
Abstract
Background
European eel is a panmictic species, whose decline has been recorded since the last 20 years. Among human-induced environmental factors of decline, the impact of water dams during species migration is questioned. The main issue of this study was to pinpoint phenotypic traits that predisposed glass eels to successful passage by water barriers. The approach of the study was individual-centred and without any a priori hypothesis on traits involved in the putative obstacles selective pressure. We analyzed the transcription level of 14,913 genes.
Results
Transcriptome analysis of three tissues (brain, liver and muscle) from individuals sampled on three successive forebays separated by water obstacles indicated different gene transcription profiles in brain between the two upstream forebays. No differences in gene transcription levels were observed in liver and muscle samples among segments. A total of 26 genes were differentially transcribed in brain. These genes encode for, among others, keratins, cytokeratins, calcium binding proteins (S100 family), cofilin, calmodulin, claudin and thy-1 membrane glycoprotein. The functional analysis of these genes highlighted a putative role of cytoskeletal dynamics and synaptic plasticity in fish upstream migration.
Conclusion
Synaptic connections in brain are solicited while eels are climbing the obstacles with poorly designed fishways. Successful passage by such barriers can be related to spatial learning and spatial orientation abilities when fish is out of the water.
Similar content being viewed by others

Background
Among anthropogenic environmental alterations, habitat loss and fragmentation are considered as a major threat to biological diversity [1] and dealing with these changes is among the greatest challenges faced by conservation biologists [2]. Habitat fragmentation of aquatic ecosystems is mainly induced by anthropogenic barriers such as dams and weirs [3]. Main effects of human-induced barriers are: (1) modification of abiotic conditions [4], (2) disruption of population and aquatic community structure in subsequent habitats [5-7] as well as (3) disruption of gene flow [8] and (4) biodiversity loss [9].

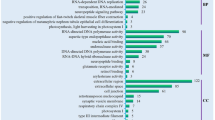
Biological functions of main genes differentially transcribed among segments 3:1 and segments 3:2. The font size is chosen according to the ratios of fold changes; i.e. FC 3:1/ FC 3:2 (Table 4). For more detailed information on each genetic sequence, BLAST statistics, and gene ontology, please see Additional file 1: Table S1.
Endemic species as well as migratory species are the most affected by water impoundment [10,11]. In the case of migratory species, habitat switch can be sometimes imperative in order to reach a particular ontogenetic stage. For diadromous species (i.e. salmonids or eels), growth and reproduction stages require different salinity environments and thus free-flowing corridors between habitats are required to fulfill their life cycle [12]. Therefore, investigating the effects of fragmentation effects on their migratory behavior is of great importance.
European eel Anguilla anguilla is a facultative catadromous fish species with a particularly complex life cycle that includes two trans-Atlantic migrations and two metamorphoses. European eel spawns in the remote Sargasso Sea [13]. After spawning, larvae of European eel drift back towards Europe and cross the Atlantic transported by the Gulf Stream system to the coasts of Europe and North Africa. Upon reaching the continental shelf, larvae metamorphose into glass eels and complete the migration into continental (fresh, estuarine and coastal) waters as yellow eels. After a highly variable feeding period in the continent, yellow eels metamorphose into silver eels that migrate back to the Sargasso Sea in a migration of 5,000-6,000 km. Upon reaching the Sargasso Sea, silver adults reproduce once and die. European eel is considered to be critically endangered of extinction with a 90-99% decline observed throughout the distribution range of the species in the last 30 years [14]. The drop in population numbers has affected both recruitment and pre-adult/adult stages and causes of the decline include both anthropogenic (overfishing, man-introduced parasites and diseases, pollution, habitat fragmentation) and natural factors. Among human-related barriers, construction of dams and the consecutive fragmentation of habitats is one of the possible factors contributing to this sharp decline, mainly because water impondment preclude (1) the upstream migration of glass eels to feeding grounds and (2) the spawning migration of adults from the feeding to the spawning grounds in the Sargasso Sea.
Studies addressing the impact of migration barriers on eel upstream migration have mainly focused on quantitative aspects such as mortality of glass eels/elvers downstream of dams due to predation, diseases or intraspecific competition [15]. Such studies also quantified barrier permeability by estimating the abundance of eels on either site thereof. Indeed, many evaluations of single-species specific fishways accounted for passage and attraction efficiency and were only based on the proportion of individuals approaching, engaging and succeeding to pass designed apparatus [16,17]. Although several phenotypic traits are expected to be associated with glass eel upstream migration [18,19], none of them has been studied in the context of passage by water obstacle. In this sense, large individual size [20], swimming speed and high energy reserves could facilitate the success of passage, whereas activity, exploratory behavior and sensibility to environmental cues [21] could increase the probability to find the fishway entrance.
Several authors have proposed a hypothesis of energy- or thyroid- dependent propensity to migrate in glass eels [18,22,23]. According to this hypothesis, the thyroid hormone metabolism is involved in the upstream migration of glass eels. This is in agreement with previous experimental studies that showed that thyroxin (T4) is involved in the migratory behavior of fish [24,25]. A role of the thyroid hormone metabolism in climbing waterfalls has also been suggested in juvenile American eels [26]. Thus, the interindividual variations in thyroid hormone metabolism could be responsible for interindividual variations in “motivation” of juvenile eels to swim against the current and to climb water obstacles.
The aim of the present study was to investigate the interindividual variation of phenotypic traits involved in the passage of water barriers. The chosen approach relied on the comparison of gene transcription (mRNA) patterns among wild glass eels collected below and above successive obstacles dispersed along the same river course.
