
Abstract
Adaptation to local host plants may impact a pollinator’s population genetic structure by reducing gene flow and driving population genetic differentiation, representing an early stage of ecological speciation. South African Rediviva longimanus bees exhibit elongated forelegs, a bizarre adaptation for collecting oil from floral spurs of their Diascia hosts. Furthermore, R. longimanus foreleg length (FLL) differs significantly among populations, which has been hypothesised to result from selection imposed by inter-population variation in Diascia floral spur length. Here, we used a pooled restriction site-associated DNA sequencing (pooled RAD-seq) approach to investigate the population genetic structure of R. longimanus and to test if phenotypic differences in FLL translate into increased genetic differentiation (i) between R. longimanus populations and (ii) between phenotypes across populations. We also inferred the effects of demographic processes on population genetic structure and tested for genetic markers underpinning local adaptation.
Results
Populations showed marked genetic differentiation (average FST = 0.165), though differentiation was not statistically associated with differences between populations in FLL. All populations exhibited very low genetic diversity and were inferred to have gone through recent bottleneck events, suggesting extremely low effective population sizes. Genetic differentiation between samples pooled by leg length (short versus long) rather than by population of origin was even higher (FST = 0.260) than between populations, suggesting reduced interbreeding between long and short-legged individuals. Signatures of selection were detected in 1119 (3.8%) of a total of 29,721 SNP markers,
Conclusions
Populations of R. longimanus appear to be small, bottlenecked and isolated. Though we could not detect the effect of local adaptation (FLL in response to floral spurs of host plants) on population genetic differentiation, short and long legged bees appeared to be partially differentiated, suggesting incipient ecological speciation. To test this hypothesis, greater resolution through the use of individual-based whole-genome analyses is now needed to quantify the degree of reproductive isolation between long and short legged bees between and even within populations.
Similar content being viewed by others

Background
Mating usually takes place among a subset of individuals of a species, typically within a portion of its distributional range, and, assuming limited dispersal, populations of that species inevitably become genetically structured [1]. The population genetic structure of populations is then determined in part by the strength of two parameters: effective population size (Ne) and the amount of gene flow among populations [1]. Low Ne is a typical feature of rare or endangered species [2] or of populations that have experienced a recent bottleneck, such as founder populations [1]. Limited gene flow may be caused by fragmentation of the landscape due to human interferences (e.g. agriculture, transport), natural barriers (e.g. waterways, mountains) or abiotic factors (e.g. climate). The environment might also exert non-negligible selection pressures upon populations, whereby adaption to ecologically different environments might reduce intraspecific gene flow and lead to reproductive barriers (e.g. selection against hybrids or immigrants, positive assortative mating) between populations or individuals varying in adaptive traits [3,4,5,6], which may represent incipient stages of ecological speciation [7].
Host plant adaptation seems to be a common feature of many insect-plant interaction systems [5, 8,9,10,11,12,13]. Adaptation to different host plant morphologies might hinder genetic exchange between insect populations and generate strong barriers to gene flow [14]. Such populations may then accumulate allele frequency differences, i.e. become genetically differentiated, often assessed via FST, which relates the amount of genetic variation among populations to the total genetic variation over all populations [15]. Initially, increased genetic differentiation is expected at loci underlying local adaptation whereas ecologically neutral loci are not subject to divergent selection and should therefore show less differentiation. This generates a heterogeneous pattern of genomic divergence characterised by ‘islands of genomic divergence’ containing FST outlier loci [16, 17]. The more adaptively divergent populations become, the greater the reduction in gene flow and the higher the genome-wide differentiation, yielding a pattern of isolation by adaptation (IBA, [18]). Host plant mediated genetic differentiation and incipient ecological speciation have been suggested for several insects [4, 5, 9, 19, 20].
South African Rediviva bees are striking examples of the bizarre morphology that host plant adaptation may generate. Females of many Rediviva species have evolved extremely elongated forelegs, sometimes longer than their entire body length [21, 22]. Forelegs are used for oil collection from oil-producing plants, whereby the bee inserts its forelegs into the host floral spurs and rubs them against the spur walls to absorb oil with specialised hairs on the tarsi [23, 24]. The extracted oil is then transported back to the nest and used to feed larvae and probably also for brood cell lining [25, 26].
