
Abstract
Background
The evolution of male pregnancy is the most distinctive characteristic of syngnathids, and their specialized life history traits make syngnathid species excellent model species for many issues in biological evolution. However, the origin of syngnathids and the evolutionary divergence time of different syngnathid species remain poorly resolved. Comprehensive phylogenetic studies of the Syngnathidae will provide critical evidence to elucidate their origin, evolution, and dispersal patterns.
Results
We sequenced the mitochondrial genomes of eight syngnathid species in this study, and the estimated divergence times suggested that syngnathids diverged from other teleosts approximately 48.8 Mya during the Eocene period. Selection analysis showed that many mitochondrial genes of syngnathids exhibited significantly lower Ka/Ks values than those of other teleosts. The two most frequently used codons in syngnathid fishes were different from those in other teleosts, and a greater proportion of the mitochondrial simple sequence repeats (SSRs) were distributed in non-coding sequences in syngnathids compared with other teleosts.
Conclusions
Our study indicated that syngnathid fishes experienced an adaptive radiation process during the early explosion of species. Syngnathid mitochondrial OXPHOS genes appear to exhibit depressed Ka/Ks ratios compared with those of other teleosts, and this may suggest that their mitogenomes have experienced strong selective constraints to eliminate deleterious mutations.
Similar content being viewed by others


Background
Members of the teleost family Syngnathidae (seahorses, pipefishes, pipehorses, and seadragons), comprising approximately 300 species, are unique among vertebrates in that they exhibit ‘male pregnancy’, where males incubate developing embryos in a brood pouch until hatching and parturition [1,2,3]. The specialized brood pouch provides protection and carries out gas exchange, osmoregulation, and limited nutrient provisioning for the developing embryos [4, 5]. In addition, members of the family Syngnathidae exhibit other unique features such as an extended body covered with an armor of bony plates instead of scales, elongated snout, and fused jaws. The unique body morphology and specialized life history traits make syngnathid species excellent flagship species for many issues in marine conservation and biological evolution [4, 6].
There has been considerable research on the phylogeny, life history and biological characteristics of syngnathid fishes [4, 7,8,9,10,11,12]. However, the biological origin and divergence history of syngnathids still unclear. The family Syngnathidae is a large and diverse clade of bony fishes, and male brood-pouch morphology was a major focus of previous evolutionary research. The brooding structures vary greatly between genera, from the simplest incubating area typical of the Nerophinae to much more complex structures, such as the sealed pouch of the Hippocampinae [13]. Previous studies hypothesized that syngnathids can be divided into five major subfamilies based on brood pouch morphology, and subsequent studies divided syngnathids into two large clades based on the position of the male brood pouch [3, 12,13,14,15]. These phylogenetic divisions have been supported in molecular phylogenetic analyses based on partial mitochondrial sequences [13, 15, 16]. However, the reliability of the hypothesis has not yet been confirmed using more representative data. To date, few studies have investigated the divergence times of the different syngnathid lineages, and this information will provide critical evidence to elucidate the origin, evolution, and dispersal patterns of the family Syngnathidae.
Mitochondrial genomes have been widely used for diverse evolutionary studies of animals, including population genetics, phylogenetics, and species identification [13, 17,18,19,20]. The circular mitogenome of teleosts is structurally conserved and contains 13 protein-coding genes, 2 rRNA genes, 22 tRNA genes, and one displacement loop (D-loop) region [21, 22], that can provide a large amount of basic data for population genetics, phylogenetics, and adaptive evolution research [13, 23]. More than one thousand complete mitochondrial DNA sequences have been determined in teleostean fishes [24], but systematic research on mitogenome structure and molecular evolution characteristics in syngnathids is still scarce. Previous studies observed gene reorganization in fish mitogenomes [24,25,26], and recent studies have shown that the accumulation of mutations in mitogenomes is influenced by life history, effective population size, and cellular energy requirements [27,28,29,30]. Given the specialized biological characteristics and extraordinary evolutionary status of syngnathid fishes, we suspected that the structure and molecular evolution characteristics of their mitochondrial genomes may exhibit significant differences compared with those in other teleosts.
In this study, the mitogenomes of eight syngnathid species were assembled to investigate the phylogenetic relationships and divergence times of syngnathid lineages. We also obtained the mitochondrial genome sequences of a further 88 teleost species for comparison of the structure and molecular evolution characteristics between syngnathids and other teleosts.