A non a priori approach was chosen to identify individual traits that could differ between downstream and upstream fish. Microarray analysis was used to detect interindividual patterns of gene transcription from a large and functionally diverse set of genes (14,913 annotated contigs [27]) from fish sampled above and directly below the barriers in a river carefully selected for its homogenous conditions.
Three different tissues were sampled: (1) muscle to provide information on fish swimming capacities [28], (2) liver as a proxy for the physiological state of organism [29], and (3) brain, to provide information on perception of environmental cues, arousal, motivation, learning and many other functions involved in behavioral patterns such as those linked to hormone metabolism [22,23].
Methods
All procedures used in this study were approved by the Aquitaine fish-birds ethic committee (a committee approved and registered by the French Ministry of Higher Education and Research under number 73).
Sampling site
Canal des Etangs, a freshwater corridor in South-Western France (44.75-44.95 N, 1.1-1.2 W) is a former artificial canal, built in 1850, linking Arcachon Basin to Lacanau Lake. The river line is linear, whereas the water flow remains homogenous and controlled by several weirs. Three successive low-distanced obstacles were built along the river length. The first weir (1,5 m height) is located at 4 km from the tidal limit and equipped with a fish ladder (6 m length, 45° slope) specifically designed for glass eels. It determines the upper limit of the most downstream 4 km section of the canal called segment 1 (Pas du Bouc; +44° 50’ 27.95”, -1° 9’ 8.09”). The second (Langouarde, +44° 51’ 32.92”, -1° 9’ 5.03”) and the third (Joncru; +44° 52’ 57.13”, -1° 8’ 11.70”) weirs are different from the first one, but similar between them; they are larger (2,5 m height) and equipped with identical fishways (rock ramp). The distances from the first weir to the 2nd and 3rd one are respectively 2.3 and 5.3 km (Table 1).
Sampling
Eels were collected using electrofishing during three consecutive days 6-8th of June 2012 under similar climatic and hydrological conditions. Individuals were sampled below the obstacle, close to the fishway entry in segment 1 and 2. Fish from the segment 3 were sampled on the fishway, as water depth before the obstacle, approximately 2 meters, precluded the use of electrofishing. Ten individuals were selected from each site according to their body size (between 83 and 155 mm) and health status (no externally visible pathogens) to minimize the potential bias. Sampled and selected fish were immediately sacrificed by decapitation and the whole brain (ca. 3.5 mg), liver (ca. 30 mg) and muscle tissues (ca. 40 mg, posterior bottom body part, skin removed) were dissected and stored in separate tubes with RNAlater buffer (1 mL, Qiagen) for gene transcription analysis. Additionally, individual weight was measured for relative condition factor (Kn) calculation [30] and otoliths were extracted for further age analysis.
Otolith analyses
Sagittal otoliths were embedded in glass slides and submerged by a drop of glue. The otolith was then polished until the core was reached, etched with 10% EDTA, stained with 5% toluidine blue to enhance the annuli and observed with optical microscope (Nikon Eclipse 90i, Japan). For each otolith, age estimation by counting the annuli around the primordium was performed by two independent readers.
Microarray analyses
Samples of brain, muscle and liver were homogenized by measn of a bead mill homogenizer (45 sec at 3000 oscillations per sec, Mixer Mill MM 200, Retsch). Total RNAs were extracted using the Absolutely RNA RT-PCR Miniprep kit (Agilent) according to the manufacturer’s instructions. A total of 9 fish were used for each sampling site, i.e. 3 pools of 3 individuals by site. RNA quality was evaluated by electrophoresis on a 1% agarose gel. RNA purity and concentration was determined using a NanoDrop spectrophotometry and Agilent 2100 Bioanalyzer. Samples were considered as of good quality RNA when showing A260/280 and A260/230 ratios close to 2 and a minimum RIN (RNA Integrity Number) of 8.
Microarray analysis was conducted using an European eel-specific array consisting of a total of 14,913 probes based on a large collection of high-throughput transcriptomic sequences [27]. Probe sequences and further details on the microarray platform can be found on the GEO database under accession number GPL15124. Sample labelling and hybridization were carried out following the Agilent One-Color Microarray-Based Gene Expression Analysis protocol (Low Input Quick Amp Labelling) [31]. For each individual, 100 ng total RNA were linearly amplified and labelled with the fluorescent dye Cy3-dCTP. In order to monitor microarray analysis work-flow, Agilent Spike-in Mix (a mixture of 10 different viral poly-adenylated RNAs) was added to each RNA sample before amplification and labelling. Labelled cRNA was purified with Qiagen RNAeasy Mini Kit and sample concentration and Cy3 specific activity were measured using a Nanodrop ND-1000 spectrophotometer.
A Cy3 specific activity between 8 and 17 pmol Cy3 per μg cRNA was considered adequate for hybridization. Prior to hybridization, a total of 600 ng of labelled cRNA was fragmented for 30 min at 60°C by adding 5 μl 10X Blocking Agent and 1 μl Fragmentation buffer, and finally diluted with 25 μl 2X GE Hybridization buffer. A volume of 40 μl was dispended into the backing slide, assembled to the microarray slide (each slide containing eight arrays) and placed in the hybridization chamber. Slides were incubated for 17 h at 65°C in an Agilent Hybridization Oven. Afterwards, slides were removed from the hybridization chamber, disassembled in GE Wash Buffer 1, and washed for 1 min in GE Wash Buffer 1 followed by one additional wash for 1 min in GE Wash Buffer 2. Hybridized slides were scanned at 5 μm resolution using an Agilent DNA microarray scanner. Slides were scanned at two different sensitivity levels (XDR Hi 100% and XDR Lo 10%) to increase the power to detect both lowly and highly expressed genes. The two linked images generated were analyzed together. Data were extracted and background subtracted using the standard procedure in Agilent Feature Extraction (FE) software v. 9.5.1. Data was normalized using a quantile normalization procedure using R (http://www.rproject.org/). Normalized fluorescence data from the arrays have been deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE56040. Differentially transcribed genes across samples were identified using the program SAM (Significance Analysis of Microarrays) version 4.0 [32], with the FDR cutoff of 5%. Groups (pools of individuals from each of three segments) were compared using the two-class unpaired test and up-and-down regulated genes were identified. A minimum fold change of 1.5 between groups was considered.
