
Abstract
Background
Laboratory evolution is an important tool for developing robust yeast strains for bioethanol production since the biological basis behind combined tolerance requires complex alterations whose proper regulation is difficult to achieve by rational metabolic engineering. Previously, we reported on the evolved industrial Saccharomyces cerevisiae strain ISO12 that had acquired improved tolerance to grow and ferment in the presence of lignocellulose-derived inhibitors at high temperature (39 °C). In the current study, we used comparative genomics to uncover the extent of the genomic alterations that occurred during the evolution process and investigated possible associations between the mutations and the phenotypic traits in ISO12.
Results
Through whole-genome sequencing and variant calling we identified a high number of strain-unique SNPs and INDELs in both ISO12 and the parental strain Ethanol Red. The variants were predicted to have 760 non-synonymous effects in both strains combined and were significantly enriched in Gene Ontology terms related to cell periphery, membranes and cell wall. Eleven genes, including MTL1, FLO9/FLO11, and CYC3 were found to be under positive selection in ISO12. Additionally, the FLO genes exhibited changes in copy number, and the alterations to this gene family were correlated with experimental results of multicellularity and invasive growth in the adapted strain. An independent lipidomic analysis revealed further differences between the strains in the content of nine lipid species. Finally, ISO12 displayed improved viability in undiluted spruce hydrolysate that was unrelated to reduction of inhibitors and changes in cell wall integrity, as shown by HPLC and lyticase assays.
Conclusions
Together, the results of the sequence comparison and the physiological characterisations indicate that cell-periphery proteins (e.g. extracellular sensors such as MTL1) and peripheral lipids/membranes are important evolutionary targets in the process of adaptation to the combined stresses. The capacity of ISO12 to develop complex colony formation also revealed multicellularity as a possible evolutionary strategy to improve competitiveness and tolerance to environmental stresses (also reflected by the FLO genes). Although a panel of altered genes with high relevance to the novel phenotype was detected, this study also demonstrates that the observed long-term molecular effects of thermal and inhibitor stress have polygenetic basis.
Similar content being viewed by others


Background
For many years, the improvement of microbial strains by non-targeted approaches, such as random mutagenesis, crossbreeding or evolutionary engineering, has been successfully used in situations where the knowledge of the biochemistry and genetics associated to the relevant pathways was limited, as well as when complex connections of multiple metabolic and regulatory pathways restricted the manipulation of the strains by targeted approaches [1]. However, once a superior phenotype has been obtained by a non-targeted approach, the identification of molecular mechanisms behind the improved traits becomes a challenging process. Such elucidation, however, is of great importance since it enables the re-introduction of only the relevant mutations to other strains, thereby avoiding the accumulation of negative mutations or trade-off events that may have occurred during the selection process [2]. In addition, the identification of genetic features and mechanisms behind a phenotype of interest is expected to improve our knowledge and understanding of complex biological systems.
Baker’s yeast Saccharomyces cerevisiae is an important protagonist in industrial biotechnology and it is considered the biocatalyst of choice for the production of ethanol from lignocellulosic biomass [3, 4]. However, the tolerance of S. cerevisiae to stressors encountered during the ethanol production process, such as heat, lignocellulose-derived inhibitors, salts, contaminants, among others, needs to be further improved.
Many aspects of the response of S. cerevisiae towards environmental stressors have already been elucidated in this model eukaryote [5–8]. Furthermore, different mechanisms by which this yeast responds to the different types of inhibitors that are found in lignocellulosic hydrolysate, such as furaldehydes, weak organic acids and phenolic compounds, have been also identified [9, 10]. In fact, the various reports of long-term adaptation experiments oriented to improve the tolerance of S. cerevisiae to a single or to a mix of hydrolysate-derived inhibitors [11–14] have helped to reveal some of the evolutionary mechanisms responsible for the superior traits in the evolved strains, notably including the up-regulation of reductases capable of the conversion of furaldehydes to less inhibitory compounds [12, 15]. In contrast, the effect of high temperature on yeast physiology has mainly been studied through heat shock experiments, that is, when cells are shortly exposed to high temperatures [16–18]. Studies on long-term adaptation of S. cerevisiae to high temperatures are scarce [19], and to our knowledge there are no reports on how S. cerevisiae evolves to survive long-term exposure to a combination of heat and inhibitors.
In the present work we further characterized an evolved industrial strain, namely ISO12, which can grow and ferment hexose sugars under a combination of stress factors (high temperature and lignocellulose-derived inhibitors) that led to complete loss of viability for the parental industrial strain Ethanol Red (ER) [20]. With the aim of uncovering possible cellular mechanisms behind the superior phenotype of ISO12, the genomic sequences of ER and ISO12 were determined and compared. In addition to this, the cell wall phenotype and lipid composition of ISO12 and ER were analysed.
Methods
Strains and reference sequences
The industrial Saccharomyces cerevisiae strain Ethanol Red [Fermentis, S.I. Lesaffre] (ER) and the derived evolved strain ISO12 [20] were used for the experiments. Cells from -80 °C glycerol stocks were maintained in YPD plates (10 g.L−1 yeast extract, 20 g. L−1 peptone, 20 g. L−1 glucose and 20 g. L−1 agar). The genome of the S. cerevisiae S288c strain [21] was used as a reference sequence during the analysis of the ER and ISO12 Next Generation Sequencing data. The R64-1-1 release of the S288c genome was downloaded from the Saccharomyces Genome Database (SGD; www.yeastgenome.org) [22] and an alternatively formatted version of this release, sacCer3, was downloaded from the UCSC Genome Browser (www.genome.ucsc.edu) [23].
Genome sequencing, variant calling and variant analysis
DNA isolation
A single colony of Ethanol Red or ISO12 was used to inoculate 10 mL of YPD in a 50 mL conical tube. The overnight culture was centrifuged (2500 RCF) for 5 min, washed with 1 mL deionized water and pelleted. The cell pellet was resuspended in 0.6 mL lysis buffer (0.1M Tris pH 8, 1 mM EDTA, 100 mM NaCl and 1 % SDS final concentration). The suspension was transferred to a ready-to-use 2 mL tube containing 500 μm beads and 0.6 mL phenol/chloroform (1:1) was added. A Precellys 24 (Bertin Technologies, France) connected to a temperature controller working with dry-ice (Cryolus, Bertin Technologies, France) was used for lysis (3 cycles of 30s and 30s pause). The resulting cell suspension was centrifuged (13,000 RCF) for 5 min., and the aqueous phase was transferred to a 1.5 mL microcentrifuge tube. 200 μL chloroform was added to the tube and was mixed and centrifuged (13,000 RCF) for 1 min. The upper layer was transferred to a new microcentrifuge tube and 1 volume of isopropanol was used to precipitate DNA by centrifugation (16,000 RCF) for 20 min. The isopropanol was decanted and the pellet was washed with 1 mL 70 % ethanol, followed by centrifugation (16,000 RCF) for 5 min. The ethanol was decanted and the pellet was allowed to dry for 30-60 min. The DNA pellet was resuspended in 100 μL TE/RNAase, incubated at 37 °C for 30 min and stored at -20 °C.
Sequencing, assembly and variant calling
The sequencing of ER and ISO12 was performed by Genewiz Inc. (South Plainfield, NJ, USA) using Illumina MiSeq 2x250bp (Illumina Inc., San Diego, CA, USA). The de novo assembly of ER and ISO12 and the reference mapping and variant calling of both strains relative to S288c were performed by Genewiz Inc. using the CLC Genomics Server (v6.5.1; CLC Bio, http://www.clcbio.com). The assemblies and raw reads were deposited online, see Availability of supporting data. The variant calling was performed with the following thresholds: Minimum coverage (read mapping) = 10×; Minimum count of a variant = 4; Minimum frequency (count/coverage) = 25.0 %. Sequence variants that did not satisfy these criteria were discarded.
Analysis of the detected variants
In order to identify the variants that were unique to either ER or ISO12, all variants that were common to both strains were disregarded according to the reference mapping results. This filtering was provided by Genewiz Inc. To further predict the protein-level effects of the unique variants, the positions of the coding region SNPs and INDELs in both ER and ISO12 were reformatted from their mapped position in S288c ORFs to their corresponding chromosomal coordinates with a custom Perl (v5.10.1) script. The variants were then analysed for their non-synonymous effect on S288c ORFs using the Variant Annotation Integrator tool at the UCSC genome browser [23]. A list of the affected ORF of every detected non-synonymous variant in ISO12 was compiled and was analysed for enrichment in Gene Ontology (GO) terms by the YeastMine tool at SGD (accessed 2015-04-23). Complementary GO analyses were performed with the AmiGO 2 database [24].
The non-synonymous to synonymous substitution rate (Ka/Ks) [25] was calculated between the ER and ISO12 strains. Every SNP was extracted from the variant calling dataset by a custom Perl script, and was then applied to the sequences of the corresponding S288c ORFs with VCFtools (v0.1.12) [26]. The Ka/Ks ratio was calculated on the modelled ER and ISO12 ORFs using the MA model of the KaKs_calculator (v1.2) [25]. The results were quality filtered to only regard values in the range of 0.01 < Ka/Ks <5. It should be stressed that the Ka/Ks model only accounts for polymorphisms and cannot accurately assess sequences that have changed in length during the evolutionary timeframe; therefore the INDELs were not considered for this analysis.
Non-reference material in the de novo assembled contigs were detected with the ABACAS software (v1.3.1) [27] coupled to the nucmer portion of MUMmer (v3.22) [28]. The unmapped contigs from both strains were aligned with each other with Blat (v34) [29] in order to detect non-reference sequences that were present in both assemblies. The contigs were blasted to the NCBI database (http://blast.ncbi.nlm.nih.gov) to find regions with similarities to non-reference S. cerevisiae strains.