Results
Mitochondrial genomes in the Syngnathidae
The complete mitochondrial genomes of the syngnathid species ranged in size from 16,462 bp to 16,961 bp, with the newly determined T. serratus, S. hardwickii, S. biaculeatus, D. boaja, D. dactyliophorus, M. manadensis, H. kelloggi, and H. mohnikei mitochondrial genomes exhibiting lengths of 16,956 bp, 16,519 bp, 16,479 bp, 16,547 bp, 16,661 bp, 16,527 bp, 16,536 bp, and 16,518 bp, respectively (Fig. 1, Table 1). The differences in genome length were largely due to variations in tandem repeats in the control regions. An approximately 200 bp non-coding insertion between 16S-rRNA and tRNA-Leu was found in T. serratus and C. flavofasciatus (Fig. 2). All the genomes shared 13 protein-coding genes, 22 tRNA genes, 2 rRNAs, and a control region, and exhibited the same gene order (Fig. 2). The AT content of the mitogenome ranged from 55.31 to 62.07% for the eight newly sequenced species, with a slight AT bias.

Map of sampling locations for syngnathid fishes

Linear comparison of mitochondrial genome organization in syngnathid fishes and other teleosts. Hm, Hippocampus mohnikei; Hk, Hippocampus kelloggi; Ts, Trachyrhampus serratus; Cf, Corythoichthys flavofasciatus; Sh, Solegnathus hardwickii; Sb, Syngnathoides biaculeatus; Pv, Pegasus volitans; Ss, Salmo salar; Tf, Takifugu flavidus
In silico analysis of simple sequence repeats (SSRs) in mitochondrial genomes
A total of 64 SSRs of different nucleotide combinations were detected in 44 species. An equal number of SSRs were detected in the syngnathids and the other teleosts: 32 SSRs were found in 18 of the 22 syngnathids, and 32 SSRs were found in 19 of the 22 other teleosts (Additional file 1). However, the distribution patterns of the SSRs were very different between the syngnathids and the other teleosts. In the syngnathids, 35.5% of the SSRs were detected in coding sequences and 64.5% in non-coding sequences. In contrast, 87.5% of the SSRs were found in coding sequences in the other teleosts, and only 12.5% of the SSRs were distributed across non-coding sequences (Fig. 3). Overall, a greater proportion of the mitochondrial SSRs were distributed in non-coding sequences in syngnathids compared with other teleosts.

Distribution of SSRs in the mitogenome of syngnathid fishes and other teleosts
Nucleotide diversity and codon usage
Sliding window analysis of sequences from 22 syngnathids and 22 other teleosts was employed to evaluate the nucleotide diversity of the mitogenomes. The nucleotide diversity exhibited similar patterns in the two groups, with Pi values ranging from 0.043 to 0.335 in syngnathids and 0.037 to 0.412 in other teleosts (Fig. 4). The ND2, ND6, and ATP8 regions showed relatively high sequence variability, while genes with relatively low sequence variability included COI, COII, COIII, and Cyt b (Fig. 4).

Nucleotide diversity (Pi) of the mitochondrial genome sequences of syngnathid fishes and other teleosts
A significant bias towards A/T was observed in the codon usage of the mitochondrial genomes of the syngnathids Hippocampus mohnikei and Doryichthys bojia, and the other teleost Takifugu flavidus (Fig. 5, Additional file 2), and G was the least common third position nucleotide in all the codon families. The most frequently used codons in H. mohnikei and D. bojia were UUA (Leu) and AUU (Ile), while the most frequently used codons in T. flavidus were UUC (Phe) and CUA (Leu) (Additional file 3). The most frequently used codons all consisted of A and T, and this may have contributed to the high A + T content in the mitogenome. There was an obvious difference between the two syngnathid fishes and T. flavidus in relative synonymous codon usage (RSCU), especially in Cys, His, Leu, and Phe. Among the 62 available codons (excluding TAA and TAG), AGA (Arg) was missing in H. mohnikei, D. bojia, and T. flavidus, while AGG (Arg) was missing in H. mohnikei and D. bojia (Fig. 5, Additional file 4).