In total, 14,913 genetic sequences were analyzed from brain and liver of 27 individuals (9 pools of three individuals) and from muscle of 18 individuals (no samples from the third segment) (Table 1).
Quantitative RT-PCR validation of microarray results
A total of 6 genes showing different transcription levels among segments (claud4, cfl1, s100a1, s100a6, s100a11, thy1) were chosen to validate the microarray results by means of quantitative real-time Reverse Transcription Polymerase Chanin Reaction (qRT-PCR). For each gene, specific primer pairs were determined using the Primer3Plus software [33] (see Additional file 1: Table S1). Gene transcription level was measured by quantitative real-time Reverse transcribed-Polymerase Chain Reaction (RT-PCR), using the β-actin gene as reference. Amplification of cDNA was monitored using the DNA intercaling dye SyberGreen. Real-time PCR reactions were performed in a MX3000P (Stratagene) following the manufacturer’s instructions (one cycle at 95°C for 10 min, and 40 amplification cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 30 s). Each 25 μL reaction contained 1 μL of reverse transcribed product template, 12.5 μL of mix including the SyberGreen fluorescent dye and the enzyme (GoTaq Probe qPCR Master Mix, Promega), 9.5 μL of sterilized pure-water and 2 μL of the gene-specific primer pair at a final concentration of 200 nM for each primer. Reaction specificity was determined for each reaction from the dissociation curve of the PCR product and by electrophoresis. The dissociation curve was obtained by following the SyberGreen fluorescence level during gradual heating of the PCR products from 60 to 95°C. Relative quantification of each gene transcription level was normalized according to the β-actin gene transcription. Hence, during our experiment, total RNAs were quantified and a same quantity was used for reverse-transcription. During the subsequent qPCR amplifications, the output cycle corresponding to β-actin was examined. This output was always obtained around the same output cycle and no significant variations were observed among conditions, demonstrating the relevance of the β-actin as reference gene in our conditions.
Statistical analyses
Comparisons among fish groups were performed by analysis of variance (ANOVA), after testing the assumptions of normality (Shapiro-Wilk test) and homoscedascity (Bartlett test) of the error terms. When assumptions were not met, the non-parametric Kruskal Wallis test was used. If significant effects were detected, a Tukey HSD test was used to determine whether means between pairs of samples were significantly different from each other. Computations were performed using R (http://www.r-project.org/).
Results
Morphometric data
First, no difference in length or weight were observed among segments (p = 0.548). In addition, no age difference was observed among segments (p = 0.497). At the opposite, the relative body condition was significantly influenced by sampling site (Table 2) and post-hoc analysis indicated significantly lower Kn values in fish sampled in the 3rd segment (Joncru) in comparison with those sampled from the 1st and the 2nd segments (p < 0.001 and p = 0.012 respectively). No difference was observed between the 1st and the 2nd segment (p = 0.12) (Table 2).
Microarray results
No differences in gene transcription levels were observed in liver and muscle samples among segments. The only differences were observed in the brain tissue (Table 3). Only few differences (n = 5 genes, FDR cutoff = 5%) were observed between segments 1 and 2 (Table 3). A larger number of genes was differentially transcribed when comparing segment 3 with the other two segments: 50 genes between segments 1-3 and 74 genes between segments 2-3 (FDR cutoff = 5%).
A total of 40 genes were common to the comparisons between segments 1-3 and segments 2-3. Moreover, all these genes were up-regulated in the most upstream segment. Indeed, these common genes showed a progressive pattern of expression, i.e. the more upstream the segment (or distanced), the more the genes were overexpressed. Differences in regulation of gene expression between the most distanced segments were up to two times higher (shown in bold, Table 4) than those determined between close-distanced segments.
qPCR validation of microarray results
To validate microarray data, the transcriptional level of 6 genes that showed strong variations in their transcription levels among sampling sites was measured by qRT-PCR method. These two independent measures, by microarray and qRT-PCR, of transcript abundance gave consistent results, i.e. similar fold changes were observed (see Additional file 1: Figure S1).
Discussion
Energetic costs of obstacle passage
Eels sampled in the upstream and downstream segments of an impounded watercourse did not show any difference in terms of age, weight or length, suggesting that the ontogenetic stage of fish was homogenous along the river. Indeed, size was previously found to be the best proxy to assess the ontogenetic stage of glass eels and its related locomotory behavior [34]. Thus, in the present study, even if an effect of ontogenic stage cannot be completely excluded, it appears unlikely that differences observed in fish brain could be explained by the life stage of fish.
In contrast, the fact that relative body condition (Kn) was lower in eels from the most upstream group in comparison to those sampled downstream could imply that passage of water barriers is an energetically and metabolically requiring event [35]. An alternative hypothesis could be that since glass eels do not feed during their upstream migration [15], the distance covered to reach upstream segments might have reduced their energy reserves [36]. Our results are contradictory to a previous study [37], where tendency to migrate was associated with higher energy reserves. In the present study, no differences in muscle and liver gene transcription among fish groups were found, which suggests that energy was not the main cue explaining the difference in passage behavior. However, it is important to notice that muscle samples from the third segment were missing (Table 3), thus precluding a full inter-segment comparison for this tissue.