Copy number variation was assessed for both the reference material and non-reference material. The ER and ISO12 reads were aligned to the reference genome and to the unmapped contigs with BWA (v0.7.12) [30], were compressed and sorted with SAMtools (v1.1) [31] and were finally analyzed for variation in copy number with CNV-seq (2014/08/12-version) [32] and R (v3.1.0) [33]. For CNV-seq, the genome size parameter was set according to the size of the reference data.
Lipidomics
Cell cultivations for lipid analysis
Cells samples for analysis of lipids were obtained from aerobic continuous cultures. Inoculum was prepared by growing the cells overnight in 500 mL shake flasks containing 50 mL defined mineral medium [34] buffered with phthalate buffer (pH 5.5). After centrifugation and a washing step with deionized water, the pellet was resuspended with 20 mL of defined mineral medium and used for inoculation of the bioreactor. Chemostat cultivations were carried out at 30 °C in 0.75 L Multifors bioreactors (Infors, Switzerland) with a working volume of 450 mL. Aerobic conditions were maintained with an air flow rate of 1 vvm and stirring at 600 rpm. The pH was controlled at 5.5 by addition of 3M KOH. Defined mineral medium with 10 g. L−1 glucose was fed at the end of the batch phase with a dilution rate of 0.1 h−1. Carbon dioxide in the exhaust gas was measured with a 1313 Fermentation monitor (Innova, AirTech Instruments, Denmark). Steady state conditions were obtained after five volume changes (confirmed by CO2 analysis in the exhaust gas). Samples for biomass determination and High Performance Liquid Chromatography (HPLC) analysis were obtained from 6 independent steady states, four of which were independent biological replicates.
Quenching of the samples was achieved by quickly adding 20 mL of cell culture to 30 mL cold methanol (83.3 % methanol in water) in pre-cooled 50 ml conical tubes. After immediate mixing, the cells were cooled in a dry ice-ethanol bath for maximum 20 s and harvested in a pre-cooled centrifuge (-9 °C; 1500 RCF) for 5 min. The supernatant was discarded and residual medium was removed with a pipette while working over dry ice-ethanol bath. A second washing of the pellet was done with 10 mL cold-buffered methanol (60 % in water). The cells were collected as before and the samples free of residual medium were frozen in liquid nitrogen and stored at -80 °C.
Cell dry weight determinations were performed at each steady state by triplicate measurements. For this, 5 mL of the culture were filtered through pre-weighted nitrocellulose filters (0.45 μm; Pall Corporation, NY, USA) washed with distilled water and dried for 8 min in a microwave (350 W). Quantification of extracellular substrates and products was done with Waters HPLC system (Mildford, MA, USA). An Aminex HPX-87H ion exchange column (Bio-Rad, Hercules, CA, USA) was used for separation and a refractive index detector (RID-6a, Shimadzu, Kyoto, Japan) was used for detection. 5mM H2SO4 was used as mobile phase at a flow rate of 0.6 mL.min−1 and the column temperature was 45 °C.
Samples of the continuous cultures were also used to inoculate 500 mL shake flasks containing 50 mL of spruce hydrolysate (whose composition is described below) diluted to 50 % (v/v) with mineral medium [34], and incubated at 39 °C. Evaluation of growth under these conditions was carried out by optical density measurements at 620 nm (OD620) as previously described [20].
Extraction and quantification of lipids
Extraction and lipid quantification were carried out at Metabolomic Discoveries GmbH (Potsdam, Germany). For lipid extraction 40 mg of quenched cells were mixed with chilled 1:2.5:1 water:methanol:chloroform. Additionally, cells were mechanically disrupted in a ball mill. Cellular debris was removed by centrifugation. 1 mL of supernatant was mixed with 750 μL water. After phase separation, the lower non-polar phase was evaporated and resuspended in methanol. The LC separation was performed using hydrophilic interaction chromatography with Accucore HILIC (Thermo Fisher Scientific Inc., Waltham, MA, USA), operated by an Agilent 1290 UPLC system (Agilent, Santa Clara, USA). The LC mobile phase was A) acetonitrile and B) 0.1 % (v/v) formic acid in water with the following gradient: 1 to 6 min increase of B) from 3 % to 15 %, 6 to 7 min increase to 50 % and flush for 1 min, subsequently equilibrate. The flow rate was 500 μL.min−1, injection volume 1 μL. The mass spectrometry was performed using a 6540 QTOF/MS Detector (Agilent, Santa Clara, USA) with electrospray ionization. The measured metabolite concentration was normalized to internal standards.
Due to the small lipid phase obtained in two of the biomass samples of ER, the analysis of the lipid content for this strain was done only with four samples. In the case of ISO12, one of the samples was identified as an outlier and excluded from the study; therefore the analysis of the lipid content of this strain was done with five samples. Significant changes of metabolites were analysed considering variance homogeneity and p-values below 0.05 were considered as significant. The difference in the concentration of the lipid species is reported as log2-ratio of ER/ISO12.
Physiological characterisation
Growth on non-fermentable carbohydrates
Mitochondrial function was evaluated by growth on glycerol and ethanol as sole carbon sources [35]. For this, YPD-grown cells of ER and ISO12 were used to inoculate YNB medium supplemented with 3 % (v/v) ethanol as carbon source or were diluted with deionized water and plated on YPG plates (10 g.L−1 yeast extract, 20 g.L−1 peptone, 3 % (v/v) glycerol, 20 g.L−1 agar) and incubated at 30 °C for 48h. YPDG plates (10 g.L−1 yeast extract, 20 g.L−1 peptone, 1 g.L−1 glucose, 3 % (v/v) glycerol, 20 g.L−1 agar) were used to determine the proportion of petite mutants that lacked or had defective mitochondrial DNA (rho − cells). Growth on YPD plates was set as control conditions.
Results and discussion
Characterisation of the genome-level effect of the ISO12-adaptation
Genome sequencing, assembly and variant calling
The genomes of the Ethanol Red (ER) and ISO12 yeast strains were sequenced using the Illumina Next Generation Sequencing (NGS) platform. This resulted in 15M and 12M reads for ER and ISO12 (NCBI Sequence Read Archive: SRR2002842 and SRR2002960), both displaying a median PHRED score of 38 (corresponding to a 0.02 % base call error rate [36]). The reads were mapped to the S. cerevisiae S288c reference genome (average coverage: ER 185×; ISO12 153×) and the sequence variants relative to the reference strain were detected for both strains. Furthermore, the reads were also used to de novo assemble the genomic sequences of ER and ISO12 in order to assess the effect of the adaptation in genomic material not found in the reference strain. The ER assembly resulted in 218 contigs (average coverage: 301×) consisting of a total of 11.5 Mbp (GenBank: JWJK00000000). For ISO12, 361 contigs were assembled (average coverage: 196×) with a total size of 11.4 Mbp (GenBank: LBNP00000000). A summary of the sequencing, reference mapping and de novo assembly results is found in Table 1.
The variant calling was set to identify two types of variations - SNPs (Single Nucleotide Polymorphisms) and INDELs (Insertions and Deletions) - and resulted in 78,925 (ER) and 78,143 (ISO12) variants relative to S288c (Table 2), using the minimum thresholds of detection stated in the Materials and Methods section. This was an expected magnitude, similar to a previous study that also mapped a diploid strain to S288c [37], and it reflected the marked differences between the genomes of different strains in general, and between the genomes of laboratory and industrial strains in particular [38, 39]. Since the adapted strain was derived from ER by laboratory adaptive evolution [20], a high number of the identified variants versus S288c (76,661) were found to be common to both strains. These SNPs and INDELs were disregarded, and further analyses focused on the remaining 2,264 variants that were found only in ER and 1,482 variants that were unique to ISO12.
Since only around 300 generations separated ISO12 from its parental strain, the number of identified variants suggested a higher mutation rate per genome than previously reported [40]. One possible reason may be an underestimation of the number of generations for ISO12, since the decrease in the number of viable cells due to the cytotoxic effects of hydrolysate and temperature was not taken into consideration every time a new batch was inoculated [20]. Also, the number of alterations reported in other evolutionary experiments using S. cerevisiae shows significant differences. For example, the number of mutations reported for S. cerevisiae CEN.PK113-7D after over 300 generations growing at high temperature (40 °C) was of only 30 SNPs distributed in 18 genes [19], while a total of 949 alterations were reported for S288c after approximately 140 generations during serial transfers with increasing concentrations of barley hydrolysate [41]. These results suggest that the mutation rate will be affected by the genomic stability of the background strain as well as by the type of stress encountered during the evolutionary experiment. Therefore, apart from a probable higher natural mutation rate in ER compared to the aforementioned strains, it is very likely that the stressful environment - given by the combination of high temperature and toxic hydrolysate-derived compounds- significantly increased the rates of random mutagenesis; an effect referred as “stress-inducible mutability” that has been discussed in previous studies [42, 43]. Also, the diploid nature of these strains possibly allows for a greater accumulation of mutants than a haploid strain would [42, 44] because undesired mutations would not be selected against and could thus be inherited and add to the accumulation of mutations as long as one functional wild type allele remains in the genome. Finally, an inevitable possibility when working with NGS data is that of erroneous data interpretation due to sequencing error. For Illumina data, a number of technical difficulties on the sequencing-level have been proposed to increase the false SNP call rate, including under-representation of GC-rich regions during the PCR-amplification step [45] and strand bias (differing genotype on the forward and the reverse strand) during read mapping [46]. In the end, we cannot rule out that phenomena like these might have affected the false positive count in the dataset. It should be mentioned that we, in addition to the reference-mapping approach, also attempted variant calling by mapping ISO12 reads to the ER de novo assembly. However, this resulted in an even higher number of variants between the two strains(~9000; data not shown), which might be due to a high rate of false positive variant calls. We thus believe that the method that is presented here (reference-mapping) was a sound choice for this study.