Relative synonymous codon usage (RSCU) of the mitochondrial genomes. Left, Hippocampus mohnikei; middle, Doryichthys bojia; right, Takifugu flavidus
Phylogenetic reconstruction and divergence time estimation
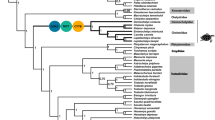
The phylogenetic relationships between syngnathids and other teleosts were determined using 13 mitochondrial gene sequences. The best substitution model for the data matrix was GTR + G + I, and maximum-likelihood (ML) analyses yielded a well-resolved phylogeny for the fishes: most of the internal branches were supported by high (> 90%) bootstrap probabilities (BPs; Fig. 6). The Syngnathidae family is a unique clade of bony fishes that exhibit special male brooding structures and our phylogenetic analysis recovered the syngnathids as a monophyletic group, with a high level of confidence (99% BP). The divergence times of syngnathids and other teleosts estimated using the MCMC tree suggest that syngnathids diverged from the other teleosts approximately 48.8 Mya (95% CI 48.4–49.0 Mya), during the Eocene period. Eleven Hippocampus species formed a monophyletic clade which constituted the largest genus (BP = 100%) that appeared 18.8 Mya (95% CI 13.7–24.0 Mya, Additional file 3), during the Miocene period. Phylogenetic reconstruction suggested that Corythoichthys flavofasciatus is the closest living relative of Trachyrhamphus serratus and a sister clade to Hippocampus. Pegasidae and Syngnathidae were the closest sister groups (BP = 100%). Indostomidae clustered with Monopterus albus (Synbranchiformes). However, Gasterosteoidei did not cluster with Syngnathoidei. The gasterosteoids Aulorhynchus flavidus, Gasterosteus aculeatus, and Hypoptychus dybowskii formed a group in the ML tree parallel to Epinephelus coioides (Perciformes).

Time-calibrated phylogeny of 96 fishes based on 13 mitochondrial genes. The black dots represent calibration points, and the five-pointed star represents a seahorse fossil. Cross-sections of the brood pouch: , fully enclosed pouch;
, fully enclosed pouch; , enclosed pouch (inverted);
, enclosed pouch (inverted); , rudimentary pouch with plates and skin-folds;
, rudimentary pouch with plates and skin-folds; , individual egg compartments;
, individual egg compartments; , unprotected eggs (free spawning)
, unprotected eggs (free spawning)
The schematic diagram of the phylogenetic relationships of syngnathid groups shows that the pipefish can be divided into two genetic lineages; one clustered with seahorses, and the other clustered independently of other syngnathids (Fig. 7a). Additionally, the phylogenetic analysis revealed that the pipehorses were paraphyletic with respect to seadragons (Fig. 7a). Phylogenetic analysis of the syngnathids with various brooding structures revealed asynchronous evolution of the brood pouch. The seahorses with a sealed pouch formed a monophyletic group, and this clustered with one lineage of bilateral-pouch syngnathids, while the other lineage of bilateral-pouch syngnathids clustered with syngnathids that have no pouch structure but egg-compartments on the abdomen (Fig. 7b).

Schematic diagram of the phylogenetic relationships of syngnathid groups (a), and phylogenetic analysis of the syngnathids with various brooding structures (b). Cross-sections of the brood pouch, fully enclosed pouch;, enclosed pouch (inverted);, rudimentary pouch with plates and skin-folds;, individual egg compartments;, unprotected eggs (free spawning)
Male-pregnant syngnathids are under strong purifying selection
Differences in the Ka/Ks ratios of fishes with different reproductive strategies were quantified by separate analyses of two groups: the male-pregnant syngnathid fishes and other teleostean fishes. The mean Ka/Ks ratio of all mitochondrial genes was slightly lower in the male-pregnant syngnathids (Ka/Ks = 0.0392) than in the other teleostean fishes (Ka/Ks = 0.0445; Fig. 8, Additional file 4). Investigation of the general pattern of Ka/Ks in the 13 mitochondrial protein-coding genes showed that five genes (COI, Cyt b, ND1, ND4, and ND6) exhibited significantly lower Ka/Ks values in the syngnathid fishes than in the other teleosts (P < 0.01; Fig. 8).

Comparison of Ka/Ks ratios for 13 individual genes and 13 gene sets between syngnathids and other fishes. “All” indicates the combined sequences of the 13 mitochondrial genes. The asterisks indicate the significance of the likelihood ratio test results from the free-ratio and one-ratio models (*, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001)
Discussion
Molecular characteristics of syngnathid mitogenomes
The mitochondrial genome organization of syngnathid fishes was quite conserved, as identified for other bony fishes [22, 31,32,33]. The differences in length among the Syngnathidae mitochondrial genome sequences are mainly due to length variation of the control region and random insertions in the intergenic regions. It is widely accepted that the mitochondrial DNA control region evolves faster than protein-coding genes [34, 35], and the pressure of purifying selection in the non-coding regions tends to be substantially weaker than that in coding regions [36]. Therefore, length variation would be more readily accumulated in the control region than in coding regions of the mitochondrial genome. In addition, random insertions of non-coding sequences between the mitochondrial genes were found in T. serratus and C. flavofasciatus, and these can be used as a basis for species identification. The insertion of non-coding sequences in the mitochondrial genome was also found in Culicoides species [37].