Differences in brain transcriptome profiles associated with upstream migration
Microarray analysis of brain tissue revealed that some genes were overexpressed in fish from the most upstream segment of the river compared to the two downstream sections.
Interestingly, most of these overexpressed genes were common to the comparisons between segments 1-3 and segments 2-3. In addition, the fold changes for most of the genes were found to increase with the number of obstacles crossed by glass eels. The analysis of the biological function of these common genes can provide new insights into the phenotypic traits that are stimulated and/or selected after obstacle passage (Figure 1). Among the 40 common overexpressed genes, 4 genes encoding for proteins belonging to the S-100 family proteins were identified.
The S-100 proteins [38,39] are known to control the intracellular homeostasis of calcium, which is one of messengers mediating the effects of neurotransmitters [40]. Moreover, S-100 proteins are also involved in microtubules and microfilaments synthesis [41,42]. They contribute to a broad spectrum of biological processes in the brain including cell migration, gene expression and neural signaling and activity [43] and even learning and memory at a higher biological level [44]. Calcium signaling is indeed an important pathway controlling neuronal activity, fast axonal flow and memory [45,46]. One of the S-100 members, S100A6 (Calcyclin), was shown to be highly expressed in rat brain neurons [47], and its suggested functions include cell proliferation, differentiation [48] as well as cytoskeletal rearrangements [49,50] and cellular signal transduction [51]. At a higher biological level, S100A6 was shown to be associated with memory formation in rats [52]. Another S-100 member is S100A11 (Calgizzarin), involved in regulating growth of cells [53] such as keratinocytes [54]. Interestingly, both S100A11 and S100A6 are specific targets of S100B [55], which is involved in neural plasticity [56]. S100A1 protein could be associated with synapsin and is involved in calcium dependent synaptic vesicle trafficking [57]. Moreover, an association between exploratory behavior and S100A1 has been suggested in mice [58]. Finally, S100P protein is involved in cytoskeletal dynamics [59] and cell proliferation [60].
Another overexpressed gene was the cytoskeletal Cofilin (or ADF). This gene encodes for a protein that is involved in actin filament destabilization [61], which in turn allows dendritic development and differentiation, as well as neural polarization in mammalian brain [62] and axonal specialization [63]. Cofilin is also involved in other similar functions, such as axogenogenesis, growth cone guidance and dendritic spine formation [64]. A role in synaptic plasticity in rats [65] and associative learning has been proposed in both rats and mice [66-68].
Another up-regulated gene was claudin 4, which is involved in epithelial tight junction [69] which in turn allows intercellular communication. Moreover, epithelial tissue is rich in intermediate filaments and cytokeratins, which could be linked with other genes overexpressed in glass eels found upstream. Thus, the genes encoding for the cytokeratin 1 and keratin 12 were found to be overexpressed in migratory eels. Keratins are structural proteins found in neurons and glial cells [70]. Keratins are also known for their transient overexpression during neural differentiation from polymorphic cells in rabbits [71]. Calmodulin is a well-studied protein involved in calcium-related [72] synaptic neurotransmission [73] and calcium-dependent gene expression [74]. Together with several transcription factors from the IEG (Immediate Early Genes) family under its regulation, calmodulin is strongly linked to learning and memory [75-78], as found in rats. Indeed, genes belonging to the IEG group are among the first genes regulated in response to environmental stimuli [79]. They are involved in long term potentiating (LTP) and in the establishment of long term memory that requires rapid de novo synthesis of proteins [80]. Among the other overexpressed genes, Thy1 encodes a neural surface glycoprotein that was shown to play a role in axogenogenesis in rats [81] and in olfactory system development in mice [82]. Fatty acid binding protein (FABP) are involved in several functions in brain, among which neural development and cognitive processes appear to be common to the functions of other overexpressed genes in this study [83].
None of the overexpressed genes were related to thyroid activity, such as iodothyronine deiodinase type I and III. Thus, the hypothesis of thyroid dependent propensity to migrate [84] or climb obstacles [26] was not supported by our results. In contrast, overexpressed genes in upstream eels were mainly involved in cellular signaling, neural development and differentiation, as well as synaptic plasticity.
Water obstacle effects on gene expression
The difference in gene transcription in fish brain between the most upstream group and the two others could be interpreted either as a difference in brain development [85] or as a difference in cognitive, learning and memory abilities between groups. Fish brain growth is allometric and development of its various parts is linked to environmental conditions [86-89]. Previous experimental investigations pointed out the association between behavioral flexibility and cognitive abilities [90]. Indeed, personality and coping style concepts were both related to individual capacities in spatial memory and learning abilities, where differential regulation of genes involved in neurogenesis was emphasized [91].
Phenotypic traits highlighted in our study seemed to be related to cognition. In our case study, passage through river impoundments would stand for a hard cognitive task as it involves spatial recognition while climbing the walls and route choice based on perception of visual cues, which is rather unusual for juvenile eels [85]. Indeed, water obstacle passage often requires to climb and to get out of the water, where the extremely developed olfactive system of eel could be less useful than in the aquatic environment, making any behavioral decision demanding higher cognitive appraisal than relying on environmental cues. Passing non-natural obstacles such as water dams could represent a real conundrum for eels and could impede the upstream migration for those with undeveloped or with no ability to develop cognitive functions.
Gene induction by obstacle crossing?
The overall results suggested a difference in brain functioning between individuals successfully crossing the water obstacles and those situated on the downstream part of water impoundment. Whether gene transcription was temporarily induced by the passage event or whether these differences pre-existed in glass eels before they met the obstacle is difficult to decipher. Indeed, transcriptomic analysis provided phenotypic data, but also represented an intermediary step from genotype towards functional phenotype [92]. Gene expression could be therefore interpreted as a physiological acclimatation or phenotypic plasticity [93,94]. Other studies used gene transcription patterns as indicators of adaptive divergence [95,96].