Analysis of the distribution and the protein-level effects of the sequence variants
The SNPs and INDELs detected in ER and ISO12 were found to be distributed throughout the extent of the S288c reference genome, albeit with varying peak density (Additional file 1: Figure S1). In fact, circa 60 % of the detected variants in both ER and ISO12 were intergenic (Table 2). Since only ~27 % of the S. cerevisiae genome is non-coding [47], this result indicated a heavy bias towards intergenic alterations during the evolution of ER into ISO12. Many investigations have shown that variations in non-coding regions have an important impact on phenotypic diversity given their influence on gene expression level [48]. Although the differences in intergenic regions would definitely merit further investigation, for the purpose of the current study we focused the analysis on variants within coding regions.
The Variant Annotation Integrator web-tool [23] was used to identify and classify the protein-level effects of the SNPs and INDELs. The non-synonymous mutations affecting coding regions (including overlapping ORFs) were distributed in the following manner: 454 non-synonymous SNPs were allocated in 221 ORFs in ER (Table 2; Additional file 2) while 306 non-synonymous SNPs were distributed among 172 ORFs in ISO12 (Table 2; Additional file 3). Forty-eight ORFs presented variants in both ER and ISO12, while 173 and 124 ORFs presented SNPs only in ER and ISO12 respectively. Altogether, ER and ISO12 presented genetic variations in a total of 347 different ORFs (Additional files 2 and 3). A majority of the non-synonymous variants were classified as missense variants, i.e. changing a single amino acid but not affecting protein length or read frame (88 % in ER; 78 % in ISO12). Furthermore there were 83 variants in both strains that were predicted to generate a premature stop codon or a frame shift, and could therefore result in loss of protein function (7 % in ER; 16 % in ISO12).
To investigate the 347 genes that were predicted to have gained non-synonymous alterations, we applied a number of complementary analyses. A smaller number of candidate genes with high relevance to the novel phenotype of ISO12 was identified by assessing functional annotation enrichment and haplotype variation (homo-/heterozygosity; cf. annotations in Additional files 2 and 3), as well as by modelling of the non-synonymous to synonymous substitution ratio (Ka/Ks) and copy number variation (CNV). These approaches are elaborated in the following sections.
Enrichment in cell periphery-related proteins
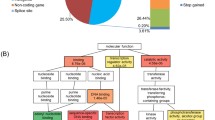
The 347 ORFs that differed between the strains were first investigated-independently from ploidy, substitution ratio and CNV-for their enrichment in Gene Ontology (GO) terms using SGD (p < 0.05, Holm-Bonferroni corrected; background: SGD default) in order to infer cellular functions and structures that were overrepresented (Fig. 1). By default, the tool at SGD assesses GO enrichment in three different categories: Biological process, Cellular component and Molecular function.

Significantly enriched Gene Ontology (GO) terms for the non-synonymous variants detected in ISO12 compared to ER (p < 0.05, Holm-Bonferroni corrected). Terms were sorted in three categories: Biological process, Cellular component and Molecular function. The enrichment analysis was performed using the YeastMine at Saccharomyces Genome Database (SGD). Note that the same ORF can figure in multiple bars; please refer to Additional file 4 for the full list of ORFs
In the Biological process ontology, transposition, RNA mediated (p = 3.74x10−6; 24 matching ORFs) and interspecies interaction between organisms (8.24x10−6; 14 matching ORFs) were the most significantly enriched terms (Fig. 1). 36 different ORFs were represented in the 9 enriched terms obtained within the category Biological process (Additional file 4). Of these 36, 24 were directed related with transposition-associated processes (transposable-element-gene) while the remaining 12 ORFs were associated with cellular processes such as flocculation (markedly the FLO gene family), agglutination, conjugation and fusion (Additional file 4).
With respect to the Cellular component category, the enrichments revealed that around 20 % of the ORFs differing between ER and ISO12 encoded proteins related with structural components of the cell such as the cell wall and the plasma membrane and with the assembly and organization of the cytoskeleton (Fig. 1). The two most enriched terms in this category corresponded to the retrotransposon nucleocapsid (p = 1.25x10−7; 23 matching ORFs) and cell periphery (p = 1.83x10−4; 65 matching ORFs); in fact, the cell periphery category was the GO term that was enriched in the highest number of ORFs (Fig. 1). Some of the molecular functions covered by the cell wall-associated ORFs included heat shock protein (HSP150), transcription factor (RPI1) and components of the cell wall integrity signalling pathway (SPA2). Sequence variations were also found in ORFs coding for various glycosylphosphatidylinositol (GPI)-anchored cell wall proteins (e.g. EGT2, YPS1, YPS6, FIT1). Moreover, ER and ISO12 differed in the genomic sequence of several ORFs coding for proteins involved in cellular surface properties such as adhesion (AGA1 and its paralogous gene FIG2), and biofilm and flocculation (FLO1, FLO5, FLO9, FLO11) (Additional file 4). SNPs related to membrane proteins included ORFs coding for various type of transporters such as the ABC-transporter PDR5, the multidrug efflux transporter ATR1, the iron transporter FTR1 and the proton-gluthatione transporter GEX1, among others. Notably, 5 of the at least 20 hexose transporters present in S. cerevisiae [49] presented genomic differences between the strains (HXT3, HXT4, HXT6, HXT13 and HXT15). Also, gene variations were found in ORFs whose transcription products are associated with the sensing and reception of nutritional signals (GPR1, KOG1) and oxidative stress (MTL1). Finally, the genomic variations identified in sequences coding for proteins involved in the assembly and organization of the cytoskeleton included SNPs in ABP1, SLA1, NUM1, BEM3 (Additional file 4).
It is well established that cell wall and membranes are highly dynamic structures that respond to various forms of environmental stress [50–52]. Heat in particular has been shown to reduce membrane potential and disrupt the membrane integrity, affecting its permeability and fluidity [53, 54]. The enrichments of genes directly related with cell wall and membrane indicate therefore that such structures were important targets during the evolutionary experiment. Although some genomic variations may be associated with the deleterious effect of the stressors over these cellular components, the direct relationship of some of the aforementioned proteins (or similar ones) with the stress tolerance response in yeasts [55, 56], suggest that variations in the regulation, structure and/or activity of these protein variants as a result of the point mutations and other genomic alterations, were necessary for the cell to adapt to the severe conditions. In fact, ISO12 exhibited an improved viability during 48h-cultivation in undiluted spruce hydrolysate while only little or no reduction of inhibitors was observed (Additional file 1). We hypothesise that this results from changes in membrane permeability that were induced by long-term exposure to stressors. Similarly, the already reported negative effects of high temperature and furfural over cytoskeleton elements and consequently over their physiological functions - including vesicular transport- may explain the alterations in more than 6 ORFs related with the cellular matrix [57–59]. Actually, according to the AmiGO 2 database [24], 20 ORFs associated with vesicle-mediated transport presented genetic variations between the strains, e.g. SEC7, SPO14 and SED4 (Additional file 5). However, bearing in mind that ISO12 is indeed capable of growing and fermenting under the combination of the stresses, the results suggest that either such alterations were phenotypically neutral or that at least some of the new variants of the actin-related proteins in ISO12 would contribute to the adaptation response, among other, by allowing the cell to carry on with crucial intracellular traffic of proteins.
The enrichment of genes in the third GO category, Molecular function covers different types of proteases. Of particular relevance are the ORFs coding for proteasome sub-units and ubiquitin-specific proteases that are present in both the nucleus and cytoplasm (RPN2, UBP8, UBP10, UBP12) (Additional file 4). Recently it was demonstrated that a single mutation in the ubiquitin-specific protease gene UPB7 was directly correlated with tolerance to spent sulphite liquor, a raw material for ethanol production with similar type of inhibitors as spruce hydrolysate [60]. Similarly, other studies have established the effect of mutations in different proteases on the response of the cell to different environmental effectors [61, 62]. The SNPs identified in ISO12 emphasize hence the relevance of the ubiquitin-mediated protein homeostasis machinery during heat and inhibitor stresses. The most enrichment terms in this ontology corresponded to aspartic-type endopeptidase activity (p = 1.42x10−5; 16 matching ORFs) and RNA-DNA hybrid ribonuclease activity (p = 7.64x10−4; 13 matching ORFs; Fig. 1).
The enrichment of transposition-related ORFs in all the three categories of the gene ontology database suggest that changes in the DNA sequences of retrotransposons are a constant feature behind the genetic differences between ER and ISO12. On the one hand, retrotransposons (Ty elements) have been clearly identified as responsible entities for evolutionary genomic and phenotypic changes in S. cerevisiae strains by causing gene disruption, promoting chromosomal rearrangements or acting as regulatory agents [63, 64]. On the other hand, the results obtained in the current study do not provide information about the transpositional activity of the Ty elements, and therefore it is not possible to establish a direct relationship between the high numbers of GO enrichments for Ty elements related events with phenotypic differences between the strains. Yet, considering the diversity of variants in the Ty coding regions (Additional files 2 and 3), we speculate that at least some of such SNPs may be responsible for a higher rate of replication of the transposon and/or for providing an adaptive advantage to ISO12 under the stress conditions.
