
Abstract
Background
Multiple familial trichoepithelioma type 1 (MFT1; MIM 601606), a rare monogenic skin disease with autosomal dominant inheritance, is characterized by the development of multiple skin-colored papules on the central area of the face, frequently occurring in the nasolabial area. The disease is associated with various mutations in the cylindromatosis (CYLD; MIM 605018) gene that are also responsible for familial cylindromatosis (FC) and Brooke-Spiegler syndrome (BSS).
Methods
Recently we have identified a Spanish MFT1 pedigree with two affected family members (father and daughter). Direct sequencing of the CYLD gene revealed a worldwide recurrent heterozygous nonsense mutation (c.2272C/T, p.R758X) in the patients.
Results
This mutation has already been detected in patients with all three clinical variants – BSS, FC and MFT1 – of the CYLD-mutation spectrum. Haplotype analysis was performed for the Spanish patients with MFT1, Dutch patients with FC and an Austrian patient with BSS, all of whom carry the same heterozygous nonsense p.R758X CYLD mutation.
Conclusions
Our results indicate that this position is a mutational hotspot on the gene and that patients carrying the mutation exhibit high phenotypic diversity.
Similar content being viewed by others

Background
Multiple familial trichoepithelioma type 1 (MFT1; MIM 601606) is an autosomal dominant condition characterized by numerous firm skin-colored papules that are trichoepitheliomas (follicular tumors). The tumors grow slowly in size and number throughout life, often producing significant cosmetic disfigurement.
MFT1, familial cylindromatosis (FC; MIM 132700) and Brooke-Spiegler syndrome (BSS, MIM 605041) have been independently mapped to chromosome 16q12-q13 by several groups [1–3]. First, FC was mapped to this region in 1995 [1], and its candidate gene, the CYLD gene, was identified in 2000 [3]. Later BSS was mapped to the same region in 2000 [4]. In the mapped region, the same causative gene was identified in 2002 [5]. Regarding MFT1, the same causative gene was identified in 2003 [6]. These genetic investigations supported the previous clinical hypothesis, that MFT1 and FC might be the consequence of the dysfunction of the same gene, since their clinical symptoms can occur in the same patient or in different patients within the same family [7].
In the mapped region, the cylindromatosis (CYLD) gene [NM_015247] was identified as the causative gene responsible for the development of these three diseases [3]. The gene encodes an enzyme with deubiquitinase activity, which is involved in the post-translational modification of its target proteins and removes Lys63-linked ubiquitin chains [8]. The protein interacts with and negatively regulates the TRAF2, TRAF6, NEMO and BCL3 proteins, affecting the NF-ĸB signaling pathway [8].
Here we report a Spanish MFT1 pedigree with an affected father and daughter, in whom we have identified the recurrent p.R758X CYLD mutation. Previously reported cases carrying the same mutation are reviewed to compare the reported clinical phenotypes and to determine the geographical distribution of the mutation. Moreover, haplotype analysis of the Spanish patients with MFT1, as well as Dutch patients with FC and an Austrian patient with BSS was performed to investigate whether the same or different mutational events are responsible for the development of these cases.
Methods
Patients
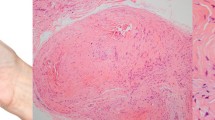
The Spanish MFT1 pedigree of Hispanic origin reported here was identified in the Levant region of Valencia, Spain. The 62-year-old father exhibited skin lesions that developed progressively on the central area of the face since the age of 14. Physical examination revealed multiple skin-colored papules measuring a few millimeters and coalescing to form plaques in both nasolabial folds (Fig. 1a), on the forehead, above the eyebrows and, to a lesser extent, on the ears, on the back of the head and on the back. Histological examination of one of the lesions from the right eyebrow revealed multiple basaloid cell aggregates with small keratinized cystic spaces surrounded by specific follicular stroma (Fig. 1b). These findings were consistent with the diagnosis of trichoepithelioma (cribriform trichoblastoma). Subsequently several biopsies were taken and all histological results supported the diagnosis of trichoepithelioma. The patient has been followed for 24 years, during which time the lesions have increased in number and size. Treatment consisted of block excision of multiple localized trichoepithelioma plaques in the nasolabial fold. Other lesions were treated mainly with electrocoagulation. The patient is currently receiving CO2 laser treatment.

