E13.5, Wt 1, Nr2e1 frc/frc 5; E15.5, Wt 1, Nr2e1 frc/frc 1; and E17.5, Wt 0, Nr2e1 frc/frc 1; P21: E13.5, Wt 0, Nr2e1 frc/frc 0; E15.5, Wt 1, Nr2e1 frc/frc 3; and E17.5, Wt 1, Nr2e1 frc/frc 2) and thus did not reach significance in terms of differential expression between Wt vs. Nr2e1 frc/frc libraries. In contrast, Nestin, a common marker of proliferating neural progenitors, which was expressed at mid to high levels, was significantly down regulated in Nr2e1 frc/frc at E13.5 when compared to Wt (−7.3 fold, P < 0.05) (Fig. 5a, Additional file 1: Table S1). This correlated with the previously published observation of reduced numbers of Nestin-positive cells in the VZ of Nr2e1-null embryos at E14.5 [11]. In addition, our data suggests that the mechanism involves a direct-up regulation by Nr2e1 in Wt, as Nestin was found within the bioinformatics enrichment analysis genes with predicted NR2E1 binding sites. Another example of our expression results being supported by the literature is the basic helix-loop-helix (bHLH) gene Neurog2, which was significantly down regulated in Nr2e1 frc/frc embryos at both E13.5 and E15.5 when compared to Wt (E13.5, −2.8 fold, P < 0.001; E15.5, −5.5 fold, P < 0.001) (Fig. 5a, Additional file 1: Table S1). These results correlated with the previously published observations of reduced expression of Neurog2 in double mutants embryos for Pax6 and Nr2e1 in the rostral granular zone during neocortex development [65]. Disruptions in Neurog2 expression are also characteristic of alterations in the pallio-subpallial boundary observed in Nr2e1-null embryos [66]. Additionally, downstream candidate genes of the pathway regulated by Neurog2 (i.e. Neurod2, and Tbr1) were found differentially expressed in our LongSAGE comparison analysis, arguing in favour of a direct role for Nr2e1 in regulating this specific pathway during neocortex development (Fig. 5a, Additional file 1: Table S1) [65].
TFBS overrepresentation analysis revealed novel-candidate-NR2E1 co-interactors
Spatial-temporal gene expression is, in general, regulated by the dual ability of transcription factors to bind specific DNA sequences and to form complexes with other regulatory proteins. NR2E1 has previously been shown to mediate gene regulation with co-interacting partners; forming regulatory complexes that lead to either direct-target-gene repression or activation [14, 21, 25, 26]. Interestingly, nuclear receptors have also been shown to mediate gene regulation via interaction with other transcription factors as co regulators [64, 67, 68]. Based on our Spearman rank ordering results, we hypothesized that the striking difference in direction of correlation for differentially abundant tags between the E13.5-E15.5, E13.5-E17.5, and E15.5-E17.5 time points, was largely due to the presence of different Nr2e1 interacting partners at different times in development. To discover novel candidate co-interactors of Nr2e1, we designed a computational experiment to identify TFBS within the vicinity of the predicted NR2E1 binding sites for each differentially-regulated gene found in the GO term category “nervous system development”. The identified binding sites were then scored for their enrichment compared to a randomized list of genes, thereby generating both a Z and Fisher score. Potential TFBSs having a Z-score value > 10 and a Fisher score value < 0.01 were considered enriched and kept for further characterization as candidate-NR2E1 co-interactors (Table 2). We further ascertained the significance of our candidate-NR2E1 co-interactors list by performing analyses on random sets of 64 genes extracted from the initial list of 770 genes obtained through the oPOSSUM-NR2E1 binding motif interrogation step. Corresponding empirical P values based on the Z-scores and Fisher scores of each of the candidate-NR2E1 co-interactors were extracted from the random sets of 64 genes and are summarized in Additional file 4: Table S4.
The relevance of these enriched TFBS was also evaluated based on the expression pattern of their corresponding transcription factors. For this, we primarily used data from the Allen Mouse Brain Atlas (http://www.brain-map.org/) at three different time points (E13.5, E15.5, and E18.5), and included data from other publically-available resources as required (Table 2) [69].
The expression data and statistical scores obtained most strongly supported the biological relevance of SOX9 (Z-score: 16.97, empirical P value: < 0.001; Fisher score 1.58E-05, empirical P value: = 0.001); a member of the SRY-box family. Examination of our LongSAGE data revealed the presence of tags corresponding to Sox9 throughout the three different time points for both genotypes (data not shown). Additionally, Sox9 has been reported to function in neural-stem/progenitor-cell regulation; as does Nr2e1. Together these data support the hypothesis that SOX9 acts as a co-interactor of NR2E1 [70]. Thus, the differentially-regulated genes found in the GO term category “nervous system development”, and the number of predicted TFBSs for both NR2E1 and SOX9 in the promoter region of these genes, are presented in Table 3 (Additional file 2: Table S2). These 40 genes represent a rich resource for the biological examination of Nr2e1 downstream targets. Here we pursue the top candidate Lhx2, a LIM-homeobox transcription factor. Lhx2 had the highest number of predicted binding sites for both NR2E1 and SOX9; 35 and 13 respectively. Visualization of the predicted binding sites within the promoter region of Lhx2 revealed a clustered distribution that was located within highly conserved DNA (Fig. 6). Localization within conserved DNA further suggested a function for these binding sites throughout evolution. Evidence from the literature highlights a spatial-temporal dynamic role for Lhx2 in the developing forebrain. Early in development (E10.5-E11.5), Lhx2 has been shown to work as a fate determinant of cortical identity [71]. Later in development, distinct roles have been described for Lhx2 depending on the forebrain structures involved; including a role in regulating progenitor cell differentiation in neocortex development (E11.5-E13.5) and a role in the neurogenic to gliogenic switch in hippocampal development (E14.5-E15.5) [72, 73]. Thus, our data, combined with the literature, support Lhx2 as a direct target of co-regulation by NR2E1 and SOX9.