Eleven genes were found to be under positive selection in ISO12
The GO analysis revealed that the dataset with the ORFs with SNPs/INDELs were enriched in terms related to cell-periphery proteins. Next, we investigated each gene in this dataset by looking at which ORFs that were considered likely to be evolved under positive selection, i.e. a directional selection driven by change in phenotype [65]. The non-synonymous to synonymous substitution ratio (Ka/Ks) [25] was calculated between the two strains based on the effect the SNPs would have on a S288c background (Additional file 6). 11 genes were found to be under positive selection (Ka/Ks >1). Although GO enrichment analysis is not applicable on this small dataset, a relationship similar to that of the above GO enrichment analysis was found. The proteins linked with these eleven ORFs were found in different locations within the cell; MTL1 (Ka/Ks = 3.2), HXT13 (Ka/Ks = 4.1), CYC3 (Ka/Ks = 3.1) and SED4 (Ka/Ks = 1.5) code for proteins associated with membranes; FLO9 (Ka/Ks = 2.3) and FLO11 (Ka/Ks = 1.3) are linked to the cell wall and SRP40 (Ka/Ks = 1.2) with the nucleus. The four other ORFs are associated with proteins of unknown/uncharacterized location and function. It can be noted that SRP40 and the four putative ORFs did not show up during the GO enrichment analysis while the other ORFs did (Additional files 4 and 5). However, it should be kept in mind that the enrichment assessment only takes functional annotations and not the number of mutations into consideration, and in that sense Ka/Ks-modelling complements the enrichment analysis.
The implications of the stress-membrane sensor Mtl1p [66] as part of the cellular response to oxidative stress are physiologically coherent with the conditions of the evolutionary experiment [20]. MTL1 is a partly redundant paralog of the major cell wall integrity (CWI) sensor MID2 and is involved in the oxidative stress response and actin cytoskeleton repolarisation [66] and has also been named a putative sensor for glucose starvation [67]. Furfural - one of the stressors that were present during the ISO12 adaptation - has been reported to cause accumulation of reactive oxygen species (ROS) that, among others, damages the actin cytoskeleton [57]. In S. cerevisiae the CWI pathway functions by a Mitogen-Activated Protein Kinase (MAPK) cascade that leads to the activation of transcription factors controlling the expression of genes responsible for adjusting cell wall structure and composition according to the environmental input, and MTL1 is one of the sensors that can initiate the cascade in response to cell wall stress [50, 68]. The apparent positive selection of MTL1 is thus highly interesting, and since the identified variants in this ORF were solely missense variants and not loss-of-function variants (such as stop and frameshift variants) we hypothesize that the CWI sensing in the adapted strain might have undergone functional alterations. However, when the CWI of ER and ISO12 were evaluated experimentally with an enzymatic cell wall lysis assay (lyticase) no significant differences in maximal lysis rate were observed (Additional file 1). From this it was concluded that there had been no changes in β 1,3-glucan integrity, which however does not rule out other changes to the CWI system. In fact, it has been reported that β-1,3-glucanase (here: lyticase) is sensed with combined signalling of the high osmolarity/glycerol pathway and the CWI, but that upstream CWI elements such as MTL1 does not contribute to the response [69]. This could explain why the detected alterations in MTL1 were not reflected in the results of the lyticase assay.
The FLO (flocculation) genes also stand out as important targets during the evolution. First, mutations in the FLO9 and FLO11 ORFs appeared to have evolved under positive selection (Additional file 6). Second, the sequence regions coding for the flocculation-related genes FLO1 and FLO11 showed homozygous SNPs in both strains (Additional files 2 and 3), and finally around 25 SNPs between the strains were found in the genome sequence of FLO5. This means that 4 out of the 5 FLO genes present in S. cerevisiae [70] differed between strains. It is known that changes in sequence, copy number transcriptional level and other regulatory mechanisms of the FLO genes affect several of the adaptive mechanisms of the cells towards detrimental environmental factors including flocculation, biofilm formation and agar invasion [71–73]. In fact, previous studies have shown direct evidence of the role of FLO genes on the adaptation capacity of yeast cells to overcome temperature or lignocellulosic-derived inhibitors [74, 75]. The genotypic variations between ISO12 and ER in the FLO genes correlated with a higher capacity in ISO12 to form biofilms (data not shown), invade agar and form novel colony morphologies under certain conditions (See Growth on non-fermentable substrates and multicellularity section below). It seems therefore very reasonable that the acquired properties of ISO12 cells to aggregate, mediated by at least some of the FLO genes variations, are part of the cellular adaptive mechanisms to resist the combination of both stresses.
Other genes predicted to have evolved under positive selective pressure were CYC3 and HXT13 (Additional file 6). CYC3 is a nuclear gene that codes for cytochrome c heme lyase. cyc3 null mutants are respiratory deficient since they lack a functional cytochrome c, and their ROS levels have been found to be lower than wild-type strains [76, 77]. Although the missense variant present in ISO12 (M45I; detected in 38 % of the mapped alleles) is most probably functionally active, the effect of such amino acid change on the decreased respiratory capacity of ISO12 (by direct effects on CYC3p or through epigenetic mechanisms) cannot be discarded [78]. In relation to HXT13, it has been proposed that down-regulation of hexose transporters as a mechanism to reduce growth could be part of the cellular response during heat-shock [79]. HXT13 is not considered a main yeast hexose transporter [80] and the impact of the changes to HXT13 is in itself not very evident, however, other hexose transporters did exhibit mutations in ISO12, including a frameshift INDEL in HXT4 (detected in 27 % of the mapped alleles).
The significance of the other six genes that appeared to have evolved under positive selection is less clear.
Haplotype- and copy number variations indicate possible gene dosage effects in ISO12
An increase in the relative content of heterozygous variants was observed in the datasets of both strains after subtraction of the common variants. Approximately 16 % of the total SNPs/INDELs were classified as heterozygous in both original datasets. This changed to 76 % in the ER dataset and to 83 % for ISO12 after the shared variants were disregarded. Due to the polyploid nature of the strains, the high content of heterozygous variants in the datasets implies that the possible novel effects of many of the strain-unique variants in ISO12 might be overshadowed by parallel wild-type alleles. On the other hand, heterozygous variants can also confer haploinsufficiency in yeast [81] (particularly affecting protein complexes due to imbalances in subunit availability [82]) and thus haplotype variations remain relevant to the ISO12 case.
It was found that loss-of-function variants such as premature stop codons and frame shifts were almost exclusively heterozygous in both strains, with no homozygous calls in ISO12 and seven in ER. It is nevertheless possible that the predicted novel stops in 15 genes in the adapted strain (Additional file 3) is contributing to the phenotype by gene dosage effects. An intriguing example in the present study is the premature stop in the external glucose sensor GPR1 (detected in 59 % of the GPR1 alleles in ISO12 through reference mapping). Gpr1p belongs to the RAS-cAMP-PKA pathway and constitutes the means of which this pathway reacts to extracellular glucose [83]. Down-regulation of the RAS-cAMP-PKA pathway has been associated to decreased heat sensitivity [84] – a prominent phenotype in ISO12- as decreased Protein Kinase A (PKA) levels induces transcription of a number of stress response genes with stress response elements (STREs) in their promoter regions [85]. The premature stop codon in Gpr1p was predicted in amino acid 251 of 961 that, in addition to the deprivation of three quarters of the protein, implicates a complete loss of the cytoplasmic tail [83] which is highly likely to cause a disruption in the signal transmission. However, to fully elucidate this, cAMP/PKA levels would have to be measured and compared between the adapted and the parental strain.
Gene dosage effects can furthermore affect phenotype not only by changes in haplotype but also by copy number variation (CNV). CNVs are, in addition to SNPs and INDELs, a common genomic evolutionary effect and have been observed both in laboratory adaptation experiments [86, 87] and in natural isolates and industrial yeast strains [38, 88]. We used a reference mapping-based method [32] to assess the changes in copy number between ER and ISO12 and detected 13 regions with a >2-fold higher CNV and 77 regions with a >2-fold decrease in ISO12 (Additional file 7). As 46 % of the CNV-regions were located either in telomere or transposon regions, they are likely to be false CNV-calls as centromeric and telomeric regions are known to be highly difficult to sequence and assemble/reference-map due to their enrichment of tandem repeats [89]. When a log2 threshold was applied, 32 of the CNV-regions were found to correspond to ORFs (excluding telomere and transposon regions), including a two-fold increase in FLO9 (p = 4.65E-31) and ENA1 (p = 3.78E-61) (Additional file 7). Three ORFs exhibited a CNV higher than three-fold: ENA2 had increased from ER to ISO12 (log2 = 3.0, p = 7.68E-206), and two ORFs, PHO12 (log2 = -2.2, p = 69E-83) and FLO1 (log2 = -3.7, p = 8.88E-126), had decreased.
Sequencing of PHO12 has been previously been proven problematic due to the presence of paralogs [39] and for this reason we cannot rule out the possibility that PHO12 is subject of a false CNV-call. But the CNVs in the FLO genes matched the above results regarding positive selection pressure and bring additional evidence to support the correlation of alterations in this gene family and the phenotype of ISO12 (cf. also the discussion regarding the effect on CNVs in FLO genes in the previous section on Ka/Ks). As for ENA1 and ENA2, they encode ATPase sodium pumps [90], and results presented by Gilbert et al. showed that an increased copy number of ENA1, ENA2 and ENA5 genes in two different lines of mutants strains obtained after evolution under increased concentrations of acetate, was associated to improved acetate and temperature robustness [91]. The similarity on the CNVs in the ENA genes in ISO12, whose adaptation was also carried out in the presence of acetic acid and at elevated temperature, supports a relationship between the function of the ENA genes and these two types of stressors and suggest that such genes have a tendency to CNVs as a common stress response in yeast.