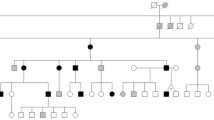
Skin symptoms, histology findings and pedigree of a Spanish family with multiple familial trichoepithelioma type 1. Two affected family members were identified. a The father presented skin-colored papules in the periorbital region, nose, nasolabial folds and upper lip. b Histological features of trichoepithelioma were islands of basaloid cells with peripheral palisading and small horny cysts (hematoxylin-eosin staining; original magnification × 10). c The daughter presented similar but smaller and fewer lesions in the nasolabial fold. d The pedigree of the investigated family. Written informed consent was obtained from all participants to publish this article and its accompanying images
The patient’s only child, a 33-year-old daughter, has lesions similar to those of her father but are fewer in number (Fig. 1c). The lesions first appeared in both nasolabial folds and, over time, began to appear on her forehead, temples, ears and scalp. Trichoepithelioma was confirmed with a biopsy of a lesion from the right temple. The daughter has been treated with electrocoagulation and cryotherapy, followed by 5 % imiquimod cream. Trichoepitheliomas in the left nasolabial fold were also treated with a single session of photodynamic therapy which had to be discontinued after a few minutes due to intense pain the area. A burn subsequently appeared in the treated area and took weeks to heal. Trichoepitheliomas were resolved but new lesions subsequently appeared in the same area. At present, the patient is receiving a CO2 laser treatment and is exhibiting good tolerance and acceptable aesthetic results. To date, no clinical or histological evidence for cylindromas or spiradenomas has been observed in either the father or the daughter. No other clinically affected member has been identified in this pedigree (Fig. 1d).
The data for Dutch and Austrian patients is publically available, and we did not take any samples from these patients ourselves. The investigated Dutch patients were previously reported by Van den Ouweland et al. [9]. Based on the development of cylindromas, these patients were diagnosed with FC. Their detailed clinical description is present in the report of Van den Ouweland et al. [9]. The investigated Austrian patient was previously reported by Grossmann et al. [10]. Based on the development of different skin appendage tumors, the diagnosis of BSS was established. The detailed clinical description of the symptoms is available in the study of Grossmann et al. [10].
Genetic investigation
The enrollment of the Spanish patients into the genetic investigations have been approved by the Clinical Research Ethics Committee (CEIC) of Consorcio Hospital General Universitario de Valencia. The performed genetic investigation was approved by the Internal Review Board of the University of Szeged, Szeged, Hungary. Written informed consents have been obtained from all the investigated subjects. The study was conducted according to the Principles of the Declaration of Helsinki.
Blood samples for genetic analyses were taken from the Spanish patients and from their clinically unaffected family members, as well as from unrelated controls. Genomic DNA was isolated with a BioRobot EZ1 DSP Workstation (QIAGEN; Godollo, Hungary). The coding regions of the CYLD gene and the flanking introns were amplified and sequenced (primer sequences were obtained from the UCSC Genome Browser, www.genome.ucsc.edu).
For haplotype analysis of the Spanish, Dutch and Austrian patients, common polymorphisms (n = 33) were genotyped from regions upstream (n = 20; rs199912760, rs375106322, rs201860550, rs149502055, rs376795685, rs144013604, rs75157714, rs201233994, rs200973965, rs77528321, rs146702654, rs6145827, rs3064638, rs73584492, rs190892314, rs200678983, rs76797023, rs77678929, rs376799359, rs201103123) and downstream (n = 13; rs370702435, rs10451132, rs201757487, rs137990687, rs368656359, rs149201712, rs185111122, rs146946436, rs141129479, rs72796392, rs111543527, rs11866167, rs35072258) of the identified mutation. A detailed list of the investigated polymorphisms is presented in Table 1. Genotypes of the investigated polymorphisms were determined with direct sequencing.
Results and discussion
Direct sequencing of the coding regions and the flanking introns of the CYLD gene from the investigated Spanish patients revealed a previously described nonsense mutation in exon 17 (c.2272C/T, p.R758X, rs121908388). This mutation results in a premature termination codon causing truncation and, thus, dysfunction of the CYLD protein. Both patients carried the mutation in heterozygous form (Fig. 2a), whereas the unaffected family members and unrelated controls carried the wild-type sequence (Fig. 2b, Additional file 1.). The identified mutation was located in the ubiquitin-specific protease domain of the CYLD protein (Fig. 2c).

