
Abstract
Background
In humans, muscle-specific nicotinergic acetylcholine receptor (AChR) is a transmembrane protein with five different subunits, coded by CHRNA1, CHRNB, CHRND and CHRNG/CHRNE. The gamma subunit of AChR encoded by CHRNG is expressed during early foetal development, whereas in the adult, the γ subunit is replaced by a ε subunit. Mutations in the CHRNG encoding the embryonal acetylcholine receptor may cause the non-lethal Escobar variant (EVMPS) and lethal form (LMPS) of multiple pterygium syndrome. The MPS is a condition characterised by prenatal growth failure with pterygium and akinesia leading to muscle weakness and severe congenital contractures, as well as scoliosis.
Results
Our whole exome sequencing studies have identified one novel and two previously reported homozygous mutations in CHRNG in three families affected by non-lethal EVMPS. The mutations consist of deletion of two nucleotides, cause a frameshift predicted to result in premature termination of the foetally expressed gamma subunit of the AChR.
Conclusions
Our data suggest that severity of the phenotype varies significantly both within and between families with MPS and that there is no apparent correlation between mutation position and clinical phenotype. Although individuals with CHRNG mutations can survive, there is an increased frequency of abortions and stillbirth in their families. Furthermore, genetic background and environmental modifiers might be of significance for decisiveness of the lethal spectrum, rather than the state of the mutation per se. Detailed clinical examination of our patients further indicates the changing phenotype from infancy to childhood.
Similar content being viewed by others

Background
Multiple pterygium syndromes (MPS) comprise a group of multiple congenital anomalies of the skin, muscles and skeleton [1, 2]. It is characterised by prenatal growth failure with webbing (pterygium) of the skin present in multiple areas and a lack of muscle movement (akinesia) leading to muscle weakness and severe congenital contractures (arthrogryposis), and scoliosis. The MPS is a clinically and genetically heterogeneous disorder but it is traditionally divided into lethal (LMPS, OMIM 253290), which is fatal before birth or very soon after birth, and non-lethal (Escobar variant, EVMPS, OMIM 26500) MPS types [3]. Escobar syndrome is characterized by short stature, pterygia of the neck, axilla, antecubital, popliteal, digital, and intercrural areas, multiple joint contractures and cleft palate [4]. Dimples at the knees and other joints might be present. Facial features include long face, downslanting palpebral fissures, ptosis, long philtrum, emotionless face, low-set ears, high-arched palate, small mouth, downturned corners of mouth, inability to fully open mouth and retrognathism. Skeletal anomalies such as fusion of cervical vertebrae, scoliosis, kyphosis, flexion contraction of fingers, rocker-bottom feet with vertical talus may be present [5]. Small penis and scrotum and cryptorchidism are seen in males. Females might have aplasia of the labia majora and small clitoris. Variable other features include intrauterine death, congenital respiratory distress, reduced foetal movement, and conductive hearing loss. Changing phenotype from birth to childhood has been observed [6].
Autosomal recessive inheritance appears to be the most common in MPS cases. However, the disease may be transmitted as an autosomal dominant or X linked trait [7–9]. Mutations in genes encoding different subunits of the nicotinergic acetylcholine receptor (AChR), an excitatory cation channel, have been associated with MPS. In humans, muscle-specific AChR is a transmembrane pentameric glycoprotein composed of four different subunits, two α subunits, one β, one δ and one γ/ε subunit, encoded by CHRNA1, CHRNB, CHRND and CHRNG/CHRNE, respectively. The AChR exists in two forms, the embryonic form, present in foetal and denervated muscle and the adult form, which is predominantly expressed postnatal [10, 11]. In humans, the switch from the embryonic AChR to the adult is apparently completed by the 33st week of gestation, in the late foetal and perinatal period [12]. The γ subunit of AChR (CHRNG) is expressed during early foetal development, whereas in the adult it is replaced by a ε subunit (CHRNE). Each AChR subunit comprises a large extracellular glycosylated N-terminal ligand-binding domain, followed by three hydrophobic transmembrane regions, which form the ionic channel, followed by an intracellular region of variable length. A fourth hydrophobic transmembrane region is located at the C-terminal domain.