Several studies on European eel considered trans-generational local adaptation hypothesis as less likely, all the more so because of random mating and absence of habitat choice at least during larval dispersal [97]. Previous studies failed to reveal a clear inter-location genetic heterogeneity of eels across Europe [98], and selection on locally adaptive traits may be too costly for eel [99]. Instead, phenotypic plasticity was hypothesized as the best strategy to deal with habitat heterogeneity in such cases [100,101]. A recent experimental study on American eel has proposed an effect of both origin and environment (salinity), as well as its interaction on gene expression [102]. However, differences in plastic responses were higher between environments within origin than gene expression variation between origins for both rearing environments, suggesting that phenotypic plasticity is not the only cause of phenotypic variation in eel, yet its contribution to the process remains overwhelming in front of (epi) genetic differences related to sampling location.
In our case study, changes in gene transcription profiles could be temporarily induced when eels cross the obstacle. The highest differences in gene transcription in brain were found for fish sampled at the third most upstream group while fish were passing the fishway. The hypothesis of temporary induction of gene transcription while crossing the obstacle could be strengthened, not only by the phenotypic plasticity of eel per se, but also by the acknowledged and functionally pertaining high plasticity of the brain [91,103,104].
Conclusion
Our results showed significant differences in gene transcription in the brain of glass eels sampled above and below the water obstacles. Although the influence of swimming distance on molecular phenotypes has to be taken into account by further analyses of non-impounded watercourse, brain plasticity and cognitive function seem to play an important role in the capacity of glass eels to cross aquatic obstacles. Two main directions for the further studies could be proposed. First, a comparison between climbing and remaining eels within the same location would allow focusing on the climbing event only. Next, the persistence of gene expression patterns could be tested by a long-term common garden experiment, thus explaining its proximate cause by separating the phenotypic plasticity and genetic components.
Availability of supporting data
Probe sequences and further details on the microarray platform can be found on the GEO database under accession number GPL15124. Normalized fluorescence data have been deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE56040.
References
Fischer J, Lindenmayer DB. Landscape modification and habitat fragmentation: A synthesis. Glob Ecol Biogeogr. 2007;16:265–80.
Soule ME. Conservation biology: the science of scarcity and diversity. Sunderland, Mass: Sinauer Associates; 1986.
Beneteau CL, Mandrak NE, Heath DD. The effects of river barriers and range expansion of the population genetic structure and stability in Greenside Darter (Etheostoma blennioides) populations. Conserv Genet. 2009;10:477–87.
Bunn SE, Arthington AH. Basic principles and ecological consequences of altered flow regimes for aquatic biodiversity. Environ Manage. 2002;30:492–507.
Hauer FR, Stanford JA, Ward JV. Serial discontinuities in a Rocky Mountain river. II distribution and abundance of trichoptera. Regul Rivers: Res Manag. 1989;3:177–82.
Araújo FG, Pinto BCT, Teixeira TP. Longitudinal patterns of fish assemblages in a large tropical river in southeastern Brazil: Evaluating environmental influences and some concepts in river ecology. Hydrobiologia. 2009;618:89–107.
Mueller M, Pander J, Geist J. The effects of weirs on structural stream habitat and biological communities. J Appl Ecol. 2011;48:1450–61.
Wofford JEB, Gresswell RE, Banks MA. Influence of barriers to movement on within-watershed genetic variation of coastal cutthroat trout. Ecol Appl. 2005;15:628–37.
Freeman MC, Pringle CM, Jackson CR. Hydrologic connectivity and the contribution of stream headwaters to ecological integrity at regional scales. J Am Water Resour Assoc. 2007;43:5–14.
Liermann CR, Nilsson C, Robertson J, Ng RY. Implications of dam obstruction for global freshwater fish diversity. BioScience. 2012;62:539–48.
Funk WC, Greene AE, Corn PS, Allendorf FW. High dispersal in a frog species suggests that it is vulnerable to habitat fragmentation. Biol Lett. 2005;1:13–6.
Postel S. Rivers for life : managing water for people and nature/Sandra Postel and Brian Richter. Washington: Island Press; 2003.
Kleckner RC, McCleave JD. The northern limit of spawning by Atlantic eels (Anguilla spp.) in the Sargasso Sea in relation to thermal fronts and surface water masses. J Mar Res. 1988;46:647–67.
Kettle AJ, Asbjørn Vøllestad L, Wibig J. Where once the eel and the elephant were together: Decline of the European eel because of changing hydrology in southwest Europe and northwest Africa? Fish Fish. 2011;12:380–411.
Tesch F-W, Thorpe JE. The eel. Oxford: UK: Blackwell Science: The Fisheries society of the British isles; 2003.
Cooke SJ, Hinch SG. Improving the reliability of fishway attraction and passage efficiency estimates to inform fishway engineering, science, and practice. Ecol Eng. 2013;58:123–32.
Kemp PS, O’Hanley JR. Procedures for evaluating and prioritising the removal of fish passage barriers: A synthesis. Fish Manag Ecol. 2010;17:297–322.
Edeline E, Dufour S, Elie P. Proximate and Ultimate Control of Eel Continental Dispersal. In: Thillart G, Dufour S, Rankin JC, editors. Spawning Migration of the European Eel. Volume 30. Netherlands: Springer; 2009. p. 433–61. Fish & Fisheries Series.
Fontaine M, Callamand O. Sur certains facteurs des migrations de l’anguille. Bulletin de la Société Zoologique de France. 1941;66:68–76.
Piper AT, Wright RM, Kemp PS. The influence of attraction flow on upstream passage of European eel (Anguilla anguilla) at intertidal barriers. Ecol Eng. 2012;44:329–36.