Direct sequencing of the CYLD gene. Direct sequencing revealed a nonsense mutation (c.2272C/T, p.R758X) in exon 17. a The affected family members carried the deletion in heterozygous form. b The unaffected family members carried the wild-type sequence. c The mutation is located in the region of the ubiquitin-specific protease domain of the CYLD protein
Previously reported Dutch and Austrian cases carrying the same mutation were also investigated in this study [9, 10]. Haplotype analysis of the Spanish patients with MFT1, the Dutch patients with FC and Austrian patients with BSS was performed (Table 1.). Our results demonstrated that the Spanish and the Dutch pedigrees carry the same haplotype, whereas the Austrian patient carries a different haplotype. Thus, it can be assumed that different mutational events are responsible for the development of the Austrian case and the Spanish and Dutch cases.
A review of all previous studies reporting the same c.2272C/T, p.R758X nonsense mutation of the CYLD gene revealed that this mutation has also been detected in patients with BSS [10–12], FC [3, 9, 13] and MFT1 [10, 14]. Thus, the c.2272C/T, p.R758X nonsense mutation of the CYLD gene can lead to the manifestation of any of the clinical variants in the disease spectrum caused by CYLD mutation, which is associated with high phenotypic diversity. Furthermore, this mutation has been detected in Caucasian American [3], South African [10], Austrian [10, 14], Czech [11], Dutch [9], Chinese [12] and Japanese patients [13] and is, thus, considered a recurrent worldwide mutation (Table 2.). These data suggest that the c.2272C/T, p.R758X nonsense mutation is located at a mutational hotspot in the CYLD gene.
To determine whether the worldwide recurrent p.R758X mutation of the CYLD gene is the result of one or more independent mutational events, we performed haplotype analysis. The haplotype analysis of the Spanish, Dutch and Austrian patients demonstrated that, although the Spanish and the Dutch patients carry the same haplotype, the clinical appearance, MFT1 and FC, respectively, is different (Table 1.). These results suggest the importance of modifying genetic and/or environmental factors. In contrast with these, the Austrian patient carried a different haplotype than the Spanish and Dutch families. Thus, we assume the presence of the same mutation is the consequence of different mutational events (Table 1.).
Conclusion
Our results support the conclusion that position 2272 in the nucleotide sequence of the CYLD cDNA [NM_015247] is a mutational hotspot on the CYLD gene. This result correlates well with our previous findings for Hungarian and Anglo-Saxon BSS families carrying the same heterozygous nonsense mutation (c.2806C > T, p.Arg936X) but different haplotypes [15]. Of note, both mutational hotspots are the location of recurrent nonsense mutations. Regarding the encoded functional domains, both of mutational hotspots affect the ubiquitin-specific protease domain of the CYLD protein (Fig. 2c). Both recurrent nonsense mutations have been reported for all three clinical variants (MFT1, BSS, FC) of the CYLD-mutation based disease spectrum and have been associated with high intra- and interfamilial phenotypic diversity [15].
These reports raise the question of how these two worldwide recurrent nonsense mutations can lead to the development of the different clinical variants of the CYLD-mutation based disease spectrum. Further studies are needed to identify putative genetic, environmental or lifestyle factors and to elucidate the mechanism leading to the enormous phenotypic differences observed in patients carrying the same c.2272C/T, p.R758X nonsense mutation.
Availability of supporting data
All the supporting data are included as additional file.
Consent to publish
Written informed consent was obtained from all participants to publish this article.
Abbreviations
- MFT1:
-
Multiple familial trichoepithelioma type 1
- FC:
-
familial cylindromatosis
- BSS:
-
Brooke-Spiegler syndrome
- CYLD:
-
cylindromatosis gene
- TRAF2:
-
TNF receptor-associated factor 2
- TRAF6:
-
TNF receptor-associated factor 6
- NEMO:
-
NF-kappa-B essential modulator
- BCL3:
-
B-cell lymphoma 3-encoded protein
- NF-κB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
References
Biggs PJ, Wooster R, Ford D, Chapman P, Mangion J, Quirk Y, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441–3.
Biggs PJ, Chapman P, Lakhani SR, Burn J, Stratton MR. The cylindromatosis gene (cyld1) on chromosome 16q may be the only tumour suppressor gene involved in the development of cylindromas. Oncogene. 1996;12:1375–7.
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160–5.