Lhx2 contains enriched clusters of NR2E1 and SOX9 binding sites in highly conserved regions. Presented is the ~17 kb of genomic sequence 5′ to, and surrounding, the Lhx2 transcription start site. NR2E1 binding sites are indicated in black, and SOX9 binding sites are indicated in red. NR2E1 binding site coordinates were derived from the position-weight matrix (PWM) described in the current manuscript; SOX9 binding sites coordinates were derived from the PWM stored in the JASPAR database (http://jaspar.binf.ku.dk/cgi-bin/jaspar_db.pl). Conservation, alignment of 100 vertebrate species from the University of California, Santa Cruz genome browser (UCSC: http://genome.ucsc.edu/); thick horizontal line, first exon of Lhx2; arrow, transcription start site and sense
Differential expression of the transcription factor Lhx2 validated our LongSAGE results
To expand our understanding of the relationship between Nr2e1 and Lhx2, and simultaneously further validate the results obtained from the LongSAGE tag libraries, we undertook two biological assays; one each in vitro and in vivo. First we retrieved with DiscoverySpace 4.0 the LongSAGE tag sequence mapping to Lhx2, and the corrected number of tags from each library (Fig. 7a). This showed that Lhx2 levels were significantly increased in Nr2e1 frc/frc libraries at two different time points (E13.5, and E15.5).

Validation of the differential abundance of Lhx2 LongSAGE tags in Wt vs. Nr2e1 frc/frc embryos. a The tag count results, mapping to Lhx2 at the three different embryonic days, are presented. Columns one to three present the tag sequence, accession number, and gene symbol corresponding to Lhx2; columns four to six, the corrected tag numbers recovered from DiscoverySpace in both Wt and Nr2e1 frc/frc embryos at each time point (E13.5, 15.5, and 17.5); column seven, the fold change between the tag numbers corresponding to Lhx2 found in Wt and Nr2e1 frc/frc embryos at each time point; and column eight, the associated P values obtained using the Audic-Claverie statistical method. According to this approach, Lhx2 expression level is significantly upregulated at both E13.5 and E15.5 in Nr2e1 frc/frc embryos. b Wt embryonic stem cells (ESC) submitted to a neurogenic differentiation protocol demonstrated expression of Nr2e1 at 12 days of differentiation (d12) whereas, as expected, Nr2e1 frc/frc ESC did not express Nr2e1 (*, P < 0.001). c Quantitative RT-PCR reveals that the Lhx2 mRNA level is upregulated by ~3.6 fold in Nr2e1 frc/frc ESC at d12 compared to Wt ESC (*, P < 0.01). d Immunofluorescence using an anti-Lhx2 antibody (green) demonstrated a similar expression pattern for the Lhx2 protein along the ventricular/subventricular zone (VZ/SVZ) of the lateral telencephalon in E15.5 Nr2e1 frc/frc embryos compared to Wt. White arrows, medial pallium; red arrows, dorsal pallium; scale bar, 200 μm. e Lhx2 protein level was increased by ~1.3 fold along the VZ/SVZ of the lateral telencephalon in E15.5 Nr2e1 frc/frc embryos compared to Wt (*, P < 0.05). (c), (d), (e) Sample Student’s t-test were performed, N = 3; error bars, standard error of the mean
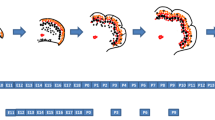
For in vitro studies, we chose a method of neurogenesis from adherent-monoculture of ESC, which sequentially mimics the development of cortical neurons over the course of 21 days of differentiation [74]. In this system: neural induction starts at day 0 (d0) of differentiation; neurogenesis starts at d6 with the generation of subplate neurons and deep layer neurons between d7 and d9, followed by upper cortical neurons around d12; finally there is a wave of gliogenesis by d21 [75]. The formation of subplate neurons corresponds to in vivo E10.5-E13.5, deep layer neurons to E11.4-E14.5, and upper cortical neurons to E13.5-E16.5 [75]. Hence, this culture system encompass the key time periods for the function of Nr2e1 in brain development. Using this method, we first detected Nr2e1 expression at d6, which then increased and peaked at d12 (data not shown). Further investigation using qRT-PCR at this latter time point not only showed a significant difference between Wt and Nr2e1 frc/frc cells for the Nr2e1 gene (Fig. 7b), but also demonstrated a significant increase in expression in Nr2e1 frc/frc cells when compared to Wt cells for the Lhx2 gene (P < 0.01) (Fig. 7c) [75]. This result was consistent with a model of Lhx2 being a direct target of, and repressed by, Nr2e1.
For in vivo studies, we examined the expression pattern of the Lhx2 protein by immunofluorescence in Wt and Nr2e1 frc/frc E15.5 embryos. The results showed similar Lhx2 protein localization when comparing Wt and Nr2e1 frc/frc embryos; along the VZ/SVZ of the developing forebrain. Furthermore, for both Wt and Nr2e1 frc/frc, expression levels varied from high in the medial pallium to low in the dorsal pallium (Fig. 7d). However, relative quantification of Lhx2 between Wt and Nr2e1 frc/frc, along the VZ/SVZ of the dorsal-lateral telencephalon, revealed a significant increase of Lhx2 protein in the Nr2e1 frc/frc embryos when compared to Wt (P < 0.01) (Fig. 7e). Thus, the significant increase at the mRNA level for the Lhx2 gene results in a significant increase at the protein level along the VZ/SVZ of the dorsal-lateral telencephalon at E15.5. These data add further support to the hypothesis that Nr2e1 directly-negatively regulates Lhx2 expression in the dorsal-lateral telencephalon during development.
Conclusions
In this work, we have generated a list of 1279 genes that are differentially expressed in response to altered Nr2e1 expression during in vivo neocortex development; this list was a critical part of our own studies, but is also an important resource for others (Additional file 1: Table and Additional file 2: Table S2). To create this list, we profiled the transcriptomes of Wt and Nr2e1 frc/frc embryos by generating LongSAGE libraries through LCM of the VZ/SVZ from the dorsal-lateral telencephalon. To further focus the work on the role of Nr2e1 during neocortex development, we chose two time points that spanned the early to mid-neurogenic stages (E13.5, E15.5), and one time point corresponding to the early switch from neurogenesis to gliogenesis (E17.5). Thus, from six LongSAGE libraries we identified 1279 candidate genes comprising both direct and indirect targets of Nr2e1 during neocortex development. This list can now be mined by us, and many other groups for the anticipated numerous co-suppressors, co-activators, and direct targets making up the molecular mechanisms of the nuclear-receptor transcription-factor Nr2e1.