Assessment of the non-reference genomic material through de novo assemblies
Industrial yeast strains often contain genomic material that is not shared with the S288c reference strain due to differences in their lineage [38, 88]. These variations in genome content can confer important strain-specific geno- and phenotypes and assessment of such regions will add to the larger picture of evolutionary effects. Again, we focused on investigating the changes between the parental and adapted strain. By mapping the contigs from the de novo assemblies to the reference genome, we found 8 contigs in ER and 13 in ISO12 that could not be mapped to the reference (listed in Additional file 8; GenBank JWJK00000000 and LBNP00000000). Alignment of the unmapped contigs with each other revealed that there were five alignment pairs that were present in both strains; in total, 81.5kb were successfully aligned (Additional file 8). The top blastx-results showed (as was hypothesised) that there were regions in these contigs with high similarities to protein-coding regions from non-S288c strains, including the wine strain AWRI796, the industrial bioethanol strain-derivative JAY291 and the sake brewing yeast Kyokai No. 7.
In contrast with the CNV-analysis of the reference-material, the majority of the CNVs detected in these contigs were higher in ISO12 than in ER: in total, 33 regions with >2-fold CNV was detected in these five pairs, of which 29 were higher in ISO12 (Additional file 8). The largest contigs in terms of size (ER contigs 43 and 90) did also contain the highest density of CNV-regions. Using blastx, we could identify that a two-fold increase for ISO12 in a region corresponding to ADH7 from the YJM1447 strain (99 % identity), and a 2-fold decrease in ISO12 in the loci with similarity to AAD10 from strain AWRI796 (97 % identity) and UIP3 from JAY291 (99 % identity). These genes are present also in S288c, however, the results of the blast search resulted in higher identities with the non-reference strains, which suggests that the lateral sequences were different. When blasted towards SGD, ER Contig 90 displayed high identity to mitochondrial material from non-S288c S.cerevisiae strains; however, none of the CNV-regions matched any annotated ORFs from the database.
The proposed increase in CNV for ADH7 is especially intriguing in the context of the novel ISO12 phenotype. Increased ADH7 activity has previously been positively correlated with HMF tolerance; upregulation of this gene has been observed in HMF tolerant yeast strains [92, 93] and overexpression of ADH7 has been shown to result in a significant increase in HMF reduction capacity with NADH as cofactor and also improved cell growth recovery in HMF [94]. However, this is a surprising find in relation to the actual ISO12 phenotype as the HMF-reduction capacity in this strain was found to be lower than in ER [20].
Lipidome comparison
Lipids play a key role on the biological functionality of membranes, by providing a permeable barrier and by maintaining its liquid crystalline state [95]. The effects of environmental stressors on cellular membranes have been extensively studied due to the essential role of these structures for proper functioning of the cell. In general, the consequences of environmental conditions on membranes are described by perturbations of the membrane fluidity, i.e. alterations on membrane composition and organization that not only affect the permeability of the barrier (e.g. leakage), but also disrupt the functionality of many membrane-associated proteins (e.g. transporters), and influence the expression of many genes (e.g. heat shock genes) [53, 96]. Lipids are of primary relevance for membrane functionality at high temperatures; in fact, it has been suggested that the reason why there are no known extreme eukaryotic thermophile can be attributed not only to their susceptibility to mRNA degradation, but also to the susceptibility of their lipids to changes in permeability and phase-state when exposed to high temperatures [97].
Alterations in lipid-membrane composition are known to take place as a response to changes in temperature and chemical stressors [98–102]. Based on the above, and in parallel to the genomic analysis, the differences in lipid composition between ER and ISO12 were analysed by a relative quantitative approach (i.e. the lipids species of one strain were compared in relation to their levels in the other strain). The lipidomes of the two strains reflected important differences in more than one lipid species (Table 3). Nine out of 119 analysed lipid species were found in relative higher concentration in ISO12 with all lipid classes represented by at least by one metabolite (p = 0.05, fold change > 2). The raw data of the lipid analysis from all the samples and the differential analysis are available in Additional file 9. Although the relevance of the particular lipids is difficult to assess, it is pertinent to note that accumulation of cerotic acid has been reported as part of a mechanism of cell resistance to oxidative stress [103]. Considering that during the evolutionary process both heat and some of the hydrolysate-derived inhibitors were a source of reactive oxygen species (ROS) [57], alterations in the content of cerotic acid may account, at least in part, for the increased resistance in ISO12. There was no significant difference in the concentration of ergosterol between both strains, but a higher concentration of ergosta-5,7,22,24(28)-tetraene-3beta-ol –the direct precursor of ergosterol– was found in ISO12. Changes in sterol composition have been found to be associated with higher thermotolerance in yeasts [19, 104, 105]. Considering that in terms of biological functionality ergosta-5,7,22,24(28)-tetraene-3beta-ol and ergosterol are very similar under normal conditions [106], the increase of ergosta-5,7,22,24(28)-tetraene-3beta-ol in ISO12 may be linked to the adaptive mechanisms developed by the cells to survive the high temperature. Similar changes in lipid composition are also likely to confer the improved cell viability in undiluted spruce hydrolysate of ISO12, that was found to be unrelated to inhibitor detoxification (Additional file 1).
The lipidomic results prompted us to look at the genomic differences associated with lipid metabolism. By matching the 414 ORFs recognized under the category lipid metabolic process (cf. AmiGO 2 database) with the 347 ORFs differing between the strains, 25 ORFs were identified (Additional file 5). The non-synonymous variants were identified in genes related with biosynthesis, regulation and modification of the main lipid classes fatty acid (EHT1), glycerophospholipid (SLC1, NTE1, GPI12, FAB1), sphingolipid (SKN1, TOR1, LCB4) and sterol (HMG1, ERG13 and MOT3) but it was not possible to directly correlate the effect of the genetic variations and the pathways associated with the metabolism of the altered lipid species. Nevertheless, both types of evidence point towards an essential readjustment in the membranes of the evolved strain. Such rearrangement in the metabolism of lipids in ISO12 would be a coherent response towards both heat and hydrolysate-derived inhibitors during the long-term adaptation process, suggesting that at least some of the alterations in lipid composition could be essential for proper membrane development and function. The distinctions in lipid-related ORFs and lipid composition could also be connected with other biological functions of lipids related to stress response such as their role as signaling molecules during heat stress [107], their involvement during the initiation of translation of heat shock proteins [108, 109] and exocytosis, a process that also largely depends on lipid-related reactions [110] and is largely affected during cellular stress [111, 112].
Physiological characterisation of ISO12
Growth on non-fermentable substrates and multicellularity
It has been established that in S. cerevisiae, the main targets of thermal death are located in the inner mitochondrial membrane [113] and that a loss of mitochondrial genes as well as the development of a mitochondria-defective (petite) phenotype are common effects of heat stress on yeasts [19, 113]. Growth evaluation of ISO12 and its parental strain ER was performed on media containing ethanol or glycerol as sole carbon sources since petite cells cannot grow in non-fermentable substrates [114, 115]. In ethanol, the biomass of ISO12 increased 6 times during the first 25 h (from OD620 = 0.6 to OD620 = 5.7 ± 0.2), while the biomass of ER increased around 9 times (from OD620 = 0.6 to OD620 = 9.3 ± 0.0). Growth was also observed on glycerol plates for the evolved strain (Fig. 2-d). The proportion of rho − cells (i.e. petite mutants lacking or with defective mitochondrial DNA) could not be determined for any of the strains since there was no difference in colony size compared with the YPDG plates. Therefore, although ISO12 did not present a true petite phenotype, the reduced growth rate in ethanol and the slower growth under mild conditions previously reported [20] hinted towards the presence of alteration(s) within the respiratory chain [115]. The correlates to the changes in cytochrome c heme lyase (CYC3), that was one of the 11 genes whose evolution was predicted to have occurred under positive selective pressure (Additional file 6). As discussed above, changes to this gene have been related to respiratory deficiencies at the benefit of decreased ROS levels [76, 77] which are two relevant aspects with respect to the ISO12 phenotype.

Colony morphology of ER (a and c)) and ISO12 (b and d) when grown on glucose (a and b) and glycerol c and d. For growth on glucose the picture shows the bottom of the colony, while the surface is presented for growth on glycerol
Additionally, the evaluation of growth on plates revealed a phenotype that has been denoted as “colony morphology response” and that is used to describe colonies with complex and organized structures [116]. This phenotype was observed for ISO12 only (Fig. 2) and the specific pattern was dependent on the carbon source. For instance, a characteristic “Y” shaped agar invasion was observed on the bottom of the glucose plates (Fig. 2-b). On glycerol, agar invasion was not perceived, but the surface of the colony had a regular and complex pattern (Fig. 2-d). The formation of biofilm after long-term cultivations was also a phenotype observed only in ISO12 strain (data not shown). As mentioned previously, agar invasion and biofilm formation have been reported as a response to different environmental stresses [117–119]. The complexity of the morphological response can be explained by the multiple and interacting signalling pathways. For instance, the filamentous growth MAPK cascade and the Ras-cAMP-PKA pathway (for a review see [120]) have been shown to be the two key signalling pathways behind the colony morphology response [116]. However, other signalling networks such as the Target of Rapamycin (TOR) pathway [121] and Cell Wall Integrity (CWI) pathway take also part in the regulation [122]. Indeed, the transcription of at least FLO11 (one of the ORFs subjected to positive selection according to the model) is regulated by the MAPK and cAMP signalling pathways [123] and changes in the expression levels of this gene strongly affect colony morphology [124]. Furthermore, the changes to MTL1 and GPR1 also emphasize that the MAPK and RAS-cAMP signalling in itself has been altered. Based on these results we hypothesize that the presence of genetic variants in different ORFs related with the aforementioned signalling pathways and possible interactions with other proteins (such as Flo11p) account for the morphological phenotypes displayed by ISO12. Moreover, the capacity of multicellular behaviour observed in ISO12 becomes an additional piece of evidence towards the relevance of multicellularity as an evolution strategy to improve competitiveness and tolerance to environmental stresses [125–127].