The mitochondrial nucleotide diversity of syngnathid fishes exhibited a similar pattern to that in other bony fishes, and the differences in sequence variability among mitochondrial genes will provide insight into their suitability for phylogenetic studies at various taxonomic levels [38, 39]. The codon usage bias was a textbook example of a weak selective pressure operating at the molecular level, and other evolutionary forces might explain its variation across different biological groups [40,41,42]. The two most frequently used codons in syngnathid fishes were different from those in other teleosts, and this may reflect a different evolutionary process for the mitochondrial genes of syngnathids compared to those of other teleosts. A significant bias towards A/T was observed in the codon usage of the mitochondrial genomes of syngnathids and other teleosts that may have contributed to the high A + T content in the mitogenome. However, codons in fish nuclear DNA sequences end predominantly in G or C, even though the coding sequences are not enriched in these nucleotides [43]. We found that G was the least common third position nucleotide in all the codon families in the mitochondrial sequences. We note that the abovementioned features are very similar to those observed in other vertebrates [44, 45].
Phylogeny of syngnathid fishes
Syngnathids are perhaps the most unusual and specialized group of fishes considering their male reproductive mode [13]. The variation in pouch structure is one of the most important phenomena to have occurred throughout the evolutionary divergence of syngnathids [14]. The estimates of divergence times obtained in this study provide new information on the evolutionary history of syngnathids that can improve our understanding of the biological adaptability of pouch structure variation. Molecular clocks are vital for reconstructing detailed timescales in the tree of life that can explain how evolutionary events have been influenced by Earth’s history [46]. Molecular dating analyses indicated that the Syngnathidae most likely evolved 48.8 Mya, and this is concordant with the oldest known syngnathid fossil (48–50 Mya) [16, 47]. Our results suggested that the divergence of Syngnathidae likely resulted from global climate change during the late Paleocene and early Eocene. The Paleocene-Eocene thermal maximum was a short interval of maximum temperatures lasting approximately 100,000 years during the late Paleocene and early Eocene epochs (roughly 55 Mya) [48, 49]. Sea surface and continental air temperatures increased by more than 5 °C at that time, and this may have had a large impact on marine fishes, especially syngnathids, considering their distinct life-history characteristics and breeding strategies. Thus, we suspect that syngnathids experienced an adaptive radiation process during their early explosion of species. Hippocampus reidi and H. ingens diverged 3.7 Mya; the calibration point that we used in this study was based on evidence from a seahorse phylogeny that indicated that the closure of the Central American Seaway during the Late Pliocene resulted in the divergence of H. reidi (West Atlantic) and H. ingens (East Pacific) from a common ancestor (3.1–3.7 Mya) [50]. The results estimated using molecular clock approaches are consistent with the divergence events in the seahorse phylogeny.
The genus Hippocampus has evolved the most complex and advanced fully enclosed brood pouches for the delivery of nutrients and oxygen to the developing embryos [4, 12]. The molecular phylogeny obtained in this study shows that the development of brood pouch was an important evolutionary innovation of the Syngnathidae and the diversification of pouch types from simple ventral gluing areas to the completely enclosed pouches emphasizes the importance of the brood pouch structure variation in the radiation of syngnathids [15, 51, 52].
Selection patterns within syngnathid mitogenomes
Nonsynonymous substitutions are generally harmful because they can cause defects in the respiratory electron transport chain and other metabolic processes [53, 54]. The conserved mitochondrial protein-coding sequences of syngnathid fishes might have undergone strong purifying selection to eliminate deleterious mutations. Syngnathid fishes appear to exhibit depressed Ka/Ks ratios for their mitochondrial OXPHOS genes compared with other teleostean fishes; this suggests that syngnathid fishes experience stronger selective constraints. Differential selection was also found in similar analyses performed in other fishes, birds, and mammals [27, 28, 30]. A study on mitogenome selection pressure in birds showed that stronger selective constraints act on highly locomotive birds and mammals with differing locomotive speeds exhibit a similar pattern [28]. Among fish species, the mitochondrial protein-coding genes of migratory fishes exhibit significantly lower Ka/Ks values than those of nonmigratory fishes [30]. The OXPHOS genes may have undergone stronger purifying selection because they play more important roles in energy metabolism than other mitochondrial genes. Moreover, the low number of SSRs in the mitochondrial coding regions of syngnathid fishes may indicate that their mitochondrial genomes are under strong purifying selection.