We have further refined this list of 1279 differentially expressed genes, culminating in a focused list of 64 candidate direct-targets of NR2E1 binding during nervous system development, for our own studies and as a resource for others (Additional file 2: Table S2). This was accomplished by performing two sequential analyses; 1) a TFBSs prediction analysis, using oPOSSUM, to identify novel direct targets of Nr2e1, and 2) a GO term overrepresentation analysis, to extract biological meaning from the latter generated list. This procedure included the generation of a novel NR2E1 PWM based on available information from the literature (Additional file 3: Table S3); the derived matrix and logo are provided (Fig. 4a). We used this matrix, in combination with the LongSAGE results, in a bioinformatic experiment to identify novel direct-target genes of Nr2e1. The resulting list of GO terms coming from this analysis (Table 1) contained genes differentially expressed, and predicted to contain NR2E1 binding sites within their promoter regions (Additional file 2: Table S2). The GO term category “nervous system development” contained 64 such genes (Fig. 5a, Additional file 2: Table S2); a list that was used in subsequent analyses.
Our approach suggested distinct roles for Nr2e1 during different neocortex developmental stages. Analyses performed on the differential-tag ratio for the 1279 Refseq accession numbers of differentially-regulated genes retrieved from the Wt and Nr2e1 frc/.frc libraries, revealed a positive correlation of the differential abundance at E13.5 and E15.5, whereas a negative correlation was obtained when comparing the two previous time points to E17.5 (Fig. 3). Thus, the differential-tag ratios found at E13.5 and E15.5 were more similar to each other than when compared to E17.5. From E13.5 to E17.5, the neocortex undergoes drastic changes, including the formation of the SVZ, a layer of cells being seeded by the VZ, and a progressive switch from neurogenesis to gliogenesis. Our results indicate that Nr2e1 has a more similar effect in the early stages of neurogenesis (E13.5 and E15.5) compared to later stages when the switch from neurogenesis to gliogenesis occurs. The mechanism driving these changes may depend on the level of Nr2e1 expression, which peaks at E13.5 and gradually decreases until birth [18].
The SOX9 transcription factor may be an important co-interactor of NR2E1 in regulating numerous target genes during nervous system development. A co-factor analysis revealed enrichment for binding sites predicted to be bound by SOX9 within the vicinity of the predicted NR2E1 binding sites (Table 2); results that remained significant after calculating empirical P values on our list of candidate co-interactors (Additional file 4: Table S4). In addition, the spatial, temporal, and strength of expression of Sox9 strongly supported a biological relationship with Nr2e1 [70]. Interestingly, others have shown that Sox9 may be involved in the acquisition of gliogenic competence of neural stem/progenitor cells during central nervous system (CNS) development [76]. Together these data suggest that co-interaction between the SOX9 transcription factor and NR2E1 may regulate the expression of 40 of the 64 genes involved in nervous system development (Additional file 2: Table S2).
The Sox family of transcription factors may generally be important co-interactors of Nr2e1 in regulating target genes during nervous system development. This family comprises 20 genes with several members expressed in neural stem/progenitor cells of the CNS, and peripheral nervous system [77–79]. They have been shown to act as either transcriptional activators or repressors by binding to similar (A/T)(A/T)CAA(A/T)G DNA motifs [78]. Recently, one of these family members, Sox2, has been shown to form a regulatory complex with Nr2e1 in adult NSC [64]. Interestingly, our co-factor TFBSs analysis revealed enrichment for the presence of associated binding sites not just for SOX9, but also three additional Sox family members (Sox17, Sox5, and SRY) within the vicinity of predicted NR2E1 binding sites for genes of the “nervous system development” GO term category (Table 2). These additional Sox family members also showed expression overlap with Nr2e1, and at least one of these members, Sox5 has been shown to bind to Fezf2-conserved-enhancer sequences, resulting in a direct repression of Fezf2 in neocortex development [80]. LongSAGE tags mapping to Sox5 were found in our libraries and Fezf2 was found significantly upregulated in the Nr2e1 frc/frc library at E15.5 (7.4 fold, P < 0.05, Fig. 5a). Hence our data suggests a specific testable hypothesis by which Nr2e1 potentially regulates Fezf2 expression through its interaction via the Sox5 protein in neocortex development. In conclusion, our data supports the hypothesis that generally the Sox family members play an important role as co-interactors of NR2E1.
Lhx2 may be an important direct-target gene of Nr2e1, with SOX9 as a co-interactor. In Nr2e1-null embryos, premature neurogenesis has been reported to occur from E9.5 to E14.5 in both the dorsal and ventral telencephalon [9]. Overexpression of Lhx2 has been reported to prolong neurogenesis in hippocampal development, resulting in generation of neurons from progenitors that would normally produce astrocytes [72]. Additionally, conditional inactivation of Lhx2 in neocortical development affects the fate of PC, resulting in a phenotype highly similar to that observed in Nr2e1-null embryos; with a reduction in the number of PC populating the VZ and premature neurogenesis in the neocortex of Lhx2-null embryos [73]. This latter phenomenon appears to involve the notch signalling pathway, with a downregulation of Hes1 being observed along the VZ of Lhx2-null embryos and aberrant expression of the Notch encoding gene along the medial to lateral dorsal telencephalon [73]. Notch pathway genes such as Notch1, Hes5, and Hes6 were also found differentially regulated in the Nr2e1 frc/frc library when compared to Wt in our LongSAGE analysis (Notch1, E13.5, −6.8 fold, P < 0.05; Hes5, E13.5, −6.8 fold, P < 0.01; E17.5, −10.3 fold, P < 0.01; Hes6, E13.5, 9 fold, P < 0.001) (Fig. 5a, Additional file 1: Table S1 and Additional file 2: Table S2). Protein regulatory networks in NSC have been demonstrated to be highly dosage dependent. For instance, phenotypic analyses of Pax6 in gain- or loss-of-function mutant cortices have shown similar phenotypic outcome, with both more or less of the protein resulting in increased neurogenesis throughout development [81]. Hence, akin to Pax6, our current results highlight a testable hypothesis in which premature neurogenesis observed in Nr2e1 frc/frc embryos [9] could be due to the upregulation of Lhx2 protein along the VZ/SVZ of the dorsal telencephalon; a phenomenon that most likely includes the concerted effect of deregulation of other Notch pathway encoding genes. Our analysis also predicted that the transcription factor pathway regulated by NR2E1 involves interaction with SOX9, which has been shown to be involved in the acquisition of gliogenic competence of neural stem/progenitor cells during CNS development [76]. Hence, our results highlight yet another testable hypothesis for the discovery of a possible novel pathway by which Nr2e1 regulates neurogenesis, which includes Lhx2 as one of the direct-target genes, and SOX9 as a co-interactor.