Foreleg length (FLL) of Rediviva females is an evolutionary highly labile trait which likely plays a role in Rediviva diversification [27]. Moreover, FLL of Rediviva varies not only between species but also between populations of the same species [21, 24, 28, 29], in which intraspecific variation in FLL has been shown to correlate with floral spur length of the main host plant Diascia [28, 29]. Since most Rediviva use a range of Diascia [21, 22] or other plant taxa (other Scrophulariaceae, Orchidaeceae, Iridaceae, Stilbaceae) as sources of oil [24, 26, 30,31,32], FLL might evolve in response to the spur length of the local community rather than to an individual host plant species, ([33], Hollens-Kuhr et al., unpublished observations), i.e. diffuse coevolution. A close match between Rediviva FLL and host plant spur lengths is, however, still necessary for successful oil extraction as the main host, Diascia, only produces oil in the distal end of the spurs (but see [34]) and thus only bees with sufficiently long forelegs are able to gather oil [24, 34]. Hence, FLL might experience strong selection to match the main host plant’s spur length.
Other factors may impact the genetic structure of Rediviva spp. populations beyond adaptation to host flower spur length. The majority of Rediviva species occur in the winter-rainfall area of South Africa [21, 22, 35], termed the Succulent Karoo biodiversity hotspot [36], which is characterised by ≥50% of the annual precipitation falling during winter [37]. Predominantly cold, rainy, and cloudy conditions during the main flowering season force winter-active bees, such as most Rediviva spp., to concentrate their foraging and brood cell provisioning activities to the short interludes of favourable weather. Hence, bees in this area likely have reduced daily activity and limited dispersal [36], which might reduce gene flow and increase genetic differentiation among populations. Furthermore, as Rediviva bees are thought to have special nesting requirements [38], regions of unsuitable habitat might further isolate Rediviva populations and reduce gene flow, as hypothesised for other ground-nesting bees in this area [39]. For example, Rediviva intermixta prefers loamy dolerite soil [25] whereas Rediviva peringueyi is unable to nest in unconsolidated, sandy soil [38]. In addition to the potential reduction in gene flow, some Rediviva bee species are probably characterised by a relatively low Ne since they occur in small and scattered populations (Kuhlmann, Hollens-Kuhr, unpublished observations).
Here, we used a restriction site-associated DNA sequencing (RAD-seq) approach to investigate the population genetic structure and demography of Rediviva longimanus. Specifically, in a population-based pooled RAD-seq dataset, we tested whether phenotypic differentiation in FLL translates into increased genetic differentiation between populations (isolation by adaptation: IBA) over the purely neutral evolutionary process of genetic drift (isolation by distance: IBD). Rediviva longimanus is among the Rediviva species (FLL = 6–23 mm) with the most extreme FLL (x̄=21 mm) and populations show noticeable differences in FLL, even over a small geographic scale [21], rendering it a particularly suitable study system with which to test for reproductive isolation related to local adaptation. We also measured differentiation between long-legged and short-legged bees within and between populations in a second pooled RAD-seq dataset, representing another test of incipient ecological speciation. We finally used an empirical FST-outlier approach as well as a PCA-based outlier detection test to identify loci underpinning local adaptation.
Methods
Study species and sampling sites
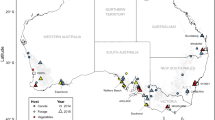
Rediviva longimanus is endemic to the Succulent Karoo in Western South Africa. Its distribution encompasses the Cederberg Mountains in the west, the Roggeveld Mountains in the east and the Nieuwoudtville area in the north [21]. Sampling of female bees was conducted at seven sites located near the towns of Nieuwoudtville, Calvinia and Clanwilliam (Fig. 1, Table 1), where R. longimanus, though rare, is abundant enough to be sampled and where we expected to find differences in FLL even across a small geographic scale (Hollens-Kuhr et al., unpublished observations).