Conclusions
The mitochondrial genome of eight syngnathid fishes were sequenced in this study, and molecular dating analyses indicated that the Syngnathidae most likely evolved 48.8 Mya, which is concordant with the oldest known syngnathid fossil. Syngnathid mitochondrial genes appear to exhibit depressed Ka/Ks ratios compared with those of other teleosts, and this may suggest that their mitogenomes have experienced strong selective constraints to eliminate deleterious mutations.
Methods
Source of data and primary treatment
The complete mitochondrial genome sequences of 88 teleost fishes available in September 2018 were downloaded from GenBank (Additional file 5). Additionally, the mitochondrial genomes of eight syngnathid fishes were newly sequenced for this study (Trachyrhamphus serratus, KJ184528; Solegnathus hardwickii, KJ184524; Syngnathoides biaculeatus, KJ184525; Doryichthys boaja, KJ184527; Doryrhamphus dactyliophorus, KP301502; Microphis manadensis, KP301501; Hippocampus kelloggi, KF703755; and Hippocampus mohnikei, KF557651). For the species to be newly sequenced, adults were collected from coastal areas of China in 2014–2017 (Fig. 1). A small amount of dorsal fin was sampled from every fish, which has no effect on the swimming and health, and then they were returned alive to the water. All samples used in this study were treated in accordance with relevant national and international guidelines.
Total genomic DNA was extracted from samples using the TIANGEN marine animal DNA kit (TIANGEN, Beijing, China) according to the manufacturer’s instructions. Degenerate primers for fragment amplification were designed based on conserved nucleotide sequences from the mitochondrial genomes of Hippocampus kuda (AP005985) and Microphis brachyurus (AP005986) using DNAssist 2.2 and Primer Premier 5.0 software [55]. The PCR amplifications, sequence assemble, and genome annotation were performed according to a slightly modified method described by Wang et al. [31].
Phylogenetic analyses and molecular dating
All protein-coding genes were aligned using MAFFT version 7 [56]. The best-fitting nucleotide substitution model was selected using ModelTest 3.06 [57]. Maximum-likelihood (ML) analyses were implemented in PhyML 3.0 [58].
Molecular dating was performed using BEAST 1.4.6 [59]. A total of 26 calibration points was used in this analysis (Additional file 6). In each case, a normal prior was used, and its mean and standard deviation were set so that the 95% confidence intervals corresponded to the upper and lower bounds of each calibration point. Thus, uncertainty concerning the exact dates of the calibration points could be accounted for.
Selection analyses
Comparison of the rates of nonsynonymous (Ka) and synonymous (Ks) substitutions can provide information on the type of selection that has acted on a given set of protein-coding sequences. The ratio of the rates of nonsynonymous to synonymous substitutions, ω (Ka/Ks), provides an indication of changes in selective pressure: Ka/Ks values > 1 indicate positive selection; Ka/Ks = 1 indicates neutral selection; and Ka/Ks < 1 indicates negative or purifying selection. The Ka/Ks ratios of all individual datasets were estimated for each branch of the phylogenetic tree using the CodeML algorithm from the PAML package [60]. The branch model was employed under two alternative assumptions: a one-ratio model, where one ω value was assumed for the entire tree, and a free-ratio model, where ω values were allowed to vary on every branch. We constructed a likelihood ratio test (LRT) to verify that the best models fitted the data. The level of significance for these LRTs was calculated using a χ2 approximation, where twice the difference in log likelihood between the models would follow a χ2 distribution, with the number of degrees of freedom corresponding to the difference in the number of parameters between the models.
Comparative analysis of the mitochondrial genomes
MISA was used to analyze SSRs in the mitochondrial genomes [61]. SSRs were detected in the mitogenome sequences of 22 syngnathids and 22 other teleosts. The nucleotide diversity of the mitochondrial protein-coding gene sequences was evaluated using sliding window analysis based on the dataset used for SSR detection (window size = 200 bp, step size = 10 bp) in DnaSP version 5.10 [62]. The base composition was calculated using BioEdit version 7.1.3.0, and the AT-skew and GC-skew were calculated according to the formulae: AT-skew = (A − T%)/(A + T%) and GC-skew = (G − C%)/(G + C%). Relative synonymous codon usage (RSCU) of all protein-coding genes was analyzed using MEGA version 6.06 [63].