Hereditary congenital myasthenic syndrome (OMIM 608931, 608930, 601462 and 254210) is predominantly caused by mutations in CHRNA1, CHRNB1, CHRND and CHRNE, the genes encoding α, β, δ and ε AChR subunits, respectively [13]. While mutations in the gene encoding the gamma subunit of the AChR (CHRNG), cause most cases of EVMPS and a smaller percentage of cases of LMPS, homozygous nonsense mutations in CHRNA1 and CHRND, are associated with LMPS [4, 14–17].
Whole-Exome Sequencing (WES) was applied in 9 Iranian cases (2 foetuses and 7 children) with clinical presentation of arthrogryposis. In 4 cases CHRNG was suspected as the most likely gene underlying the disease and the association of CHRNG with the disease was confirmed in three out of four cases. Here, we report one novel and two previously reported deletions of two nucleotides in CHRNG, causing a frameshift predicted to result in premature termination of the foetally expressed gamma subunit of the AChR in three families of Iranian descent with EVMPS. We also emphasize the changing phenotype from infancy to childhood and intrafamilial variability.
Cases
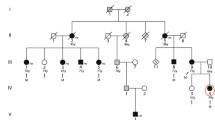
In family 1 (Fig. 1a), a female (IV:3) was born to healthy consanguineous (first-cousin) parents of Iranian descent. The mother had history of two spontaneous abortions at 6 weeks and 7 weeks of gestation. Pregnancy was uneventful. Amniocentesis was performed because of high risk of trisomy 21 in the first trimester screening. Chromosomal study was normal. Elective cesarean section was performed at term. At birth, her weight was 2800 g (10th centile), and her length and head circumference were 44 cm (<3rd centile) and 35 cm (50th centile), respectively. Physical examination at 1 month of age disclosed a very short neck, mild pterygia in the axillae, elbows and knees, contracture of joints (elbows, wrists, fingers, knees and ankles) clenched hands with thumbs held across palm and club feet (varus). The elbows and knees were held in flexed position and had limitation of movement (Fig. 2a, b). She had rockerbottom feet, with almost no movement in ankles (Fig. 2c). Facial dysmorphism included hemangioma over forehead and nose, strabismus, flat nasal bridge, downturned corners of mouth. Whole body X-rays did not show major abnormality.

Pedigrees for families included in this study. Affected individuals are represented with shaded symbols and probands are indicated with an arrow. Identified mutation is indicated in each family

Clinical features of subjects with CHRNG mutations and non-lethal variant of MPS. Case IV:1 from family 1, demonstrates a very short neck, mild pterygia in the axillae, elbows and knees, contracture of joints, clenched hands with thumbs held across palm and club feet, at 1 month of age. The elbows and knees were held in flexed position and had limitation of movement (a and b). She had rockerbottom feet (c). In family 2, individual II:1 has multiple joint contractures in the neck, shoulders, elbows, wrist, fingers, knees and halluces were noted (d) and dimpling in the elbows (e). She has a capillary hemangioma on nasal nip and forehead, and micrognathia in the face (f). In family 3, individual IV:3 at the age of 7 years demonstrates facial dysmorphism including posterior and anterior low hairline, down-slanting palpebral fissures, epicanthal folds, broad nose, high nasal bridge, long philtrum, high-arched palate, rockerbottom feet, and micrognathia (g). He has camtodactyly of all fingers and skin syndactyly of fingers (h) and the teeth are small and malpositioned (i). The micropenis is apparent (j)
In family 2 (Fig. 1b), individual II:1 was the first and only child of a unrelated couple. The mother noted reduced foetal movement during pregnancy. Delivery was at 39 weeks of gestation by cesarean section because of breech position. Birth weight, length and head circumference were 3150 g (<50th centile), 43 cm (<3rd centile) and 36 cm (75th centile), respectively. When assessed at two and a half months, her weight was 4500 g (<10th centile), height 53 cm (<3rd centile) and head circumference 41.5 cm (<75th centile). Multiple joint contractures in the neck (torticolli), shoulders, elbows, wrist, fingers, knees and halluces were noted (Fig. 2d). The hands were clenched and thumb held across palm. The shoulders were rounded, sloping and decreased in muscle mass. There was dimpling in the elbows (Fig. 2e). Pterygium was noted in the axillary region. She had a capillary hemangioma on nasal nip and forehead, and micrognathia in the face (Fig. 2f). Full body X-ray showed scoliosis. Echocardiography was normal. Ultra-sound examination of the brain, abdomen (liver, gall bladder, pancreas and spleen), kidneys and hip joint appeared normal.