Methods
Ethics statement
All procedures involving animals were in accordance with the Canadian Council on Animal Care and UBC Animal Care Committee (Protocol A11-0412).
LongSAGE libraries generation
Libraries were generated from tissue samples obtained by LCM of dorsal VZ/SVZ from the telencephalon of wild-type (Wt) and Nr2e1 frc/frc embryos at E13.5, E15.5, and E17.5 as described by us previously [31]. Briefly, one embryo per genotype at each developmental time point was sectioned at 20 μm thickness to generate the tissue samples. Sections from each embryo underwent LCM, and the isolated tissue was pooled and RNA extracted using an RNeasy Micro Kit (Qiagen Inc., Mississauga, Ontario, Canada). The LongSAGE-lite method was used to construct the libraries using 15 to 86 ng of high quality RNA from each embryo [31, 37, 82]. Each library was sequenced to a depth of >100,000 raw tags and the processed data is accessible on the Mouse Atlas of Gene Expression project website (http://www.mouseatlas.org/) and the NIH SAGEmap data repository http://www.ncbi.nlm.nih.gov/projects/SAGE/) [83].
LongSAGE data analysis
LongSAGE libraries were analysed using the DiscoverySpace 4.0 application (http://www.bcgsc.ca/platform/bioinfo/software/ds) [44]. The library data were electronically filtered based on procedures previously described by us [45, 84]. Briefly, duplicated ditags (identical copies of a ditag) and singletons (tags counted only once) were retained for analysis. Sequence data were filtered for bad tags (tags with one N-base call), and linker-derived tags (artefact tags). Only tags with a sequence quality factor greater than 99 % were included in the analysis. Sequence tag comparisons between Wt and Nr2e1 frc/frc libraries were performed and a P-value cutoff < 0.05 using the Audic-Claverie statistical method was used [46]. LongSAGE tags exhibiting differential expression levels were mapped to transcripts in the NCBI Reference Sequence (Refseq) collection (version 52, released March 8th 2012) and Ensembl gene collection (version 66, released February 2012).
NR2E1 binding site profile construction
No position-weight matrix (PWM) was available in public databases to model NR2E1 transcription factor binding site (TFBS) specificity. Thus, a literature review was conducted and the sequences reported to be bound by NR2E1 were compiled. Next, the MEME motif discovery tool (http://meme-suite.org/) was applied, with default parameter settings, to identify a DNA sequence pattern within the data [85].
oPOSSUM promoter analysis
A pooled list of RefSeq accession numbers for transcripts exhibiting differential expression between Wt and Nr2e1 frc/frc genotypes, at least at one of the three different time points, was subjected to an oPOSSUM TFBS analysis. The oPOSSUM software was run using default settings with both the constructed NR2E1 PWM and the JASPAR core vertebrate PWM collection (http://www.cisreg.ca/oPOSSUM/) [39, 40]. Briefly, for each Refseq accession number, oPOSSUM automatically retrieved the genomic DNA sequences around annotated transcription start sites (TSS) in Ensembl (plus 5000 bp of both upstream and downstream sequence), performed an alignment of the orthologous sequences (human to mouse), and extracted non-coding DNA sequences that are conserved above a predefined threshold (default: top 10 % of conserved regions, minimum conservation 70 %). oPOSSUM results include the positions of predicted TFBSs, and the scores of the sites.
GO term enrichment analysis
Refseq accession numbers for those genes predicted to contain NR2E1 binding sites in the oPOSSUM database were submitted to the DAVID service (http://david.abcc.ncifcrf.gov/summary.jsp) for GO term annotation enrichment analysis [50, 51]. The Refseq identifiers were converted to DAVID identifiers (IDs), using the DAVID knowledgebase [59]. GO biological-process term enrichment was assessed relative to the entire set of mouse genes as provided by DAVID. Results were filtered to exclude those enriched GO terms associated with 2 or less submitted genes. A significant P-value threshold was applied using a multiple testing correction (Bonferroni, P value < 0.05).
Clustering-correlation
Expression clustering was performed on the differential-tag ratios of the initial list of 1279 differentially expressed genes, and the genes annotated within the enriched GO term “nervous system development” category using the Gene Cluster software [47]. Hierarchical clustering was performed on both the gene list and the embryonic stages using Spearman correlation with complete linkage clustering. Tag counts were corrected to account for library sizes; (observed tag counts/total useful tags) X 100,000, and tags having a count value of “0” (no expressed tags) were adjusted to a value of “1” for fold change calculations only. Spearman rank correlation analyses on the differential-tag ratios were performed using STATISTICA 12.0 (Statsoft, Inc., Tulsa, OK, USA). For these latter analyses, genes were considered as valid when their differential-tag ratios between Wt and Nr2e1 frc/frc were not equal to zero.