Fenske C, Banerjee P, Holden C, Carter N. Brooke-Spiegler syndrome locus assigned to 16q12-q13. J Invest Dermatol. 2000;114:1057–8.
Gutierrez PP, Eggermann T, Holler D, Jugert FK, Beermann T, Grussendorf-Conen E-I, et al. Phenotype diversity in familial cylindromatosis: a frameshift mutation in the tumor suppressor gene CYLD underlies different tumors of skin appendages. J Invest Dermatol. 2002;119:527–31.
Hu G, Onder M, Gill M, Aksakal B, Oztas M, Gurer MA, et al. A novel missense mutation in CYLD in a family with Brooke-Spiegler syndrome. J Invest Dermatol. 2003;121:732–4.
Gerretsen AL, Beemer FA, Deenstra W, Hennekam FAM, van Vloten WA. Familial cutaneous cylindromas: investigations in five generations of a family. J Am Acad Dermatol. 1995;33:199–206.
Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappa-B signalling by deubiquitination. Nature. 2003;424:801–5.
Van den Ouweland AM, Elfferich P, Lamping R, van de Graaf R, van Veghel-Plandsoen MM, Franken SM, et al. Identification of a large rearrangement in CYLD as a cause of familial cylindromatosis. Fam Cancer. 2011;10:127–32.
Grossmann P, Vanecek T, Steiner P, Kacerovska D, Spagnolo DV, Cribier B, et al. Novel and recurrent germline and somatic mutations in a cohort of 67 patients from 48 families with Brooke-Spiegler syndrome including the phenotypic variant of multiple familial trichoepitheliomas and correlation with the histopathologic findings in 379 biopsy specimens. Am J Dermatopathol. 2013;35:34–44.
Kazakov DV, Thoma-Uszynski S, Vanecek T, Kacerovska D, Grossmann P, Michal M. A case of Brooke-Spiegler syndrome with a novel germline deep intronic mutation in the CYLD gene leading to intronic exonization, diverse somatic mutations, and unusual histology. Am J Dermatopathol. 2009;31:664–73.
Zhang G, Huang Y, Yan K, Li W, Fan X, Liang Y, et al. Diverse phenotype of Brooke-Spiegler syndrome associated with a nonsense mutation in the CYLD tumor suppressor gene. Exp Dermatol. 2006;15:966–70.
Oiso N, Mizuno N, Fukai K, Nakagawa K, Ishii M. Mild phenotype of familial cylindromatosis associated with an R758X nonsense mutation in the CYLD tumour suppressor gene. Br J Dermatol. 2004;151:1084–6.
Kazakov DV, Vanecek T, Zelger B, Carlson JA, Spagnolo DV, Schaller J, et al. Multiple (familial) trichoepitheliomas: a clinicopathological and molecular biological study, including CYLD and PTCH gene analysis, of a series of 16 patients. Am J Dermatopathol. 2011;33:251–65.
Nagy N, Rajan N, Farkas K, Kinyó A, Kemény L, Széll M. A mutational hotspot in CYLD causing cylindromas: a comparison of phenotypes arising in different genetic backgrounds. Acta Derm Venereol. 2013;93:743–5.
Acknowledgements
This research was supported by the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of TÁMOP-4.2.4.A/ 2-11/1-2012-0001 “National Excellence Program.” Nikoletta Nagy was also supported by the Hungarian Scientific Research Fund (OTKA) PD104782 grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KF carried out the mutation and haplotype analysis and the first draft of the manuscript. BKD and JV participated in the mutation and haplotype analysis. LCS, AMVM, JJVC, AMB, GMB and RRL cared for Spanish patients and perform the histological examination. TV and DVK cared for Austrian patient and carried out the mutation analysis. JNRK and AMWO cared for Dutch patients and carried out the mutation analysis. MS and NN were mentors who designed and guided the research study. All authors read and approved the final manuscript.
Additional file
Additional file 1:
Sequencing data of Patient III/1 demonstrated the same heterozygous mutation, which has been detected in case of Patient II/2. (JPG 163 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Farkas, K., Deák, B.K., Sánchez, L.C. et al. The CYLD p.R758X worldwide recurrent nonsense mutation detected in patients with multiple familial trichoepithelioma type 1, Brooke-Spiegler syndrome and familial cylindromatosis represents a mutational hotspot in the gene. BMC Genet 17, 36 (2016). https://doi.org/10.1186/s12863-016-0346-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-016-0346-9