In family 3 (Fig. 1c), individual IV:3 was born to first-cousin parents of Iranian descent. The first pregnancy was a boy (IV:1) with multiple contractures and pterygium in joints, who died at the age of 6 months. The second pregnancy was a twin pregnancy, the proband (IV:3) and a similarly affected boy (IV:2) who died at the age of 1.5 years. The third pregnancy resulted in intra-uterine-foetal death at 20 weeks of gestation (IV:4). Delivery was by caesarean section at 9 months of pregnancy. Birth weight was 2500 g (5th centile). Pterygium in knees and elbows was noticed from birth. The infant had multiple episode of epistaxis, which led to the diagnosis of von Willebrand type III hemophilia. Milestones were delayed; he held his head at age 1 year, he stood at 2 years of age, and walked at age 2.5 years. Cognition was normal and he started saying words at age 10 months and he had normal speech at 7 years of age. Bilateral inguinal hernia was treated surgically at the age of 3 years old. At 7 years of age, his height, weight and head circumference were 117 cm (<25th centile), 16 kg (<3rd centile) and 49.5 cm (<50th centile), respectively. Detailed review at the age of 7 years revealed facial dysmorphism including posterior and anterior low hairline, down-slanting palpebral fissures, epicanthal folds, broad nose, high nasal bridge, long philtrum, high-arched palate, and micrognathia (Fig. 2g). The teeth were small and malpositioned (Fig. 2i). Pterygia were present in neck, axillae, antecubital and in intercrural region. Contracture at elbows, limitation of elbow extension, limited movement of flexion and extension in wrists, camtodactyly of all fingers and skin syndactyly of fingers, rockerbottom feet, and second toe overlapping first and third in both feet, limited movement in ankles, limited flexion and extension of knees and micropenis were apparent (Fig. 2g, h, j). Intellectual development appeared normal. Clinical features in the investigated families are summarised in Table 1.
Methods
DNA isolation
Blood samples were collected from probands, parents and siblings. Extraction of genomic DNA was performed from whole blood from patients and parents, using DNeasy Blood & Tissue kit (Qiagen, Hilden Germany), according to the manufacturer’s instructions. In addition, genomic DNA was extracted from 120 Iranian blood donors, who served as controls.
Genetic analysis
Exome sequence analysis
The WES was performed on DNA from patients and their unaffected parents, as previously described [18]. Briefly, target enrichment was performed with 3 μg genomic DNA using the Sure SelectXT Human All Exon kit version 5 (Agilent Technologies, Santa Clara, CA, USA) to generate barcoded whole-exome sequencing libraries. Libraries were sequenced on the HiSeq2000 platform (Illumina, San Diego, CA, USA) as paired-end 2 × 100-bp reads with 60x coverage. Quality assessment of the sequence reads was performed by generating QC statistics with FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). Read alignment to the reference human genome (hg19, UCSC assembly, February 2009) was done using BWA [19] with default parameters. After removal of polymerase chain reaction (PCR) duplicates (Picard tools, http://picard.sourceforge.net) and file conversion (samtools [20]) quality score recalibration, indel realignment and variant calling were performed with the HaplotypeCaller algorithm in the GATK package [21] based on established best practices [22].
Variant annotation and selection
Variants were annotated with ANNOVAR [23] using a wide range of databases such as dbSNP build 135, dbNSFP, KEGG, the Gene Ontology project and tracks from the UCSC. A filtering strategy, directed to disease gene candidates, was performed by QIAGEN’s Ingenuity® Variant Analysis™ software (www.qiagen.com/ingenuity) from QIAGEN Redwood City. We focused on exonic variants where the mutation produced a missense change, stop gain or stop loss. We required at least two mutations in the same gene for further analysis. Only those changes that were predicted to be damaging or with unknown impact were kept. We excluded SNPs that were shared with our control dataset (>1 % in dbSNP [24], the Exome Variant Server (NHLBI)(http://evs.gs.washington.edu/EVS/), the 1000 Genome Project Database and the human Background Variant Database (http://neotek.scilifelab.se/hbvdb/)) as well as those labeled as compound heterozygous.