Co-factor TFBS enrichment analysis and transcription factor candidate evaluation
A customized bioinformatics analysis, based on the oPOSSUM combination site analysis feature, was performed to identify TFBS patterns that were significantly enriched in the vicinity of predicted NR2E1 binding sites for the 64 candidate genes found in the “nervous system development” GO term category. Sites within 100 bp of predicted NR2E1 binding sites were retrieved from the oPOSSUM database. Sites overlapping an NR2E1 motif were excluded. Both the NR2E1 sites and proximal sites were subject to the default oPOSSUM parameters of conservation level (top 10 % of conserved regions with a minimum percentage identity of 70 %), threshold level (default matrix score threshold of 80 %), and search region level (5000 bp upstream and downstream of TSS). The analysis was performed against a background of 500 genes selected randomly from the oPOSSUM database. Over-representation results were considered significant based on a Z score (>10) and a Fisher score (<0.01) according to the literature [40]. To further validate the significance of these results, additional oPOSSUM co-factor analyses were performed on 1000 sets of 64 genes selected randomly from the list of 770 genes enriched for GO terms, using the same analysis parameters and the same set of 500 background genes as the one described above for the “nervous system development” gene set. The significance of the Z and Fisher score for each of the co-factors was determined by empirical P value, computed as “n/N” where “n” is the number of times the Z and Fisher score from set of the random trials for the co-factor was more significant than the Z and Fisher score from the “nervous system development” set for that co-factor, divided by the total number “N” of random trials (in this case 1000). These results are shown in Additional file 4: Table S4.
Transcription factors with enriched binding site predictions were additionally assessed for their expression pattern at E13.5, 15.5, and 18.5 using images from the Allen Mouse Brain Atlas (ABA, http://www.brain-map.org/) [69]. Expression results from transcription factors with enriched binding site predictions that were unavailable from the ABA were evaluated using other publicly available resources; Eurexpress (http://www.eurexpress.org/ee/), GenePaint (http://www.genepaint.org/Frameset.html), and the primary literature [86, 87]. The expression pattern was summarized according to the specificity and strength of the expression along the VZ/SVZ, and other forebrain structures. The relevance of the expression pattern for each transcription factor was scored as absent (−), low (+), moderate (++), or high (+++); where absence of VZ/SVZ expression, or ubiquitous expression in the entire embryo forebrain, was scored as +, whereas strong and restricted expression along the VZ/SVZ was scored as +++. The transcription factor having a high score (+++) was retained as the most interesting candidate. For the highest-scoring transcription factors we cross-validated the expression pattern by looking for the number of corresponding tags in the LongSAGE libraries.
Embryos preparation
Timed-pregnant mice were euthanized by cervical dislocation, and embryos at E15.5 were dissected and fixed in 4 % paraformaldehyde (PFA) with 0.1 M PO buffer (0.1 M Na2HPO4, pH 8.0) for 6 h at 4 °C. The embryos were then cryoprotected as described in the literature, and embedded in optimal cutting temperature (OCT) compound (Tissue-tek, Torrance, California, USA) on dry ice [11]. Embryos were sectioned at 20 μm using a Cryo Star HM550 cryostat (MICROM International, Kalamazoo, Michigan, USA), and mounted for immunofluorescence.
Immunofluorescence and imaging analysis
For antibody staining, 20 μm sagittal cryosections from embryos were rehydrated in sequential washes of 1x phosphate buffered saline (PBS), permeabilized in PBS with 0.3 % triton, and blocked with 1 % BSA (bovine serum albumin) in PBS triton 0.3 % for 1 h at room temperature. Goat anti-Lhx2 primary antibody (1:1000) (Santa Cruz, Dallas, TX, USA, sc-19344) was incubated overnight at 4 °C. Rabbit anti-goat Alexa 488 (1:1000) (Invitrogen, Burlington, Ontario, Canada, A21222) was incubated for 2 h at room temperature in the dark. Tiled images were retrieved with an Olympus BX61 motorized fluorescence microscope at 20X magnification (Olympus America Inc., Center Valley, Pennsylvania, USA). Intensity quantification was performed using Image-Pro (Media Cybernetics Inc., Bethesda, Maryland, USA). The relative intensity level of Lhx2 was calculated as described in the literature [88]. Briefly, the sum of the signal intensity was divided by the area selected and multiplied by the thickness of the section and the number of sections. A background correction was applied using the signal intensity resulting from Hoechst staining for each sections quantified. A total of 28 different sections were assessed on six embryos, three different animals for each genotype (Wt and Nr2e1 frc/frc). All values represent the mean ± standard error of the mean (SEM). Statistical analysis was performed using Student’s t-test.
Embryonic stem cells culture
ESC from Wt and Nr2e1 frc/frc blastocysts were derived, and maintained in culture as described in the literature [89]. The two cell lines used were mEMS1239 (B6129F1-Nr2e1 frc/frc, Hprt1 b-m3/Y), and mEMS1271 (B6129F1-Nr2e1 +/+, Hprt1 b-m3/Y).
The ESC differentiation procedure involved the use of an adapted method of neurogenesis from adherent monoculture [74, 75]. Briefly, the cells were seeded at low density (~10,000 cells/mm2) on gelatin coated dishes, in a chemically defined medium exempt of cyclopamine, and maintained in culture for 12 days (fresh media every two days). RNA aliquots were prepared on day 12 and were used for quantitative RT-PCR (qRT-PCR).
Quantitative RT-PCR
RNA from ESC grown in an adapted method of neurogenesis from adherent monoculture, collected on day 12, was extracted using Qiagen RNA Mini Plus kit (Qiagen Inc., Mississauga, Ontario, Canada). RNA was treated with Qiagen DNase kit (Qiagen Inc., Mississauga, Ontario, Canada), and cDNA was generated using Superscript III Master Mix kit (Invitrogen, Burlington, Ontario, Canada). cDNA quantification was performed using ABI Taqman® assays specifically designed for Nr2e1 (Mm00455855_m1), and Lhx2 (Mm00839783_m1) (Applied Biosystems Inc., Foster city, USA). The 7500 fast real-time PCR system and Taqman® fast universal PCR Master Mix were used (Applied Biosystems Inc., Foster city, USA). The cycle threshold (Ct) value was defined as the number of cycles required for the fluorescent signal to cross a threshold above the background signal, and is inversely proportional to the amount of target cDNA. All values represent the mean ± SEM. Statistical analysis was performed using Student’s t-test.