Conclusions
From the results presented in the current study, the improved tolerance to combined stresses in ISO12 is shown to reside in multiple mechanisms requiring more than just a few mutations. The results indicate that the adaptive evolution that was used to generate ISO12 gave rise to non-synonymous variants significantly enriched in GO terms related to cell periphery-related proteins. It was also found that, besides the improved thermotolerance of ISO12 reported in the previous study, the evolved strain has also a higher capability than ER to withstand the toxic effects of high concentrations of spruce hydrolysate-derived inhibitors and that such capability is not related with furaldehyde-reduction capacity (Additional file 1). The analysis of the lipid composition of the cells indicates that significant rearrangements in the lipidome may also account for the superior phenotype of ISO12 under conditions of stress. Membranes would be feasible and convenient targets for adaptation since the lethal effect of both types of stressors (heat and hydrolysate) could be counteracted by a common mechanism. Thus, the ISO12 genotype and phenotype converges to suggest that the membranes and lipidome of S. cerevisiae may play an important role during evolutionary adaptation to high temperature in the presence of hydrolysate-derived inhibitors. The key mutations, however, are difficult to pinpoint due to the high degree of strain mutability imposed by the conditions of the evolution process. Based on a predictive model of genes under positive selection and on other genomic variations investigated, the genes MTL1, FLO1/5/9/11, CYC3, GPR1, ADH7 and ENA1-2 are strong candidates for being directly responsible for the phenotypic changes between the strains. Further sequencing of different clones from the adaptation experiment displaying the improved phenotype could be done in the future to narrow the number of key mutations related with the superior phenotype. However, from an applied point of view it would seem more reasonable to attempt to replace the mutations that are responsible for negative traits accumulated in ISO12: e.g. its slower growth rate and its reduced capacity for NADPH-dependent reduction.
Availability of supporting data
The sequence reads from this project have been deposited at the NCBI Sequence Read Archive under the accessions SRR2002842 (ER) and SRR2002960 (ISO12). The assembly data set supporting the results of this article has been deposited at DDBJ/EMBL/GenBank under the accessions JWJK00000000 (ER) and LBNP00000000 (ISO12). The versions described in this paper are version JWJK01000000 and LBNP01000000.
Abbreviations
- CNV:
-
Copy number variation
- CWI:
-
Cell wall integrity
- ER:
-
Ethanol Red
- GO:
-
Gene ontology
- HMF:
-
5-(hydroxymethyl) furfural
- HPLC:
-
High performance liquid chromatography
- INDEL:
-
Insertion or deletion
- MAPK:
-
Mitogen-activated protein kinase
- MLR:
-
Maximal lysis rate
- NGS:
-
Next generation sequencing
- OD:
-
Optical density
- ORF:
-
Open reading frame
- SGD:
-
Saccharomyces genome database
- SNP:
-
Single nucleotide polymorphism
References
Çakar ZP, Turanli-Yildiz B, Alkim C, Yilmaz U. Evolutionary engineering of Saccharomyces cerevisiae for improved industrially important properties. Fems Yeast Res. 2012;12(2):171–82.
Oud B, van Maris AJA, Daran JM, Pronk JT. Genome-wide analytical approaches for reverse metabolic engineering of industrially relevant phenotypes in yeast. Fems Yeast Res. 2012;12(2):183–96.
Jeffries TW. Engineering yeasts for xylose metabolism. Curr Opin Biotech. 2006;17(3):320–6.
Laluce C, Schenberg ACG, Gallardo JCM, Coradello LFC, Pombeiro-Sponchiado SR. Advances and Developments in Strategies to Improve Strains of Saccharomyces cerevisiae and Processes to Obtain the Lignocellulosic Ethanol-A Review. Appl Biochem Biotech. 2012;166(8):1908–26.
Toone WM, Jones N. Stress-activated signalling pathways in yeast. Genes Cells. 1998;3(8):485–98.
Gasch AP. The environmental stress response: a common yeast response to diverse environmental stresses. In: Yeast stress responses. Springer; 2003. p. 11–70.
Rodicio R, Heinisch JJ. Together we are strong - cell wall integrity sensors in yeasts. Yeast. 2010;27(8):531–40.
Berry DB, Guan Q, Hose J, Haroon S, Gebbia M, Heisler LE, et al. Multiple means to the same end: the genetic basis of acquired stress resistance in yeast. Plos Genet. 2011;7(11), e1002353.
Almeida JRM, Runquist D, Sànchez i Nogué V, Lidén G, Gorwa-Grauslund MF. Stress-related challenges in pentose fermentation to ethanol by the yeast Saccharomyces cerevisiae. Biotechnol J. 2011;6(3):286–99.
Liu ZL. Molecular mechanisms of yeast tolerance and in situ detoxification of lignocellulose hydrolysates. Appl Microbiol Biot. 2011;90(3):809–25.
Liu ZL, Slininger PJ, Gorsich SW. Enhanced biotransformation of furfural and hydroxymethylfurfural by newly developed ethanologenic yeast strains. Appl Biochem Biotech. 2005;121:451–60.
Martin C, Marcet M, Almazan O, Jönsson LJ. Adaptation of a recombinant xylose-utilizing Saccharomyces cerevisiae strain to a sugarcane bagasse hydrolysate with high content of fermentation inhibitors. Bioresource Technol. 2007;98(9):1767–73.
Heer D, Sauer U. Identification of furfural as a key toxin in lignocellulosic hydrolysates and evolution of a tolerant yeast strain. Microb Biotechnol. 2008;1(6):497–506.
Almario MP, Reyes LH, Kao KC. Evolutionary Engineering of Saccharomyces cerevisiae for Enhanced Tolerance to Hydrolysates of Lignocellulosic Biomass. Biotechnol Bioeng. 2013;110(10):2616–23.
Heer D, Heine D, Sauer U. Resistance of Saccharomyces cerevisiae to High Concentrations of Furfural Is Based on NADPH-Dependent Reduction by at Least Two Oxireductases. Appl Environ Microb. 2009;75(24):7631–8.
Morano KA, Grant CM, Moye-Rowley WS. The Response to Heat Shock and Oxidative Stress in Saccharomyces cerevisiae. Genetics. 2012;190(4):1157–95.
Morano KA, Liu PCC, Thiele DJ. Protein chaperones and the heat shock response in Saccharomyces cerevisiae. Curr Opin Microbiol. 1998;1(2):197–203.
Verghese J, Abrams J, Wang YY, Morano KA. Biology of the Heat Shock Response and Protein Chaperones: Budding Yeast (Saccharomyces cerevisiae) as a Model System. Microbiol Mol Biol R. 2012;76(2):115–58.
Caspeta L, Chen Y, Ghiaci P, Feizi A, Buskov S, Hallström BM, et al. Altered sterol composition renders yeast thermotolerant. Science. 2014;346(6205):75–8.
Wallace-Salinas V, Gorwa-Grauslund MF. Adaptive evolution of an industrial strain of Saccharomyces cerevisiae for combined tolerance to inhibitors and temperature. Biotechnol Biofuels. 2013;6(1):151.
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, et al. Life with 6000 genes. Science. 1996;274(5287):546.
Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, Costanzo MC, et al. The Reference Genome Sequence of Saccharomyces cerevisiae: Then and Now. G3-Genes Genom Genet. 2014;4(3):389–98.
Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42(D1):D764–70.
Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, et al. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25(2):288–9.
Zhang Z, Li J, Zhao X-Q, Wang J, Wong GK-S, Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics. 2006;4(4):259–63.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009;25(15):1968–9.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12.
Kent WJ. BLAT - The BLAST-like alignment tool. Genome Res. 2002;12(4):656–64.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10.
Team RC. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. http://www.R-project.org/.
Verduyn C, Postma E, Scheffers WA, Vandijken JP. Effect of Benzoic-Acid on Metabolic Fluxes in Yeasts - a Continuous-Culture Study on the Regulation of Respiration and Alcoholic Fermentation. Yeast. 1992;8(7):501–17.
Sherman F. Suppression in the yeast Saccharomyces cerevisiae. Cold Spring Harbor Monograph Archive. 1982;11:463–86.
Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8(3):186–94.
Wohlbach DJ, Rovinskiy N, Lewis JA, Sardi M, Schackwitz WS, Martin JA, et al. Comparative Genomics of Saccharomyces cerevisiae Natural Isolates for Bioenergy Production. Genome Biol Evol. 2014;6(9):2557–66.
Borneman AR, Desany BA, Riches D, Affourtit JP, Forgan AH, Pretorius IS, et al. Whole-Genome Comparison Reveals Novel Genetic Elements That Characterize the Genome of Industrial Strains of Saccharomyces cerevisiae. Plos Genet. 2011;7(2):e1001287.
Nijkamp JF, van den Broek M, Datema E, de Kok S, Bosman L, Luttik MA, et al. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact. 2012;11:36.
Drake JW. A Constant Rate of Spontaneous Mutation in DNA-Based Microbes. P Natl Acad Sci USA. 1991;88(16):7160–4.
Heer D. Molecular basis of stress resistance in Saccharomyces cerevisiae, PhD thesis. Zürich: ETH; 2009.
Szafraniec K, Borts RH, Korona R. Environmental stress and mutational load in diploid strains of the yeast Saccharomyces cerevisiae. P Natl Acad Sci USA. 2001;98(3):1107–12.
Goho S, Bell G. Mild environmental stress elicits mutations affecting fitness in Chlamydomonas. P Roy Soc B-Biol Sci. 2000;267(1439):123–9.
Lada AG, Stepchenkova EI, Waisertreiger ISR, Noskov VN, Dhar A, Eudy JD, et al. Genome-Wide Mutation Avalanches Induced in Diploid Yeast Cells by a Base Analog or an APOBEC Deaminase. Plos Genet. 2013;9(9):e1003736.
Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011;39(13):e90.
Guo Y, Li J, Li CI, Long JR, Samuels DC, Shyr Y. The effect of strand bias in Illumina short-read sequencing data. BMC Genomics. 2012;13.
Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB. Annotating non-coding regions of the genome. Nat Rev Genet. 2010;11(8):559–71.
Connelly CF, Skelly DA, Dunham MJ, Akey JM. Population Genomics and Transcriptional Consequences of Regulatory Motif Variation in Globally Diverse Saccharomyces cerevisiae Strains. Mol Biol Evol. 2013;30(7):1605–13.
Kruckeberg AL. The hexose transporter family of Saccharomyces cerevisiae. Arch Microbiol. 1996;166(5):283–92.
Levin DE. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol R. 2005;69(2):262–91.
Horvath I, Multhoff G, Sonnleitner A, Vigh L. Membrane-associated stress proteins: More than simply chaperones. BBA-Biomembranes. 2008;1778(7-8):1653–64.
Moskvina E, Imre EM, Ruis H. Stress factors acting at the level of the plasma membrane induce transcription via the stress response element (STRE) of the yeast Saccharomyces cerevisiae. Mol Microbiol. 1999;32(6):1263–72.
Vigh L, Maresca B, Harwood JL. Does the membrane's physical state control the expression of heat shock and other genes? Trends Biochem Sci. 1998;23(10):369–74.
Zhang M, Shi J, Jiang L. Modulation of mitochondrial membrane integrity and ROS formation by high temperature in Saccharomyces cerevisiae. Electron J Biotechno. 2015;18(3): 202-209.
de Thozée CP, Cronin S, Goj A, Golin J, Ghislain M. Subcellular trafficking of the yeast plasma membrane ABC transporter, Pdr5, is impaired by a mutation in the N-terminal nucleotide-binding fold. Mol Microbiol. 2007;63(3):811–25.
Panaretou B, Piper PW. Plasma-Membrane Atpase Action Affects Several Stress Tolerances of Saccharomyces cerevisiae and Schizosaccharomyces Pombe as Well as the Extent and Duration of the Heat-Shock Response. J Gen Microbiol. 1990;136:1763–70.
Allen SA, Clark W, McCaffery JM, Cai Z, Lanctot A, Slininger PJ, et al. Furfural induces reactive oxygen species accumulation and cellular damage in Saccharomyces cerevisiae. Biotechnol Biofuels. 2010;3.
Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: From small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radical Bio Med. 2001;31(12):1624–32.
Farah ME, Amberg DC. Conserved actin cysteine residues are oxidative stress sensors that can regulate cell death in yeast. Mol Biol Cell. 2007;18(4):1359–65.
Pinel D, Colatriano D, Jiang H, Lee H, Martin VJJ. Deconstructing the genetic basis of spent sulphite liquor tolerance using deep sequencing of genome-shuffled yeast. Biotechnol Biofuels. 2015;8.
Hernandez-Lopez MJ, Garcia-Marques S, Randez-Gil F, Prieto JA. Multicopy Suppression Screening of Saccharomyces cerevisiae Identifies the Ubiquitination Machinery as a Main Target for Improving Growth at Low Temperatures. Appl Environ Microb. 2011;77(21):7517–25.
Hanna J, Leggett DS, Finley D. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol Cell Biol. 2003;23(24):9251–61.
Jordan IK, McDonald JF. Tempo and mode of Ty element evolution in Saccharomyces cerevisiae. Genetics. 1999;151(4):1341–51.
Lesage P, Todeschini AL. Happy together: the life and times of Ty retrotransposons and their hosts. Cytogenet Genome Res. 2005;110(1-4):70–90.
Mitchell-Olds T, Willis JH, Goldstein DB. Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat Rev Genet. 2007;8(11):845–56.
Vilella F, Herrero E, Torres J, de la Torre-Ruiz MA. Pkc1 and the upstream elements of the cell integrity pathway in Saccharomyces cerevisiae, Rom2 and Mtl1, are required for cellular responses to oxidative stress. J Biol Chem. 2005;280(10):9149–59.
Petkova MI, Pujol-Carrion N, Arroyo J, Garcia-Cantalejo J, de la Torre-Ruiz MA. Mtl1 Is Required to Activate General Stress Response through Tor1 and Ras2 Inhibition under Conditions of Glucose Starvation and Oxidative Stress. J Biol Chem. 2010;285(25):19521–31.
Jendretzki A, Wittland J, Wilk S, Straede A, Heinisch JJ. How do I begin? Sensing extracellular stress to maintain yeast cell wall integrity. Eur J Cell Biol. 2011;90(9):740–4.
Bermejo C, Rodriguez E, Garcia R, Rodriguez-Pena JM, de la Concepcion MLR, Rivas C, et al. The sequential activation of the yeast HOG and SLT2 pathways is required for cell survival to cell wall stress. Mol Biol Cell. 2008;19(3):1113–24.
Teunissen AWRH, Steensma HY. Review - the Dominant Flocculation Genes of Saccharomyces cerevisiae Constitute a New Subtelomeric Gene Family. Yeast. 1995;11(11):1001–13.
Fidalgo M, Barrales RR, Ibeas JI, Jimenez J. Adaptive evolution by mutations in the FLO11 gene. P Natl Acad Sci USA. 2006;103(30):11228–33.
Smukalla S, Caldara M, Pochet N, Beauvais A, Guadagnini S, Yan C, et al. FLO1 Is a Variable Green Beard Gene that Drives Biofilm-like Cooperation in Budding Yeast. Cell. 2008;135(4):726–37.
Watanabe J, Uehara K, Mogi Y. Adaptation of the Osmotolerant Yeast Zygosaccharomyces rouxii to an Osmotic Environment Through Copy Number Amplification of FLO11D. Genetics. 2013;195(2):393–405.
Westman JO, Mapelli V, Taherzadeh MJ, Franzen CJ. Flocculation Causes Inhibitor Tolerance in Saccharomyces cerevisiae for Second-Generation Bioethanol Production. Appl Environ Microb. 2014;80(22):6908–18.
Hodgson JA, Berry DR, Johnston JR. Discrimination by heat and proteinase treatments between flocculent phenotypes conferred on Saccharomyces cerevisiae by the genes FLO1 and FLO5. J Gen Microbiol. 1985;131(12):3219–27.
Dumont ME, Ernst JF, Hampsey DM, Sherman F. Identification and Sequence of the Gene Encoding Cytochrome-C Heme Lyase in the Yeast Saccharomyces cerevisiae. Embo J. 1987;6(1):235–41.
Ocampo A, Liu J, Schroeder EA, Shadel GS, Barrientos A. Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 2012;16(1):55–67.
Merz S, Westermann B. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol. 2009;10(9):R95.
Jones DL, Petty J, Hoyle DC, Hayes A, Oliver SG, Riba-Garcia I, et al. Genome-wide analysis of the effects of heat shock on a Saccharomyces cerevisiae mutant with a constitutively activated cAMP-dependent pathway. Comp Funct Genom. 2004;5(5):419–31.
Reifenberger E, Freidel K, Ciriacy M. Identification of Novel Hxt Genes in Saccharomyces cerevisiae Reveals the Impact of Individual Hexose Transporters on Glycolytic Flux. Mol Microbiol. 1995;16(1):157–67.
Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, et al. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169(4):1915–25.
Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424(6945):194–7.
Kraakman L, Lemaire K, Ma PS, Teunissen AWRH, Donaton MCV, Van Dijck P, et al. A Saccharomyces cerevisiae G-protein coupled receptor, Gpr1, is specifically required for glucose activation of the cAMP pathway during the transition to growth on glucose. Mol Microbiol. 1999;32(5):1002–12.
Engelberg D, Zandi E, Parker CS, Karin M. The Yeast and Mammalian Ras Pathways Control Transcription of Heat-Shock Genes Independently of Heat-Shock Transcription Factor. Mol Cell Biol. 1994;14(7):4929–37.
Martínez-Pastor MT, Marchler G, Schuller C, MarchlerBauer A, Ruis H, Estruch F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress-response element (STRE). Embo J. 1996;15(9):2227–35.
Dunham MJ, Badrane H, Ferea T, Adams J, Brown PO, Rosenzweig F, et al. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. P Natl Acad Sci USA. 2002;99(25):16144–9.
Brown CJ, Todd KM, Rosenzweig RF. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol. 1998;15(8):931–42.
Bergström A, Simpson JT, Salinas F, Barre B, Parts L, Zia A, et al. A High-Definition View of Functional Genetic Variation from Natural Yeast Genomes. Mol Biol Evol. 2014;31(4):872–88.
Cohn M, Liti G, Barton DB. Telomeres in fungi. In: Comparative Genomics. Springer; 2006: 101-130.
Ruiz A, Arino J. Function and regulation of the Saccharomyces cerevisiae ENA sodium ATPase system. Eukaryot Cell. 2007;6(12):2175–83.
Gilbert A, Sangurdekar DP, Srienc F. Rapid Strain Improvement Through Optimized Evolution in the Cytostat. Biotechnol Bioeng. 2009;103(3):500–12.
Sehnem NT, Machado AD, Leite FCB, Pita WD, de Morais MA, Ayub MAZ. 5-Hydroxymethylfurfural induces ADH7 and ARI1 expression in tolerant industrial Saccharomyces cerevisiae strain P6H9 during bioethanol production. Bioresource Technol. 2013;133:190–6.
Liu ZL, Ma MG, Song MZ. Evolutionarily engineered ethanologenic yeast detoxifies lignocellulosic biomass conversion inhibitors by reprogrammed pathways. Mol Genet Genomics. 2009;282(3):233–44.
Liu ZL, Moon J, Andersh BJ, Slininger PJ, Weber S. Multiple gene-mediated NAD(P)H-dependent aldehyde reduction is a mechanism of in situ detoxification of furfural and 5-hydroxymethylfurfural by Saccharomyces cerevisiae. Appl Microbiol Biot. 2008;81(4):743–53.