Sampling locations of R. longimanus populations in South Africa. Population labels with an asterisk correspond to the four population pools (AP, LC, LI, LF) used for population genetic analysis in this study. For the two leg pools, we included individuals from LC, LI and LF as well as from three other populations (LA, LB, LG) to obtain two pools comprising either individuals with the longest or the shortest foreleg length (FLL), irrespective of population of origin. Sample sizes and mean relative FLL are given in brackets
DNA extraction and RAD-seq
DNA was extracted from the thorax, legs or head of females using a DTAB protocol (modified from [40]), which consists of a digestion step with proteinase K in DTAB buffer, followed by extraction with chloroform:isoamyl alcohol 24:1. DNA quality and quantity were assessed using an Epoch spectrophotometer (BioTek, Winooski, USA), by agarose gel electrophoresis and with a Qubit 3.0 fluorometer (Thermo Fisher Scientific, Waltham, USA). Only non-degraded and intact DNA samples were further processed. We first DNA barcoded each individual bee by sequencing the mitochondrial cytochrome oxidase I ‘animal barcode’ region [41] and were able to confirm species identity, i.e. Rediviva longimanus, for each sample included.
We then pooled individual DNA extracts according to two pooling schemes for restriction site-associated DNA sequencing (RAD-seq). In order to infer the population genetic structure and demography of R. longimanus populations, we pooled individuals into four population pools corresponding to four of seven sampling locations (Fig. 1, Table 1): AP (N = 11), LC (N = 24), LF (N = 22), LI (N = 28). These sites encompassed the range of FLL in R. longimanus and differed significantly in mean relative FLL (except LI versus LF, see Additional files 1 and 2), calculated as in [27], i.e. foreleg length divided by head width. We decided to use relative rather than absolute FLL to account for variation in FLL that might be due to variation in overall body size. We note, however, that head width, a proxy for body size [42], varied little between individuals and results of our study (e.g. multiple matrix regression, see below) did not change qualitatively when using absolute rather than relative FLL (data not shown).
In the second pooling scheme, we used samples from six of seven sites to generate two pools according to FLL, to which we refer as leg pools (Table 1, Fig. 1, see Additional file 3). One pool consisted of the twenty R. longimanus individuals with the overall longest relative FLL and the other pool consisted of twenty individuals showing the shortest relative FLL. Mean relative FLL of the long leg pool (x̄ = 7.1 ± 0.16 SD) was significantly different (LM, two-tailed test, t = − 14.2, P < 0.01) from the short leg pool (x̄=6.4 ± 0.13 SD).
In the leg pools, we pooled bees showing the most extreme foreleg lengths, i.e. shortest and longest, independent of their population of origin, because we were interested in testing for genetic differentiation with respect to FLL across populations, which might be indicative of the initial stage of ecological speciation. Pooling individuals from different populations but with the same morphology, as in our two leg pools, has been recognised as a valid and highly useful approach, especially for the identification of candidate genes for host adaptation [4, 5]. Although each of our leg pools comprised individuals from five of seven populations (Additional file 2), we lacked sufficient long or short legged bees from some populations to allow a balanced sampling design. A caveat of our approach, then, is that a component of the genetic differentiation we detected among the leg pools dataset might be due to population differentiation.
For each pool, 1.5 μg of genomic DNA, normalised to a final DNA concentration of 60 ng/μl, was sent to Floragenex, Inc. (Eugene, USA) for RAD-seq. RAD-seq is an increasingly used [43,44,45,46] Next Generation Sequencing approach that yields a reduced representation of the whole genome. By using restriction enzymes that cut DNA at restriction enzyme-specific sites, which occur randomly over the genome, one obtains DNA fragments that are sheared to generate sizes appropriate for sequencing [47]. Subsequent sequencing of homologues fragments in several individuals or pools is able to reveal thousands of single nucleotide polymorphisms (SNPs), which can be analysed in a population genomic framework [47]. RAD-seq was carried out according to the original RAD-seq protocol [47, 48]. DNA was digested using the restriction enzyme PstI, randomly sheared and adapters with unique multiplex identifier (MID) ‘barcodes’ (10 bp) for each pool sample were attached to the DNA fragments prior to sequencing. Pooled libraries were run on an Illumina HiSeq2500 platform (Illumina, San Diego, USA) to generate 125 bp single-end reads.
Data processing and SNP calling
Following a quality control in FASTQC v. 0.11.5 [49], sequence reads were demultiplexed, filtered for quality and trimmed of 10 bp MID sequences using STACKS v. 1.42 [50] under default settings. Since there is no reference genome available for R. longimanus or a close relative, we identified RAD loci de novo using denovo_map.pl in STACKS (m = 5, M = 2, n = 0). Moreover, we remove highly repetitive stacks, loci with a log-likelihood below − 20 and confounding loci, i.e. multiple genomic loci matching a single catalogue locus.