Availability of supporting data
LongSAGE processed data is accessible on the Mouse Atlas of Gene Expression project website (http://www.mouseatlas.org/) and the NIH SAGEmap data repository (http://www.ncbi.nlm.nih.gov/projects/SAGE/). All additional supporting data is included as additional files.
Abbreviations
- ABA:
-
Allen Mouse Brain Atlas
- BSA:
-
Bovine Serum Albumin
- CNS:
-
Central Nervous System
- ESC:
-
Embryonic Stem Cells
- GO:
-
terms Gene Ontology terms
- LCM:
-
Laser Capture Microdissection
- NSC:
-
Neural Stem Cells
- OCT:
-
Optimal Cutting Temperature
- PBS:
-
Phosphate Buffered Saline
- PC:
-
Progenitor Cells
- PFA:
-
Paraformaldehyde
- PO:
-
Na2HPO4, pH 8.0
- PWM:
-
Position Weight Matrix
- TFBS:
-
Transcription Factor Binding Site
- SAGE:
-
Serial Analysis of Gene Expression
- SELEX:
-
Systematic Evolution of Ligands by Exponential Enrichment
- TSS:
-
Transcription Start Sites
- SVZ:
-
Subventricular Zone
- UCSC:
-
University of California, Santa Cruz
- UTR:
-
Untranslated Region
- VZ:
-
Ventricular Zone
- Wt:
-
Wild-Type
References
Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–6.
Angevine Jr JB, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–8.
de Carlos JA, Lopez-Mascaraque L, Valverde F. Dynamics of cell migration from the lateral ganglionic eminence in the rat. J Neurosci. 1996;16:6146–56.
Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–50.
Tamamaki N, Fujimori KE, Takauji R. Origin and route of tangentially migrating neurons in the developing neocortical intermediate zone. J Neurosci. 1997;17:8313–23.
Bayer SA, Altman J. Neocortical development. New York: Raven; 1991.
Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–69.
Job C, Tan SS. Constructing the mammalian neocortex: the role of intrinsic factors. Dev Biol. 2003;257:221–32.
Roy K, Kuznicki K, Wu Q, Sun Z, Bock D, Schutz G, et al. The Tlx gene regulates the timing of neurogenesis in the cortex. J Neurosci. 2004;24:8333–45.
Land PW, Monaghan AP. Expression of the transcription factor, tailless, is required for formation of superficial cortical layers. Cereb Cortex. 2003;13:921–31.
Li W, Sun G, Yang S, Qu Q, Nakashima K, Shi Y. Nuclear receptor TLX regulates cell cycle progression in neural stem cells of the developing brain. Mol Endocrinol. 2008;22:56–64.
Shi Y, Chichung Lie D, Taupin P, Nakashima K, Ray J, Yu RT, et al. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature. 2004;427:78–83.
Zhang CL, Zou Y, He W, Gage FH, Evans RM. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature. 2008;451:1004–7.
Sun G, Yu RT, Evans RM, Shi Y. Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc Natl Acad Sci U S A. 2007;104:15282–7.
Biddie SC, John S. Minireview: conversing with chromatin: the language of nuclear receptors. Mol Endocrinol. 2014;28:3–15.
Lonard DM, O’Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700.
Lonard DM, Lanz RB, O’Malley BW. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28:575–87.
Monaghan AP, Grau E, Bock D, Schutz G. The mouse homolog of the orphan nuclear receptor tailless is expressed in the developing forebrain. Development. 1995;121:839–53.
Monaghan AP, Bock D, Gass P, Schwager A, Wolfer DP, Lipp HP, et al. Defective limbic system in mice lacking the tailless gene. Nature. 1997;390:515–7.
Young KA, Berry ML, Mahaffey CL, Saionz JR, Hawes NL, Chang B, et al. Fierce: a new mouse deletion of Nr2e1; violent behaviour and ocular abnormalities are background-dependent. Behav Brain Res. 2002;132:145–58.
Yokoyama A, Takezawa S, Schule R, Kitagawa H, Kato S. Transrepressive function of TLX requires the histone demethylase LSD1. Mol Cell Biol. 2008;28:3995–4003.
Zhao C, Sun G, Li S, Shi Y. A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat Struct Mol Biol. 2009;16:365–71.
Sun G, Ye P, Murai K, Lang MF, Li S, Zhang H, et al. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat Commun. 2011;2:529.
Zhao C, Sun G, Ye P, Li S, Shi Y. MicroRNA let-7d regulates the TLX/microRNA-9 cascade to control neural cell fate and neurogenesis. Sci Rep. 2013;3:1329.
Iwahara N, Hisahara S, Hayashi T, Horio Y. Transcriptional activation of NAD + −dependent protein deacetylase SIRT1 by nuclear receptor TLX. Biochem Biophys Res Commun. 2009;386:671–5.
Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, Shimohama S, et al. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:15599–604.
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–7.
Hanriot L, Keime C, Gay N, Faure C, Dossat C, Wincker P, et al. A combination of LongSAGE with Solexa sequencing is well suited to explore the depth and the complexity of transcriptome. BMC Genomics. 2008;9:418.
Gunnersen JM, Augustine C, Spirkoska V, Kim M, Brown M, Tan SS. Global analysis of gene expression patterns in developing mouse neocortex using serial analysis of gene expression. Mol Cell Neurosci. 2002;19:560–73.
Koehl A, Schmidt N, Rieger A, Pilgram SM, Letunic I, Bork P, et al. Gene expression profiling of the rat superior olivary complex using serial analysis of gene expression. Eur J Neurosci. 2004;20:3244–58.
D’Souza CA, Chopra V, Varhol R, Xie YY, Bohacec S, Zhao Y, et al. Identification of a set of genes showing regionally enriched expression in the mouse brain. BMC Neurosci. 2008;9:66.
Popesco MC, Frostholm A, Rejniak K, Rotter A. Digital transcriptome analysis in the aging cerebellum. Ann N Y Acad Sci. 2004;1019:58–63.