Abbreviations
- BP:
-
Bootstrap probability
- D-loop:
-
Displacement loop
- LRT:
-
Likelihood ratio test
- ML:
-
Maximum-likelihood
- RSCU:
-
Relative synonymous codon usage
- SSRs:
-
Simple sequence repeats
References
Lin Q, Fan S, Zhang Y, Xu M, Zhang H, Yang Y, et al. The seahorse genome and the evolution of its specialized morphology. Nature. 2016;540:395.
Stölting KN, Wilson AB. Male pregnancy in seahorses and pipefish: beyond the mammalian model. Bioessays. 2007;29:884–96.
Wilson AB, Vincent A, Ahnesjo I, Meyer A. Male pregnancy in seahorses and pipefishes (family Syngnathidae): rapid diversification of paternal brood pouch morphology inferred from a molecular phylogeny. J Hered. 2001;92:159–66.
Foster SJ, Vincent ACJ. Life history and ecology of seahorses: implications for conservation and management. J Fish Biol. 2004;65:1–61.
Linton JR, Soloff BL. The physiology of the brood pouch of the male sea horse Hippocampus erectus. B Mar Sci. 1963;14:45–61.
Qin G, Zhang Y, ALFC H, Zhang Y, Lin Q, Qin G, et al. Seasonal distribution of seahorses employing a social reproductive strategy and supporting for a closed fishing season. ICES J Mar Sci. 2017;74:2170-9.
Bergert BA, Wainwright PC. Morphology and kinematics of prey capture in the syngnathid fishes Hippocampus erectus and Syngnathus floridae. Mar Biol. 1997;127:563–70.
Kendrick AJ, Hyndes GA. Patterns in the abundance and size-distribution of syngnathid fishes among habitats in a seagrass-dominated marine environment. Estuar Coast Shelf S. 2003;57:631–40.
Gurkan Ş, Taşkavak E. Length–weight relationships for Syngnathid fishes of the Aegean Sea. Turkey Belg J Zool. 2007;137:219–22.
Neutens C, Adriaens D, Christiaens J, De Kegel B, Dierick M, Boistel R, Van Hoorebeke L. Grasping convergent evolution in syngnathids: a unique tale of tails. J Anat. 2014;224:710–23.
Bannikov AF, Carnevale G, Popov YA. An extraordinary pipefish (Teleostei, Syngnathidae) with fully developed anal fin from the Oligocene of the North Caucasus (SW Russia). Boll Soc Paleontol Ital. 2017;56:79–88.
Hamilton H, Saarman N, Short G, Sellas AB, Moore B, Hoang T, Grace CL, Gomon M, Crow K, Brian Simison W. Molecular Phylogeny and Patterns of Diversification in Syngnathid Fishes. Mol Phylogen Evol. 2016;107:388-403.
Zhang YH, Qin G, Zhang HX, Wang X, Lin Q. DNA barcoding reflects the diversity and variety of brooding traits of fish species in the family Syngnathidae along China’s coast. Fish Res. 2017;185:137–44.
Wilson AB, Ahnesjo I, Vincent ACJ, Meyer A. The dynamics of male brooding, mating patterns, and sex roles in pipefishes and seahorses (family Syngnathidae). Evolution. 2003;57:1374–86.
Wilson AB, Orr JW. The evolutionary origins of Syngnathidae: pipefishes and seahorses. J Fish Biol. 2011;78:1603–23.
Teske PR, Beheregaray LB. Evolution of seahorses' upright posture was linked to Oligocene expansion of seagrass habitats. Biol Lett. 2009;5:521–3.
Tiratha RS. Mitochondrial gene rearrangements: new paradigm in the evolutionary biology and systematics. Bioinformation. 2008;3:95–7.
Galtier N, Nabholz B, Glémin S, Hurst G. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol. 2009;18:4541–50.
Hewitt GM. Speciation, hybrid zones and phylogeography. Mol Ecol. 2010;10:537–49.
Karl SA, Toonen RJ, Grant WS, Bowen BW. Common misconceptions in molecular ecology: echoes of the modern synthesis. Mol Ecol. 2012;21:4171–89.
Miya M, Nishida M. Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): first example of transfer RNA gene rearrangements in bony fishes. Mar Biotechnol. 1999;1:416–26.
Broughton R, Milam J, Roe B. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001;11:1958–67.