Koga Y. Thermal Adaptation of the Archaeal and Bacterial Lipid Membranes. Archaea. 2012;2012:789652.
de Marañón IM, Chaudanson N, Joly N, Gervais P. Slow heat rate increases yeast thermotolerance by maintaining plasma membrane integrity. Biotechnol Bioeng. 1999;65(2):176–81.
Hickey DA, Singer GAC. Genomic and proteomic adaptations to growth at high temperature. Genome Biol. 2004;5(10):117.
Hunter K, Rose AH. Lipid Composition of Saccharomyces cerevisiae as Influenced by Growth Temperature. BBA-Lipid Lipid Met. 1972;260(4):639–53.
Klose C, Surma MA, Gerl MJ, Meyenhofer F, Shevchenko A, Simons K. Flexibility of a Eukaryotic Lipidome - Insights from Yeast Lipidomics. Plos One. 2012;7(4):e35063.
Xia JM, Yuan YJ. Comparative Lipidomics of Four Strains of Saccharomyces cerevisiae Reveals Different Responses to Furfural, Phenol, and Acetic Acid. J Agr Food Chem. 2009;57(1):99–108.
Lindberg L, Santos AXS, Riezman H, Olsson L, Bettiga M. Lipidomic Profiling of Saccharomyces cerevisiae and Zygosaccharomyces bailii Reveals Critical Changes in Lipid Composition in Response to Acetic Acid Stress. Plos One. 2013;8(9):e73936.
Balogh G, Peter M, Glatz A, Gombos I, Torok Z, Horvath I, et al. Key role of lipids in heat stress management. Febs Lett. 2013;587(13):1970–80.
Matias AC, Pedroso N, Teodoro N, Marinho HS, Antunes F, Nogueira JMF, et al. Down-regulation of fatty acid synthase increases the resistance of Saccharomyces cerevisiae cells to H(2)O(2). Free Radical Bio Med. 2007;43(10):1458–65.
Swan TM, Watson K. Stress tolerance in a yeast sterol auxotroph: role of ergosterol, heat shock proteins and trehalose. Fems Microbiol Lett. 1998;169(1):191–7.
Wijeyaratne SC, Ohta K, Chavanich S, Mahamontri V, Nilubol N, Hayashida S. Lipid-Composition of a Thermotolerant Yeast, Hansenula Polymorpha. Agr Biol Chem Tokyo. 1986;50(4):827–32.
Daum G, Lees ND, Bard M, Dickson R. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast. 1998;14(16):1471–510.
Dickson RC. Roles for sphingolipids in Saccharomyces cerevisiae. In: Sphingolipids as signaling and regulatory molecules. Springer; 2010: 217-231.
Meier KD, Deloche O, Kajiwara K, Funato K, Riezman H. Sphingoid base is required for translation initiation during heat stress in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17(3):1164–75.
Cowart LA, Gandy JL, Tholanikunnel B, Hannun YA. Sphingolipids mediate formation of mRNA processing bodies during the heat-stress response of Saccharomyces cerevisiae. Biochem J. 2010;431:31–8.
Johansen J, Ramanathan V, Beh CT. Vesicle trafficking from a lipid perspective: Lipid regulation of exocytosis in Saccharomyces cerevisiae. Cell Logist. 2012;2:151–60.
Saris N, Makarow M. Transient ER retention as stress response: conformational repair of heat-damaged proteins to secretion-competent structures. J Cell Sci. 1998;111:1575–82.
Stein K, Chiang HL. Exocytosis and Endocytosis of Small Vesicles across the Plasma Membrane in Saccharomyces cerevisiae. Membranes. 2014;4(3):608–29.
Van Uden N. Temperature Profiles of Yeasts. Adv Microb Physiol. 1985;25:195.
Ephrussi B, Hottinguer H, Chimenes AM. Action De Lacriflavine Sur Les Levures.1. La Mutation Petite Colonie. Ann I Pasteur Paris. 1949;76(4):351–67.
Sherman F. Getting started with yeast. Method Enzymol. 2002;350:3–41.
Granek JA, Magwene PM. Environmental and Genetic Determinants of Colony Morphology in Yeast. Plos Genet. 2010;6(1):e1000823.
Gagiano M, Bauer FF, Pretorius IS. The sensing of nutritional status and the relationship to filamentous growth in Saccharomyces cerevisiae. Fems Yeast Res. 2002;2(4):433–70.
Verstrepen KJ, Klis FM. Flocculation, adhesion and biofilm formation in yeasts. Mol Microbiol. 2006;60(1):5–15.
Zupan J, Raspor P. Invasive growth of Saccharomyces cerevisiae depends on environmental triggers: a quantitative model. Yeast. 2010;27(4):217–28.
Lengeler KB, Davidson RC, D'Souza C, Harashima T, Shen WC, Wang P, et al. Signal transduction cascades regulating fungal development and virulence. Microbiol Mol Biol R. 2000;64(4):746–85.
Vinod PK, Sengupta N, Bhat PJ, Venkatesh KV. Integration of Global Signaling Pathways, cAMP-PKA, MAPK and TOR in the Regulation of FLO11. Plos One. 2008;3(2):e1663.
Birkaya B, Maddi A, Joshi J, Free SJ, Cullen PJ. Role of the Cell Wall Integrity and Filamentous Growth Mitogen-Activated Protein Kinase Pathways in Cell Wall Remodeling during Filamentous Growth. Eukaryot Cell. 2009;8(8):1118–33.
Rupp S, Summers E, Lo HJ, Madhani H, Fink G. MAP kinase and cAMP filamentation signaling pathways converge on the unusually large promoter of the yeast FLO11 gene. Embo J. 1999;18(5):1257–69.
Voordeckers K, De Maeyer D, van der Zande E, Vinces MD, Meert W, Cloots L, et al. Identification of a complex genetic network underlying Saccharomyces cerevisiae colony morphology. Mol Microbiol. 2012;86(1):225–39.
Sànchez i Nogué V, Bettiga M, Gorwa-Grauslund MF. Isolation and characterization of a resident tolerant Saccharomyces cerevisiae strain from a spent sulfite liquor fermentation plant. AMB Express. 2012;2(1):1–11.
Oud B, Guadalupe-Medina V, Nijkamp JF, de Ridder D, Pronk JT, van Maris AJA, et al. Genome duplication and mutations in ACE2 cause multicellular, fast-sedimenting phenotypes in evolved Saccharomyces cerevisiae. P Natl Acad Sci USA. 2013;110(45):E4223–31.
Weitao T. Multicellularity of a unicellular organism in response to DNA replication stress. Res Microbiol. 2009;160(1):87–8.
Acknowledgements
We would like to thank Lina Lindahl for her support during the analysis of the lipidome data. The Swedish Energy Agency (Energimyndigheten) is gratefully acknowledged for funding this research. The computations were performed on resources provided by SNIC (Swedish National Infrastructure for Computing) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under project b2013228, using the UPPNEX bioinformatics resources.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VWS conceived the study, performed the sample preparation for the genomics and lipidomics, and performed the alternative-carbon source experiment. DB designed and performed the in-house bioinformatics as well as the viability- and cell wall lysis experiments. DA participated in the design of the bioinformatics analysis. VWS and DB drafted the manuscript. MGG participated in the design of the study and revised the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Supplementary Information. This file contains supplementary Materials & Methods and Results for the physiological characterisation of ISO12 and for the distribution of sequence variants in the two strains.
Additional file 2:
Synonymous and non-synonymous variants in Ethanol Red. This file contains a list of the synonymous and non-synonymous SNPs and INDELs that were found to be unique to Ethanol Red compared to ISO12 (with a S288c background). The analysis was performed using the Variant Annotation Integrator the UCSC Genome Browser.
Additional file 3:
Synonymous and non-synonymous variants in ISO12. This file contains a list of the synonymous and non-synonymous SNPs and INDELs that were found to be unique to ISO12 compared to Ethanol Red (with a S288c background). The analysis was performed using the Variant Annotation Integrator the UCSC Genome Browser.
Additional file 4:
ORFs that were matched to the significantly enriched Gene Ontology terms. This file contains a list of all the ORFs that were matched to the significantly enriched Gene Ontology (GO) terms (analysed with the YeastMine function at Saccharomyces Genome Database).
Additional file 5:
ORFs related to vesicle mediated transport, lipid metabolic processes and mitochondria. This file contains a list of all the ORFs that were matched to these three Ontology (GO) terms (analysed with the AmiGO 2 database).
Additional file 6:
K a /K s modelling of ISO12 versus ER. This file contains the results of the modelling of the non-synonymous to synonymous substitution ratio (Ka/Ks).
Additional file 7:
Copy Number Variation assessment of the reference strain regions in ER and ISO12. This file contains the results of the read mapping-based Copy Number Variation (CNV) analysis of the reference strain (S288c) regions of ER and ISO12.
Additional file 8:
Assessment of the non-reference strain regions in ER and ISO12, including Copy Number Variations. This file contains the results of the CNV-analysis of the non-reference strain regions of ER and ISO12 based on the de novo assemblies of both strains, as well as a list of the unmapped contigs and their alignments.
Additional file 9:
Metabolome and differential analysis of the lipid species compared between ER and ISO12. This file contains the dataset of the relative concentration of each lipid species (119 metabolites) on each biological sample of ER and ISO12, together with the differential analysis.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wallace-Salinas, V., Brink, D.P., Ahrén, D. et al. Cell periphery-related proteins as major genomic targets behind the adaptive evolution of an industrial Saccharomyces cerevisiae strain to combined heat and hydrolysate stress. BMC Genomics 16, 514 (2015). https://doi.org/10.1186/s12864-015-1737-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1737-4