Ouchi Y, Kubota Y, Ito C. Serial analysis of gene expression in methamphetamine- and phencyclidine-treated rodent cerebral cortices: are there common mechanisms? Ann N Y Acad Sci. 2004;1025:57–61.
Guipponi M, Li QX, Hyde L, Beissbarth T, Smyth GK, Masters CL, et al. SAGE analysis of genes differentially expressed in presymptomatic TgSOD1G93A transgenic mice identified cellular processes involved in early stage of ALS pathology. J Mol Neurosci. 2010;41:172–82.
Mazarei G, Neal SJ, Becanovic K, Luthi-Carter R, Simpson EM, Leavitt BR. Expression analysis of novel striatal-enriched genes in Huntington disease. Hum Mol Genet. 2010;19:609–22.
George AJ, Gordon L, Beissbarth T, Koukoulas I, Holsinger RM, Perreau V, et al. A serial analysis of gene expression profile of the Alzheimer’s disease Tg2576 mouse model. Neurotox Res. 2010;17:360–79.
Peters DG, Kassam AB, Yonas H, O’Hare EH, Ferrell RE, Brufsky AM. Comprehensive transcript analysis in small quantities of mRNA by SAGE-lite. Nucleic Acids Res. 1999;27:e39.
Wahl MB, Heinzmann U, Imai K. LongSAGE analysis significantly improves genome annotation: identifications of novel genes and alternative transcripts in the mouse. Bioinformatics. 2005;21:1393–400.
Ho Sui SJ, Fulton DL, Arenillas DJ, Kwon AT, Wasserman WW. oPOSSUM: integrated tools for analysis of regulatory motif over-representation. Nucleic Acids Res. 2007;35:W245–52.
Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, et al. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 2005;33:3154–64.
Das MK, Dai HK. A survey of DNA motif finding algorithms. BMC Bioinformatics. 2007;8 Suppl 7:S21.
Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–87.
Stenman JM, Wang B, Campbell K. Tlx controls proliferation and patterning of lateral telencephalic progenitor domains. J Neurosci. 2003;23:10568–76.
Robertson N, Oveisi-Fordorei M, Zuyderduyn SD, Varhol RJ, Fjell C, Marra M, et al. DiscoverySpace: an interactive data analysis application. Genome Biol. 2007;8:R6.
Romanuik TL, Wang G, Holt RA, Jones SJ, Marra MA, Sadar MD. Identification of novel androgen-responsive genes by sequencing of LongSAGE libraries. BMC Genomics. 2009;10:476.
Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7:986–95.
de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–4.
Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20:3246–8.
Portales-Casamar E, Arenillas D, Lim J, Swanson MI, Jiang S, McCallum A, et al. The PAZAR database of gene regulatory information coupled to the ORCA toolkit for the study of regulatory sequences. Nucleic Acids Res. 2009;37:D54–60.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Dennis Jr G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3.
Portales-Casamar E, Kirov S, Lim J, Lithwick S, Swanson MI, Ticoll A, et al. PAZAR: a framework for collection and dissemination of cis-regulatory sequence annotation. Genome Biol. 2007;8:R207.
Yu RT, Chiang MY, Tanabe T, Kobayashi M, Yasuda K, Evans RM, et al. The orphan nuclear receptor Tlx regulates Pax2 and is essential for vision. Proc Natl Acad Sci U S A. 2000;97:2621–5.
Zhang CL, Zou Y, Yu RT, Gage FH, Evans RM. Nuclear receptor TLX prevents retinal dystrophy and recruits the corepressor atrophin1. Genes Dev. 2006;20:1308–20.
Qu Q, Sun G, Li W, Yang S, Ye P, Zhao C, et al. Orphan nuclear receptor TLX activates Wnt/beta-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nat Cell Biol. 2010;12:31–40. sup pp 31–39.
Yu RT, McKeown M, Evans RM, Umesono K. Relationship between Drosophila gap gene tailless and a vertebrate nuclear receptor Tlx. Nature. 1994;370:375–9.
Hubisz MJ, Pollard KS, Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform. 2011;12:41–51.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50.
Sherman BT, Huang da W, Tan Q, Guo Y, Bour S, Liu D, et al. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007;8:426.
da Huang W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–75.
Hosack DA, Dennis Jr G, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70.
da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13.
Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–8.
Shimozaki K, Zhang CL, Suh H, Denli AM, Evans RM, Gage FH. SRY-box-containing gene 2 regulation of nuclear receptor tailless (Tlx) transcription in adult neural stem cells. J Biol Chem. 2012;287:5969–78.
Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, et al. Sequential phases of cortical specification involve Neurogenin-dependent and -independent pathways. Embo J. 2004;23:2892–902.
Stenman J, Yu RT, Evans RM, Campbell K. Tlx and Pax6 co-operate genetically to establish the pallio-subpallial boundary in the embryonic mouse telencephalon. Development. 2003;130:1113–22.
Peng GH, Ahmad O, Ahmad F, Liu J, Chen S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum Mol Genet. 2005;14:747–64.
Aranguren XL, Beerens M, Coppiello G, Wiese C, Vandersmissen I, Lo Nigro A, et al. COUP-TFII orchestrates venous and lymphatic endothelial identity by homo- or hetero-dimerisation with PROX1. J Cell Sci. 2013;126:1164–75.
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–76.
Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, Gomez Gaviro MV, et al. SOX9 induces and maintains neural stem cells. Nat Neurosci. 2010;13:1181–9.
Chou SJ, Perez-Garcia CG, Kroll TT, O’Leary DD. Lhx2 specifies regional fate in Emx1 lineage of telencephalic progenitors generating cerebral cortex. Nat Neurosci. 2009;12:1381–9.
Subramanian L, Sarkar A, Shetty AS, Muralidharan B, Padmanabhan H, Piper M, et al. Transcription factor Lhx2 is necessary and sufficient to suppress astrogliogenesis and promote neurogenesis in the developing hippocampus. Proc Natl Acad Sci U S A. 2011;108:E265–74.
Chou SJ, O’Leary DD. Role for Lhx2 in corticogenesis through regulation of progenitor differentiation. Mol Cell Neurosci. 2013;56:1–9.