Yu L, Peng D, Liu J, Luan P, Liang L, Hang L, et al. On the phylogeny of Mustelidae subfamilies: analysis of seventeen nuclear non-coding loci and mitochondrial complete genomes. BMC Evol Biol. 2011;11:92.
Shi W, Miao XG, Kong XY. A novel model of double replications and random loss accounts for rearrangements in the Mitogenome of Samariscus latus (Teleostei: Pleuronectiformes). BMC Genomics. 2014;15:352.
Kong X, Dong X, Zhang Y, Shi W, Wang Z, Yu Z. A novel rearrangement in the mitochondrial genome of tongue sole, Cynoglossus semilaevis: control region translocation and a tRNA gene inversion. Genome. 2009;52:975–84.
Saitoh K, Hayashizaki K, Yokoyama Y, Asahida T, Toyohara H, Yamashita Y. Complete nucleotide sequence of Japanese flounder (Paralichthys olivaceus) mitochondrial genome: structural properties and cue for resolving teleostean relationships. J Hered. 2000;91:271–8.
Chong RA, Mueller RL. Low metabolic rates in salamanders are correlated with weak selective constraints on mitochondrial genes. Evolution. 2013;67:894–9.
Shen YY, Shi P, Sun YB, Zhang YP. Relaxation of selective constraints on avian mitochondrial DNA following the degeneration of flight ability. Genome Res. 2009;19:1760–5.
Strohm JH, Gwiazdowski RA, Hanner R. Fast fish face fewer mitochondrial mutations: patterns of dN/dS across fish mitogenomes. Gene. 2015;572:27–34.
Sun YB, Shen YY, Irwin DM, Zhang YP. Evaluating the roles of energetic functional constraints on teleost mitochondrial-encoded protein evolution. Mol Biol Evol. 2011;28:39–44.
Wang X, Han X, Zhang Y, Liu S, Lin Q. Phylogenetic analysis and genetic structure of the seahorse, Hippocampus fuscus from the Arabian and Red Sea based on mitochondrial DNA sequences. Mitochondrial DNA A. 2019;30:165–71.
Teacher AG, André C, Merilä J, Wheat CW. Whole mitochondrial genome scan for population structure and selection in the Atlantic herring. BMC Evol Biol. 2012;12:248.
Smith DR, Lee RW. Nucleotide diversity in the mitochondrial and nuclear compartments of Chlamydomonas reinhardtii: investigating the origins of genome architecture. BMC Evol Biol. 2008;8:156.
Wilkinson M, Cotton JA, Lapointe F, Pisani D, Page R. Properties of Supertree methods in the consensus setting. Syst Biol. 2007;56:330–7.
Tang DL, Di BP, Wei GF, Ni IH, Oh IS, Wang SF. Spatial, seasonal and species variations of harmful algal blooms in the South Yellow Sea and East China Sea. Hydrobiologia. 2006;568:245–53.
Resch AM, Carmel L, Mariñoramírez L, Ogurtsov AY, Shabalina SA, Rogozin IB, et al. Widespread positive selection in synonymous sites of mammalian genes. Mol Biol Evol. 2007;24:1821–31.
Matsumoto Y, Yanase TT, Noda H. Species-specific mitochondrial gene rearrangements in biting midges and vector species identification. Med Vet Entomol. 2010;23:47–55.
Kartavtsev YP. Divergence at Cyt-b and co-1 mtDNA genes on different taxonomic levels and genetics of speciation in animals. Mitochondrial DNA. 2011;22:55.
Thai BT, Si VN, Phan PD, Austin CM. Phylogenetic evaluation of subfamily classification of the Cyprinidae focusing on Vietnamese species. Aquat Living Resour. 2007;20:143–53.
Galtier N, Roux C, Rousselle M, Romiguier J, Figuet E, Glémin S, et al. Codon usage bias in animals: disentangling the effects of natural selection, effective population size and GC-biased gene conversion. Mol Biol Evol. 2018;35:1092–103.
Pouyet F, Mouchiroud D, Duret L, Sémon, M. Recombination, meiotic expression and human codon usage. Elife. 2017;6:e27344.
Rudolph KLM, Bianca MS, Diego V, Robert JW, John CM, Claudia K, et al. Data files for downstream analysis of the “mammalian codon usage” manuscript. 2016.
Fitzgerald LM, Rodríquez A, Smutzer G. Codon usage in bony fishes. Mol Mar Biol Biotechnol. 1993;2:112–9.