Polymerase chain reaction (PCR) and Sanger sequencing
The variants found by WES in the candidate genes were examined in the individuals by PCR and Sanger sequencing using an ABI 3730XL (GATC Biotech, Constance, Germany and Eurofins MWG Operon, Ebersberg, Germany) if they had either a variant frequency <1 % in the healthy population or a minor allele frequency (MAF) below the normal MAF in the European population in EVS. PCR was performed on DNA samples from patients and their unaffected parents. PCR primers are available on request.
Results
Genetic findings
Data from WES on DNA from patients and their parents were analysed through the use of the Ingenuity Variant Analysis (IVA) software (Qiagen, Hilden Germany). The filtering strategy narrowed the starting variants to 16, 11 and 12 genes in case IV:3 (family 1), II:1 (family 2) and IV:3 (family 3), respectively. This approach allowed the identification of homozygous deletion of two nucleotides in exons of CHRNG in these cases. All other related disease-causing genes were excluded in these cases. We identified a novel homozygous deletion of two nucleotides in exon 9 (c.1009-1010delCA) (ID SUB1128405) of CHRNG in case IV:3 of family 1, leading to a frameshift predicted to result in premature termination (p.His337fs60Ter) (Fig. 3). In case II:1 of family 2, the filtering strategy narrowed the variants to 11 genes, including a homozygous previously reported deletion of two nucleotides in exon 7 (c.753-754delCT) of CHRNG, leading to a frameshift predicted to result in a premature termination (p.Val253fs44Ter) (Fig. 3). In case IV:3 of family 3, a previously reported homozygous deletion of two nucleotides in exon 5 (c.401-402delCT) of CHRNG was identified, leading to a frameshift. The c.401-402delCT mutation was predicted to result in premature termination (p.Pro134fs43Ter) (Fig. 3).

CHRNG mutations in the investigated families with EVMPS
Putative deleterious variants in CHRNG exons 5, 7 and 9, were confirmed in the children and their parents by PCR and Sanger sequencing analysis. The unaffected parents were heterozygous for the two nucleotide deletions. None of the mutations was reported in public databases as a polymorphism. However, in order to define the frequency of the identified variants in population-matched controls, PCR and Sanger sequencing of CHRNG exons 5, 7 and 9 was performed in 120 Iranian blood donors, who served as controls. The results from sequencing analysis excluded the CHRNG sequence variants in 120 Iranian control individuals.
The recurrent homozygous deletion of two nucleotides in exon 7 (c.753-754delCT) was identified in the only child of a unrelated couple (in case II:1 of family 2). Haplotyping was not available so it is not known if this was due to a founder mutation. However, the percentage of homozygosity in exome sequencing of this case compared to two other probands born to the first-cousin parents was notably lower (5.6 %).
Discussion
The mammalian muscle-specific acetylcholine receptor is a transmembrane glycoprotein with two alpha, one beta, one delta and one gamma (expressed in foetal and denervated muscle) or epsilon (expressed in adult skeletal muscle) subunit [12]. The gamma subunit of AChR, encoded by CHRNG, is expressed prior to the thirty-third week of gestation in humans [10, 25, 26]. It helps to establish the primary encounter of muscle and axon and has thus, a pivotal role in neuromuscular organogenesis and ligand binding [27]. Mutations in gamma subunits cause MPS, either the severe and fatal form (LMPS), or the milder and non-lethal Escobar variant [4, 14, 15]. Previous study has demonstrated that disruption of gamma subunit expression prevents the correct localization of the receptor in cell membranes [14]. The ultimate result of the impaired γ subunit structure is reduced prenatal muscle strength and movement, explaining the contractures and pterygia [14].
Here, we report three families with EVMPS associated with one novel and two previously reported frameshift mutations in CHRNG. These mutations are anticipated to result in premature mRNA nonsense-mediated decay followed by a γ subunit deficiency or predicted to result in premature termination of transcription of the foetally expressed gamma subunit of the AChR. All patients had clinical features consistent with MPS Escobar variant, including arthrogryposis multiplex congenita, multiple pterygia, short stature and dysmorphic facial features, corresponding to the developmental deformities in utero.