Kemp PS, Anderson JJ, Vowles AS. Quantifying behaviour of migratory fish: Application of signal detection theory to fisheries engineering. Ecol Eng. 2012;41:22–31.
Edeline E, Bardonnet A, Bolliet V, Dufour S, Elie P. Endocrine control of Anguilla anguilla glass eel dispersal: Effect of thyroid hormones on locomotor activity and rheotactic behavior. Horm Behav. 2005;48:53–63.
Bureau du Colombier S, Bolliet V, Lambert P, Bardonnet A. Energy and migratory behaviour in glass eels (Anguilla anguilla). Physiol Behav. 2007;92:684–90.
Fontaine M. Physiological Mechanisms in the Migration of Marine and Amphihaline Fish. Advances in Marine Biology. 1976;13:241–355.
Woodhead AD. Endocrine physiology of fish migration. G. Allen and Ungin; 1975.
Castonguay M, Dutil J-D, Audet C, Miller R. Locomotor Activity and Concentration of Thyroid Hormones in Migratory and Sedentary Juvenile American Eels. Trans Am Fish Soc. 1990;119:946–56.
Coppe A, Pujolar JM, Maes GE, Larsen PF, Hansen MM, Bernatchez L, et al. Sequencing, de novo annotation and analysis of the first Anguilla anguilla transcriptome: EeelBase opens new perspectives for the study of the critically endangered european eel. BMC Genomics. 2010;11:635.
Sébert P, Mortelette H, Nicolas J, Amérand A, Belhomme M, Moisan C. In vitro aerobic and anaerobic muscle capacities in the European eel, Anguilla anguilla: Effects of a swimming session. Respir Physiol Neurobiol. 2011;176:118–22.
Larsson Å, Lewander K. Metabolic effects of starvation in the eel, Anguilla anguilla L. Comp Biochem Physiol A Physiol. 1973;44:367–74.
Cren L. The Length-Weight Relationship and Seasonal Cycle in Gonad Weight and Condition in the Perch (Perca fluviatilis). J Anim Ecol. 1951;20:201.
Pujolar JM, Marino IAM, Milan M, Coppe A, Maes GE, Capoccioni F, et al. Surviving in a toxic world: transcriptomics and gene expression profiling in response to environmental pollution in the critically endangered European eel. BMC Genomics. 2012;13:507.
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–4.
Imbert H, Arrowsmith R, Dufour S, Elie P. Relationships between locomotor behavior, morphometric characters and thyroid hormone levels give evidence of stage-dependent mechanisms in European eel upstream migration. Horm Behav. 2008;53:69–81.
Cui Y, Wootton RJ. Effects of ration, temperature and body size on the body composition, energy content and condition of the minnow, Phoxinus phoxinus (L.). J Fish Biol. 1988;32:749–64.
Edeline E, Lambert P, Rigaud C, Elie P. Effects of body condition and water temperature on Anguilla anguilla glass eel migratory behavior. J Exp Mar Biol Ecol. 2006;331:217–25.
Bureau du Colombier S, Lambert P, Bardonnet A. Is feeding behaviour related to glass eel propensity to migrate? Estuar Coast Shelf Sci. 2008;80:323–9.
Zimmer DB, Eubanks JO, Ramakrishnan D, Criscitiello MF. Evolution of the S100 family of calcium sensor proteins. Cell Calcium. 2013;53:170–9.
Shang X, Cheng H, Zhou R. Chromosomal mapping, differential origin and evolution of the S100 gene family. Genet Sel Evol. 2008;40:449–64.
Andressen C, Blumcke I, Celio MR. Calcium-binding proteins: Selective markers of nerve cells. Cell Tissue Res. 1993;271:181–208.
Donato R. Perspectives in S-100 protein biology. Cell Calcium. 1991;12:713–26.
Donato R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001;33:637–68.
Di Donato V, Auer TO, Duroure K, Del Bene F. Characterization of the Calcium Binding Protein Family in Zebrafish. PLoS One. 2013;8:1.
Hermann A, Donato R, Weiger TM, Chazin WJ. S100 calcium binding proteins and ion channels. Front Pharmacol. 2012;3:67.
Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26.
Heizmann CW. Calcium signaling in the brain. Acta Neurobiol Exp. 1993;53:15–23.
Jastrzebska B, Filipek A, Nowicka D, Kaczmarek L, Kuznicki J. Calcyclin (S100A6) binding protein (CacyBP) is highly expressed in brain neurons. J Histochem Cytochem. 2000;48:1195–202.
Nowotny M, Bhattacharya S, Filipek A, Krezel AM, Chazin W, Kuznicki J. Characterization of the interaction of calcyclin (S100A6) and calcyclin-binding protein. J Biol Chem. 2000;275:31178–82.
Schneider G, Filipek A. S100A6 binding protein and Siah-1 interacting protein (CacyBP/SIP): Spotlight on properties and cellular function. Amino Acids. 2011;41:773–80.
Leśniak W, Słomnicki ŁP, Filipek A. S100A6 - New facts and features. Biochem Biophys Res Commun. 2009;390:1087–92.
Li H, Guan HW. Research progress on calcium-binding protein S100A6. J Dalian Med Univ. 2012;34:194–9.
Gruden MA, Storozheva ZI, Sewell RDE, Kolobov VV, Sherstnev VV. Distinct functional brain regional integration of Casp3, Ascl1 and S100a6 gene expression in spatial memory. Behav Brain Res. 2013;252:230–8.
He H, Li J, Weng S, Li M, Yu Y. S100A11: Diverse Function and Pathology Corresponding to Different Target Proteins. Totowa, NJ: Etats-Unis: Cell biochemistry and biophysics; 2009.