Liu QN, Xin ZZ, Bian DD, Chai XY, Zhou CL, Tang BP. The first complete mitochondrial genome for the subfamily Limacodidae and implications for the higher phylogeny of Lepidoptera. Sci Rep. 2016;6:35878.
Sarvani RK, Parmar DR, Tabasum W, Thota N, Sreenivas A, Gaur A. Characterization of the complete mitogenome of Indian mouse deer, Moschiola indica (Artiodactyla: Tragulidae) and its evolutionary significance. Sci Rep. 2018;8:2697.
Dietrich LEP, Tice MM, Newman DK. The co-evolution of life and earth. Curr Biol. 2006;16:395–400.
Bellwood DR. The Eocene fishes of Monte Bolca: the earliest coral reef fish assemblage. Coral Reefs. 1996;15:11–9.
Röhl U, Westerhold T, Bralower TJ, Zachos JC. On the duration of the Paleocene-Eocene thermal maximum (PETM). Geochem Geophy Geosy. 2007;81:229–47.
Torfstein A, Winckler G, Tripati A. Productivity feedback did not terminate the Paleocene-Eocene thermal maximum (PETM). Clim Past. 2010;6:265–72.
Duque-Caro H. Neogene stratigraphy, paleoceanography and paleobiogeography in Northwest South America and the evolution of the Panama seaway. Palaeogeogr Palaeocl. 1990;77:203–34.
Dawson CE. Indo-Pacific pipefishes (Red Sea to the Americas): Gulf Coast research laboratory; 1985.
Herald ES. From pipefish to seahorse - a study of phylogenetic relationships. San Francisco California Academy of Sciences; 1959.
Brown MD, Trounce IA, Jun AS, Allen JC, Wallace DC. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 leber's hereditary optic neuropathy mitochondrial DNA mutation. J Biol Chem. 2000;275:39831–6.
Weber K, Wilson JN, Taylor L, Brierley E, Johnson MA, Turnbull DM, et al. A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am J Hum Genet. 1997;60:373–80.
Singh VK, Mangalam AK, Dwivedi S, Naik S. Primer premier: program for design of degenerate primers from a protein sequence. Biotechniques. 1998;24:318–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91.
Thiel T, Michalek W, Varshney RK, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106:411–22.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Acknowledgements
We thank Shao-Hua Fan and Peng Shi who provided excellent comments and suggestions on the manuscript.
Funding
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA13020103), Youth Talent Program Supported by Laboratory for Marine Fisheries Science and Food Production Processes, Pilot National Laboratory for Marine Science and Technology (Qingdao)(2018-MFS-T04), the National Key Research and Development Program (2017YFC0506302), and the National Natural Science Foundation of China (41576145). The funders had no role in the study design, data collection and analysis, interpretation of data, or writing of the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. All sequence data have been deposited in GenBank, DNA sequences are available under GenBank accessions: Trachyrhamphus serratus, KJ184528; Solegnathus hardwickii, KJ194524; Syngnathoides biaculeatus, KJ184525; Doryichthys boaja, KJ184527; Doryrhamphus dactyliophorus, KP301502; Microphis manadensis, KP301501; Hippocampus kelloggi, KF703755; and Hippocampus mohnikei, KF557651.
Author information
Authors and Affiliations
Contributions
QL and XW designed the experiments; HXZ and GQ collected specimens; XW and YHZ analysed the data; XW, QL, and YHZ wrote the paper; All authors contributed to the editing and gave final approval for publication.
Corresponding author
Ethics declarations
Ethics approval
All samples used in this study were with Animal Ethics approval for experimentation granted by the Chinese Academy of Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
List of the SSRs in the mitochondrial genome of 44 teleost fishes. (DOCX 27 kb)
Additional file 2:
Statistics of the codon usage in the mitochondrial genes. (XLSX 13 kb)
Additional file 3:
95% confidence interval of dating analysis. (JPG 797 kb)
Additional file 4:
Selection analysis of the mitochondrial genes. (XLSX 53 kb)
Additional file 5:
List of the species used in this study. (XLSX 15 kb)
Additional file 6:
Calibration points used in the divergence time analysis by BEAST List of the species used in this study. (XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Wang, X., Zhang, Y., Zhang, H. et al. Complete mitochondrial genomes of eight seahorses and pipefishes (Syngnathiformes: Syngnathidae): insight into the adaptive radiation of syngnathid fishes. BMC Evol Biol 19, 119 (2019). https://doi.org/10.1186/s12862-019-1430-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1430-3