Gaspard N, Bouschet T, Herpoel A, Naeije G, van den Ameele J, Vanderhaeghen P. Generation of cortical neurons from mouse embryonic stem cells. Nat Protoc. 2009;4:1454–63.
Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, et al. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature. 2008;455:351–7.
Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 2003;17:1677–89.
Castillo SD, Sanchez-Cespedes M. The SOX family of genes in cancer development: biological relevance and opportunities for therapy. Expert Opin Ther Targets. 2012;16:903–19.
Kiefer JC. Back to basics: sox genes. Dev Dyn. 2007;236:2356–66.
Pevny L, Placzek M. SOX genes and neural progenitor identity. Curr Opin Neurobiol. 2005;15:7–13.
Kwan KY, Lam MM, Krsnik Z, Kawasawa YI, Lefebvre V, Sestan N. SOX5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. Proc Natl Acad Sci U S A. 2008;105:16021–6.
Sansom SN, Griffiths DS, Faedo A, Kleinjan DJ, Ruan Y, Smith J, et al. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009;5:e1000511.
Khattra J, Delaney AD, Zhao Y, Siddiqui A, Asano J, McDonald H, et al. Large-scale production of SAGE libraries from microdissected tissues, flow-sorted cells, and cell lines. Genome Res. 2007;17:108–16.
Lash AE, Tolstoshev CM, Wagner L, Schuler GD, Strausberg RL, Riggins GJ, et al. SAGEmap: a public gene expression resource. Genome Res. 2000;10:1051–60.
Siddiqui AS, Khattra J, Delaney AD, Zhao Y, Astell C, Asano J, et al. A mouse atlas of gene expression: large-scale digital gene-expression profiles from precisely defined developing C57BL/6 J mouse tissues and cells. Proc Natl Acad Sci U S A. 2005;102:18485–90.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8.
Visel A, Thaller C, Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32:D552–6.
Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011;9:e1000582.
Singaraja RR, Huang K, Sanders SS, Milnerwood AJ, Hines R, Lerch JP, et al. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet. 2011;20:3899–909.
Yang GS, Banks KG, Bonaguro RJ, Wilson G, Dreolini L, de Leeuw CN, et al. Next generation tools for high-throughput promoter and expression analysis employing single-copy knock-ins at the Hprt1 locus. Genomics. 2009;93:196–204.
Acknowledgements
We thank the entire Mouse Atlas of Gene Expression Project members; with special thanks to Dr. Robert A. Holt and the Genome Science Centre Sequencing Team for their work on the LongSAGE libraries. We also thank all the Pleiades Promoter Project members. Lastly, we thank Dr. Charles N. de Leeuw and Katrina Bepple for aid in manuscript preparation, as well as Marina Campbell for administrative assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SJMJ, MAM, and EMS initiated the project. YYX and SB performed the laser capture microdissections. SJMJ and MAM oversaw the LongSAGE libraries generation. KGB, RJB, and SHW derived the ESC from Wt and Nr2e1 frc/frc blastocysts. JFS and XCD developed the ESC differentiation procedure. JFS, DA, XCD, EMS, and WWW designed the NR2E1 position weight matrix. JFS, DA, EMS, and WWW designed the bioinformatics pipeline. JFS and DA performed the bioinformatics analyses. JFS performed the immunofluorescences and quantitative RT-PCR experiments. JFS performed all data analyses, wrote most of the text, and created all of the figures for this manuscript. XCD, DA, and WWW contributed to the text of this manuscript. DA, XCD, KGB, SJMJ, MAM, EMS, and WWW revised the manuscript prior to submission. All authors read and approved the final manuscript.
Elizabeth M. Simpson and Wyeth W. Wasserman contributed equally to this work.
Additional files
Additional file 1: Table S1.
List of the 1387 differentially abundant tag sequences mapping to the 1279 genes, differentially expressed in response to altered Nr2e1 levels during in vivo neocortex development and the corresponding tag numbers, calculated fold changes and P values at E13.5, E15.5, and E17.5. Columns one and two present the gene ID/symbol, associated tag sequence(s) and corresponding Refseq ID for each gene. Columns three and four, seven and eight, eleven and twelve present the number of tags found in Wt and Nr2e1 frc/frc libraries at E13.5, E15.5, and E17.5 inclusively. Column five, nine, and thirteen present the fold change values resulting from calculations based on the tag number values found in the previous columns (details, see Methods). Column six, ten, and fourteen present the associated P values calculated using the Audic-Claverie statistical test for each corresponding tags at the three different time points (E13.5, E15.5 and E17.5).
Additional file 2: Table S2.
List of the 1279 genes, differentially expressed in response to altered Nr2e1 levels during in vivo neocortex development, and the summary of significant findings for each. Columns one and two present the gene ID/symbol and associated transcript ID for each gene. Columns three to five present the genes having significantly differentially abundant tags at E13.5, E15.5, and E17.5, respectively. Columns six to nine present the genes retained after performing subsequent bioinformatics analyses. *, Multiple tags that were either up- or down-regulated but mapped to the same gene; details of the tag sequences and the direction of change are depicted. NA, not applicable.
Additional file 3: Table S3.
Nr2e1 binding sites containing sequences extracted from the literature and used to generate a position weight matrix. Columns one and two present the gene names and species origin of the DNA sequence analyzed. Column three presents the DNA sequence analyzed. Column four presents the PAZAR ID matching the DNA sequence presented in column three. Column five presents the PubMed ID associated with the manuscript from which the sequences were derived. NA, not applicable.
Additional file 4: Table S4.
Empirical P values extracted from the Z-scores and Fisher scores of each candidate-NR2E1 co-interactors. Column one presents the transcription factor symbol. Column two and three present the calculated empirical P values for both the Z-score and Fisher score of the corresponding transcription factor.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Schmouth, JF., Arenillas, D., Corso-Díaz, X. et al. Combined serial analysis of gene expression and transcription factor binding site prediction identifies novel-candidate-target genes of Nr2e1 in neocortex development. BMC Genomics 16, 545 (2015). https://doi.org/10.1186/s12864-015-1770-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1770-3