Sakaguchi M, Sonegawa H, Murata H, Kitazoe M, Futami J, Kataoka K, et al. S100A11, an dual mediator for growth regulation of human keratinocytes. Mol Biol Cell. 2008;19:78–85.
Deloulme JC, Assard N, Mbele GO, Mangin C, Kuwano R, Baudier J. S100A6 and S100A11 are specific targets of the calcium- and zinc-binding S100B protein in vivo. J Biol Chem. 2000;275:35302–10.
Nishiyama H, Knopfel T, Endo S, Itohara S. Glial protein S100B modulates long-term neuronal synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99(6):4037–42.
Benfenati F, Ferrari R, Onofri F, Arcuri C, Giambanco I, Donato R. S100A1 codistributes with synapsin I in discrete brain areas and inhibits the F-actin-bundling activity of synapsin I. J Neurochem. 2004;89:1260–70.
Ackermann GE, Marenholz I, Wolfer DP, Chan WY, Schäfer B, Erne P, et al. S100A1-deficient male mice exhibit increased exploratory activity and reduced anxiety-related responses. Biochim Biophys Acta, Mol Cell Res. 2006;1763:1307–19.
Tóthová V, Gibadulinová A. S100P, a peculiar member of S100 family of calcium-binding proteins implicated in cancer. Acta Virol. 2013;57:238–46.
Arumugam T, Simeone DM, Schmidt AM, Logsdon CD. S100P Stimulates Cell Proliferation and Survival via Receptor for Activated Glycation End Products (RAGE). J Biol Chem. 2004;279:5059–65.
Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25:457–69.
Garvalov BK, Flynn KC, Neukirchen D, Meyn L, Teusch N, Wu XW, et al. Cdc42 regulates cofilin during the establishment of neuronal polarity. J Neurosci. 2007;27:13117–29.
Bradke F, Dotti CG. Differentiated neurons retain the capacity to generate axons from dendrites. Curr Biol. 2000;10:1467–70.
Sarmiere PD, Bamburg JR. Regulation of the Neuronal Actin Cytoskeleton by ADF/Cofilin. J Neurobiol. 2004;58:103–17.
Racz B, Weinberg RJ. Spatial organization of cofilin in dendritic spines. Neuroscience. 2006;138:447–56.
Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Görlich A, et al. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010;29:1889–902.
Yuen EY, Liu W, Kafri T, van Praag H, Yan Z. Regulation of AMPA receptor channels and synaptic plasticity by cofilin phosphatase Slingshot in cortical neurons. J Physiol. 2010;588:2361–71.
Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–15.
Eckelhoefer HA, Rajapaksa TE, Wang J, Hamer M, Appleby NC, Ling J, Lo DD: Claudin-4: Functional studies beyond the tight junction. Methods in Molecular Biology. 2011;762:115–128.
Chua KL, Lim TM. Type I and type II cytokeratin cDNAs from the zebrafish (Danio rerio) and expression patterns during early development. Differentiation. 2000;66:31–41.
Iwatsuki H, Suda M. Transient expression of keratin during neuronal development in the adult rabbit spinal ganglion. Anat Sci Int. 2010;85:46–55.
Gnegy ME. Ca2+/calmodulin signaling in NMDA-induced synaptic plasticity. Crit Rev Neurobiol. 2000;14:91–129.
Delorenzo RJ. Calmodulin in neurotransmitter release and synaptic function. Fed Proc. 1982;41:2265–72.
Greenberg ME, Thompson MA, Sheng M. Calcium regulation of immediate early gene transcription. J Physiol Paris. 1992;86:99–108.
Zheng F, Luo Y, Wang H. Regulation of brain-derived neurotrophic factor-mediated transcription of the immediate early gene Arc by intracellular calcium and calmodulin. J Neurosci Res. 2009;87:380–92.
Davis S, Bozon B, Laroche S. How necessary is the activation of the immediate early gene zif 268 in synaptic plasticity and learning? Behav Brain Res. 2003;142:17–30.
Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J Neurobiol. 1994;25:294–303.
Igaz LM, Bekinschtein P, Vianna MMR, Izquierdo I, Medina JH. Gene expression during memory formation. Neurotox Res. 2004;6:189–203.
Miyashita T, Kubik S, Lewandowski G, Guzowski JF. Networks of neurons, networks of genes: An integrated view of memory consolidation. Neurobiol Learn Mem. 2008;89:269–84.
Lanahan A, Worley P. Immediate-Early Genes and Synaptic Function. Neurobiol Learn Mem. 1998;70:37–43.
Xue GP, Rivero BP, Morris RJ. The surface glycoprotein Thy-1 is excluded from growing axons during development - a study of the expression of Thy-1 during axogenesis in hippocampus and hindbrain. Development. 1991;112:161–76.
Xue GP, Calvert RA, Morris RJ. Expression of the neuronal syrface glycoprotein Thy-1 is under posttranscirptional control, and is spatially regulated in the developping olfactory system. Development. 1990;109:851–4.
Moullé VS, Cansell C, Luquet S, Cruciani-Guglielmacci C. The multiple roles of Fatty Acid Handling Proteins in brain. Front Physiol. 2012;3:385.
Edeline E, Dufour S, Briand C, Fatin D, Elie P. Thyroid status is related to migratory behavior in Anguilla anguilla glass eels. Mar Ecol Prog Ser. 2004;282:261–70.
Tomoda H, Uematsu K. Morphogenesis of the Brain in Larval and Juvenile Japanese Eels, Anguilla japonica. Brain Behav Evol. 1996;47:33–41.
Gonda A, Herczeg G, Merilä J. Population variation in brain size of nine-spined sticklebacks (Pungitius pungitius) - Local adaptation or environmentally induced variation? BMC Evol Biol. 2011;11:75.