The novel homozygous deletion (c.1009-1010delCA; p.His337fs60Ter) in family 1 is located in exon 9 of CHRNG. In the absence of nonsense-mediated decay, the mutation is predicted to result a frameshift followed by a truncated gamma AChR subunit lacking intracellular region and the fourth hydrophobic transmembrane domain. Consequently, the mutation will lead to a dysfunctional or deficient foetal AChR, which in turn will cause impaired prenatal neuromuscular transmission and organogenesis followed by developmental defects. It is likely that this homozygous mutation caused the history of abortions in this family. Since the abortions occurred very early in pregnancy (at 7 and 8 weeks of gestation, respectively), the fetopathological examination could not be performed and therefore, there are no further clinical details. It is thus uncertain whether the typical clinical features corresponding to the lethal MPS were present in these cases.
The recurrent c.753-754delCT, pVal253Alafs44Ter mutation identified in family 2 was previously reported in patients from different ethnic backgrounds affected with both the lethal and non-lethal phenotypes [15, 17]. It was found in the homozygous state and in combination with a second CHRNG heterozygous mutation. The previously reported homozygous c.753-754delCT, pVal253Alafs44Ter mutation was found in cases with the lethal and non-lethal MPS, born to consanguineous parents of Turkish descent [15, 17]. It was also found together with a second CHRNG heterozygous mutation in two compound heterozygous individuals with EVMPS phenotype [15]. Thus, the c.753-754delCT mutation may cause both lethal and non-lethal MPS, and the homozygous state of the mutation is not decisive of the lethal spectrum. Furthermore, there is no apparent correlation between mutation position and clinical phenotype. Therefore, genetic background and interindividual differences, and environmental modifiers might be of significance.
As previously reported, severity of the phenotype varies significantly both within and between families with MPS [6, 15]. This is further supported in family 3, in which the first and second siblings with non-lethal MPS were severely affected and died in the early infancy. However, it is unclear how the lethality in the two affected siblings was influenced by the severity of the congenital deformities. As previously suggested [15] the chance of a non-lethal MPS phenotype arising in further affected siblings within families associated with homozygous c.401-402delCT; p.Pro134Argfs34Ter mutation is more likely. The c.401-402delCT; p.Pro134Argfs34Ter mutation was previously reported in a 14-year-old boy with clinical features closely resembling individual IV:3 of family 3 [4]. Contracture and pterygia in all major joints and facial features characteristic for Escobar syndrome was present in both cases. However, bilateral inguinal hernia and micropenis observed in our patient, was not reported in the previous case. This homozygous deletion mutation was further reported in an individual with EVMPS phenotype born to a non-consanguineous couple from the UK [15]. The frequently observed CHRNG c.753-754delCT; pVal253Alafs44Ter and c.401-402delCT; p.Pro134Argfs34Ter mutations indicate the mutational hotspot of these residues [4, 15, 17].
It has been suggested that there is a high concordance between siblings regarding the severity of clinical findings in families with EVMPS [15]. However, the history of increased frequency of abortions and stillbirth in reported families with EVMPS [4, 14, 15] and the presence of two abortions in family 1 and two perinatal deaths in family 3 suggest that variable severity of disease could exist in the same family.
Given the fact that CHRNG is expressed only during early foetal development and in denervated cells and that the γ subunit is not a component of the adult acetylcholine receptor [10–12], postnatal muscle biopsy from affected individuals with EVMPS phenotype is inapplicable to assess the effect of CHRNG mutations on transcripts and protein levels. Likewise, it is unlikely that postnatal muscle biopsy discloses specific muscle histopathology, as postnatal muscle weakness is not a feature of CHRNG associated MPS Escobar variant. Although myasthenic features and abnormal muscle histopathology is not expected in patients with CHRNG mutations, congenital diaphragmatic muscle weakness, diffuse myopathy and myasthenic-like features have frequently been reported in some patients [14]. This can be due to the role of γ subunit AChR in muscle organogenesis.