Kihslinger RL, Lema SC, Nevitt GA. Environmental rearing conditions produce forebrain differences in wild Chinook salmon Oncorhynchus tshawytscha. Comp Biochem Physiol A Mol Integr Physiol. 2006;145:145–51.
von Krogh K, Sørensen C, Nilsson GE, Øverli Ø. Forebrain cell proliferation, behavior, and physiology of zebrafish, Danio rerio, kept in enriched or barren environments. Physiol Behav. 2010;101:32–9.
Gonda A, Välimäki K, Herczeg G, Merila J. Brain development and predation: Plastic responses depend on evolutionary history. Biol Lett. 2012;8:249–52.
Salvanes AGV, Moberg O, Ebbesson LOE, Nilsen TO, Jensen KH, Braithwaite VA. Environmental enrichment promotes neural plasticity and cognitive ability in fish. Proc R Soc B Biol Sci. 2013;280:13.
Sorensen C, Johansen IB, Overli O. Neural plasticity and stress coping in teleost fishes. Gen Comp Endocrinol. 2013;181:25–34.
Fay JC, Wittkopp PJ. Evaluating the role of natural selection in the evolution of gene regulation. Heredity. 2008;100:191–9.
Giger T, Excoffier L, Day PJR, Champigneulle A, Hansen MM, Powell R, et al. Life history shapes gene expression in salmonids. Curr Biol. 2006;16:R281–2.
Aubin-Horth N, Renn SCP. Genomic reaction norms: Using integrative biology to understand molecular mechanisms of phenotypic plasticity. Mol Ecol. 2009;18:3763–80.
Derome N, Duchesne P, Bernatchez L. Parallelism in gene transcription among sympatric lake whitefish (Coregonus clupeaformis Mitchill) ecotypes. Mol Ecol. 2006;15:1239–49.
Larsen PF, Nielsen EE, Williams TD, Hemmer-Hansen J, Chipman JK, Kruhøffer M, et al. Adaptive differences in gene expression in European flounder (Platichthys flesus). Mol Ecol. 2007;16:4674–83.
Pacariz S, Westerberg H, Björk G. Climate change and passive transport of European eel larvae. Ecol Freshw Fish. 2014;23:86–94.
Dannewitz J, Maes GE, Johansson L, Wickström H, Volckaert FAM, Järvi T. Panmixia in the European eel: A matter of time. Proc R Soc B Biol Sci. 2005;272:1129–37.
Lenormand T. Gene flow and the limits to natural selection. Trends Ecol Evol. 2002;17:183–9.
Sultan SE, Spencer HG. Metapopulation structure favors plasticity over local adaptation. Am Nat. 2002;160:271–83.
Edeline E. Adaptive phenotypic plasticity of eel diadromy. Mar Ecol Prog Ser. 2007;341:229–32.
Côté CL, Castonguay M, Kalujnaia MS, Cramb G, Bernatchez L. In absence of local adaptation, plasticity and spatially varying selection rule: A view from genomic reaction norms in a panmictic species (Anguilla rostrata). BMC Genomics. 2014;15:403.
Ebbesson LOE, Braithwaite VA. Environmental effects on fish neural plasticity and cognition. J Fish Biol. 2012;81:2151–74.
Oliveira RF. Social plasticity in fish: integrating mechanisms and function. J Fish Biol. 2012;81:2127–50.
Acknowledgments
We thank Lorenzo Zane, Stefania Bortoluzzi, Alessandro Coppe and all members of AGC (Anguilla Genomics Consortium) for their contribution to the ca. 15.000 contigs microarray of European eel and their permission to use the EELBASE 2.0 (The European eel transcriptome database version 2, http://compgen.bio.unipd.it/eeelbase/). We are very grateful to Alice Laharanne and other members of Fédération de Pêche de Gironde, Nicolas Deligne, Romaric le Barh (Irstea Bordeaux) and Sebastien Dufour (Syndicat Intercommunal d’Aménagement des Eaux du Bassin Versant des Etangs du Littoral Girondin) for their help in sampling, and we truly acknowledge Christian Rigaud (Irstea Bordeaux) for his field work help and many valuable remarks throughout this study. This work has been supported through the “HYNES” collaborative project between Irstea and the French Electric Company (EDF).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The data of this paper are original and no part of this manuscript has been published or submitted for publication elsewhere. The authors have no competing interests in this study.
Authors’ contributions
TP, FD and FP have performed sampling, otolith and tissues exraction, RNA extraction and retrotranscription to cDNA. MM and MP have performed all the steps of microarray analysis. GM and EO participated in study design and interpretation of results. LB provided the access to ca. 15000 contigs microarray of European eel. All the authors read and appoved the final manuscript.
Fabien Pierron and Francoise Daverat contributed equally to this work.
Additional file
Additional file 1: Figure S1.
Comparison of fold changes (FC) between the transcription levels of brain genes encoding for Claudin 4, Cofilin 1, S100A1, S100A11 and S100A6 proteins and Thy-1, obtained by microarray (white bars) and RT-qPCR analysis (black bars). Two fold changes are compared: A) between the segments 3:1 and B) between the segments 3:2. Table S1. Sequences of specific primers pairs used in quantitative RT-PCR analyses. Table S2. Details about the genes that were differentially transcribed in eels from segment 3 as compared to individuals from segment 2 (FC 3:2) or from segment 1 (FC 3:1).
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Podgorniak, T., Milan, M., Pujolar, J.M. et al. Differences in brain gene transcription profiles advocate for an important role of cognitive function in upstream migration and water obstacles crossing in European eel. BMC Genomics 16, 378 (2015). https://doi.org/10.1186/s12864-015-1589-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1589-y