Clinical examination of our patients indicates that the complete clinical features of MPS Escobar variant develop past infancy. This is supported by previous study describing detail clinical features of individuals with EVMPS [6]. While certain clinical features are not present at birth and become apparent, as the infant grows older, other features improve with time. For example, the clear pterygia is not apparent in cases IV:3 (family 1) and II:1 (family 2), at the age of one and two and a half months, respectively, whereas it is completely evident in case IV:3 (family 3), at the age of seven years. Infants have the severe joint contractures and limited movement at birth, which persists but might improve as the child grows older. Many infants born with Escobar syndrome have clenched hands with the thumb held across the palm. However, they can open their fist and have improvement of the position of the thumb as they grow older [6]. Individuals IV:3 and II:1 in family 1 and 2, respectively, show clenched hands with the thumbs held across thumb. Likewise, individual IV:3 in family 3 is said to have had the similar clinical finding at infancy. However, this clinical condition has improved and he only has camptodactyly at the age of 7 years. Moreover, the characteristic facial appearance of Escobar syndrome including down-slanting palpebral fissures, ptosis, long philtrum, sad and emotionless face, observed in the older child is not present in the infants with Escobar syndrome. Facial features dominantly present at birth include epicanthal fold, low-set ears, and micrognathia. Capillary hemangioma observed in individuals IV:3 and II:1 in family 1 and 2, respectively, is additional clinical feature that has been reported in infants with Escobar syndrome, but it is absent in the older child with Escobar syndrome. Taking together, accurate diagnosis and prognosis require follow-up of infants presenting multiple joint contractures or arthrogryposis multiplex congenital at birth, as many of them develop the characteristic features of Escobar syndrome MPS over the first few years of life.
Conclusions
In conclusion, we present three families affected by non-lethal MPS and a novel and two previously reported homozygous frameshift truncated mutations in CHRNG predicted to result in truncations. We suggest that there is no apparent correlation between mutation position and clinical phenotype. Our data suggest the changing phenotype from infancy to childhood in individuals affected by MPS Escobar variant and that severity of the phenotype varies both within and between families with MPS.
Web resources
Following Databases were used in this study:
-
The Exome Variant Server: NHLBI Exome Sequencing Project (ESP), Seattle, WA;
-
1000 Genome Project Database: http://browser.1000genomes.org/index.html
-
Human Background Variant DataBase: http://neotek.scilifelab.se/hbvdb/
Abbreviations
AChR, acetylcholine receptor; EVMPS, escobar variant of multiple pterygium syndrome; LMPS, lethal form of multiple pterygium syndrome; MPS, multiple pterygium syndromes; MAF, minor allele frequency; PCR, polymerase chain reaction; WES, whole-exome sequencing; EVS: exome Variant Server
References
Hall JG. The lethal multiple pterygium syndromes. Am J Med Genet. 1984;17(4):803–7.
Gillin ME, Pryse-Davis J. Pterygium syndrome. J Med Genet. 1976;13(3):249–51.
Hall JG, Reed SD, Rosenbaum KN, Gershanik J, Chen H, Wilson KM. Limb pterygium syndromes: a review and report of eleven patients. Am J Med Genet. 1982;12(4):377–409.
Morgan NV, Brueton LA, Cox P, Greally MT, Tolmie J, Pasha S, Aligianis IA, van Bokhoven H, Marton T, Al-Gazali L, et al. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am J Hum Genet. 2006;79(2):390–5.
Chen H, Chang CH, Misra RP, Peters HA, Grijalva NS, Opitz JM. Multiple pterygium syndrome. Am J Med Genet. 1980;7(2):91–102.
Thompson EM, Donnai D, Baraitser M, Hall CM, Pembrey ME, Fixsen J. Multiple pterygium syndrome: evolution of the phenotype. J Med Genet. 1987;24(12):733–49.
McKeown CM, Harris R. An autosomal dominant multiple pterygium syndrome. J Med Genet. 1988;25(2):96–103.
Prontera P, Sensi A, Merlo L, Garani G, Cocchi G, Calzolari E. Familial occurrence of multiple pterygium syndrome: expression in a heterozygote of the recessive form or variability of the dominant form? Am J Med Genet A. 2006;140(20):2227–30.
Tolmie JL, Patrick A, Yates JR. A lethal multiple pterygium syndrome with apparent X-linked recessive inheritance. Am J Med Genet. 1987;27(4):913–9.
Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, Methfessel C, Sakmann B. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature. 1986;321(6068):406–11.
Bouzat C, Bren N, Sine SM. Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron. 1994;13(6):1395–402.
Hesselmans LF, Jennekens FG, Van den Oord CJ, Veldman H, Vincent A. Development of innervation of skeletal muscle fibers in man: relation to acetylcholine receptors. Anat Rec. 1993;236(3):553–62.
Engel AG, Sine SM. Current understanding of congenital myasthenic syndromes. Curr Opin Pharmacol. 2005;5(3):308–21.
Hoffmann K, Muller JS, Stricker S, Megarbane A, Rajab A, Lindner TH, Cohen M, Chouery E, Adaimy L, Ghanem I, et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet. 2006;79(2):303–12.
Vogt J, Morgan NV, Rehal P, Faivre L, Brueton LA, Becker K, Fryns JP, Holder S, Islam L, Kivuva E, et al. CHRNG genotype-phenotype correlations in the multiple pterygium syndromes. J Med Genet. 2012;49(1):21–6.
Michalk A, Stricker S, Becker J, Rupps R, Pantzar T, Miertus J, Botta G, Naretto VG, Janetzki C, Yaqoob N, et al. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet. 2008;82(2):464–76.
Bayram Y, Karaca E, Coban Akdemir Z, Yilmaz EO, Tayfun GA, Aydin H, Torun D, Bozdogan ST, Gezdirici A, Isikay S, et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest. 2016;126(2):762–78.
Olive M, Abdul-Hussein S, Oldfors A, Gonzalez-Costello J, van der Ven PF, Furst DO, Gonzalez L, Moreno D, Torrejon-Escribano B, Alio J, et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum Mol Genet. 2015;24(13):3638–50.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Missias AC, Chu GC, Klocke BJ, Sanes JR, Merlie JP. Maturation of the acetylcholine receptor in skeletal muscle: regulation of the AChR gamma-to-epsilon switch. Dev Biol. 1996;179(1):223–38.
Yumoto N, Wakatsuki S, Sehara-Fujisawa A. The acetylcholine receptor gamma-to-epsilon switch occurs in individual endplates. Biochem Biophys Res Commun. 2005;331(4):1522–7.
Koenen M, Peter C, Villarroel A, Witzemann V, Sakmann B. Acetylcholine receptor channel subtype directs the innervation pattern of skeletal muscle. EMBO Rep. 2005;6(6):570–6.
Acknowledgements
We thank the family members who provided samples and clinical information for this study. We would like to thank the Iranian Blood Transfusion Organisation (IBTO) for assistance with the collection of blood samples from Iranian blood donors. We thank the Bioinformatics Core Facility platform, at the Sahlgrenska Academy, University of Gothenburg for assistance with the bioinformatics analyses.
Funding
The study was supported by grants from the Swedish Research Council (H.T.) and the Swedish Society of Medicine (H.T.). The funders had no role in the design of the study and collection, analysis, decision to publish, interpretation of data or preparation of the manuscript.
Authors’ contributions
All authors conceived the study. AK and HT participated in study design and collected the data. AK, NA and AK contributed to clinical assessment of the patients. BO, A-RM and HT analysed the WES data. BO and A-RM participated in computational and bioinformatics design and analysis. AK, HT and BO wrote the manuscript. All authors contributed to the conception and design of the study, analysis and interpretation of data, drafting the manuscript and provided critical revisions and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
The parents in this study provided written informed consent to publish their family trees and family data.
Ethics and consent to participate
The parents in this study provided written informed consent to participate in this study. The study was approved by the ethical standards of the relevant institutional review board, the Kariminejad-Najmabadi Pathology & Genetics ethics committee and the ethics review committee in the Gothenburg region (Dn1: 842–14).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kariminejad, A., Almadani, N., Khoshaeen, A. et al. Truncating CHRNG mutations associated with interfamilial variability of the severity of the Escobar variant of multiple pterygium syndrome. BMC Genet 17, 71 (2016). https://doi.org/10.1186/s12863-016-0382-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-016-0382-5