Since STACKS was not specifically designed for the use of data from pooled samples and its SNP calling algorithm is therefore likely to miss low frequency variants in the pool, we used POPOOLATION2 [51] for SNP calling. We mapped all our RAD reads against the reference catalogue created in STACKS using the bwa mem algorithm of BWA v. 0.7.12 [52]. Mapping results were filtered for a minimum Phred quality score of 20 and converted into mpileup format in SAMTOOLS v. 0.1.19 [53]. For each population pair, we then calculated the allele frequency difference at each position with a minimum coverage of 10 and a minimum minor allele count of 2 using the snp-frequency-diff.pl script of POPOOLATION2 (also see Additional file 4).
Genome-wide variation and population genetic structure
Genome-wide patterns of genetic diversity were assessed by calculating the population mutation rate (Watterson’s θ) and nucleotide diversity (Tajima’s Π) in NPSTAT v.1.0 [54]. The accuracy of allele frequency estimates in pooled samples can be increased not only by high sequence coverage but also by using large sliding windows as this avoids incorrect estimates due to stochastic error [55]. To do so, we concatenated all RAD tags and calculated genetic diversity measures over this continuous sequence stretch (one window) for positions with a minimum coverage of 10, minimum count of the minor allele of 2 and a minimum Phred score of 20.
Pairwise and overall genetic differentiation were estimated as the fixation index FST [1] in POPOOLATION2, only considering positions with a minimum coverage of ten and a minimum minor allele count of two per RAD locus i.e. using windows of 115 bp (125 bp reads minus 10 bp MID). However, we also checked the robustness of our estimates by repeating the calculations under even more conservative settings (minimum coverage = 20, minimum minor allele count = 6); results did not qualitatively change. In order to exclude repetitive regions, we set a maximum coverage threshold to exclude those loci with the 2% highest coverage (> 73x for AP, > 58x for LC, > 60x for LF, > 51x for LI) from genetic diversity and FST calculations. All other parameters were left as default. In addition, we recalculated FST after removing loci potentially under selection, i.e. loci identified in either PCADAPT or the tails of the FST distribution (see below), to account for a potential bias in our FST estimates due to selection. Confidence intervals (CI’s) for the FST estimates were inferred by bootstrapping 1000 times in the R package BOOTSTRAP v. 2017.2 [56]. In addition to FST, we also assessed population genetic structure by principal component analysis (PCA) in the R package PCADAPT v. 3.0.4 [57].
We then investigated if population genetic differentiation in the population pools could be explained by differences in FLL (isolation by adaptation, IBA) or geographic distance (isolation by distance, IBD). We regressed the matrices of pairwise population genetic differentiation, transformed to FST/(1-FST), on relative FLL and on log-transformed geographic distances using a multiple matrix regression with randomisation (MMRR) analysis [58] with 999 permutations in the R package ECODIST v. 1.2.2 [59] to avoid pseudoreplication because of the non-independence of FST values within a dataset. Geographic distances between population pairs were inferred via the shortest path in GOOGLE EARTH v. 6.2 (Table 3).
Demographic history of Rediviva longimanus
Since estimates for Watterson’s θ and Tajima’s Π suggested very low genomic diversity for all population pools (see Results below), we tested for a bottleneck in each population using FASTSIMCOAL2 v. 2.5.2.21 [60]. We first excluded RAD tags with SNPs potentially under selection (see below) using a custom bash script, and then produced the folded (i.e. based on the allele frequencies of only the minor allele) site frequency spectrum (SFS) in POOL-HMM v. 1.4.3 [61].
In FASTSIMCOAL2 we first estimated model parameters using sequential Markov coalescence simulations and a conditional maximization algorithm (ECM, [60]). In addition to a bottleneck scenario, we also modelled a constant population size scenario and a population expansion scenario. Model comparisons were performed according to the Akaike Information Criterion (AIC) and Akaike’s weight of evidence (w), as suggested by Excoffier et al. ([59], see Additional file 4 for more details).
Genetic differentiation by FLL using leg pools
We also computed FST between the leg pools dataset to measure the effect of FLL on genetic differentiation and to infer potential reproductive isolation due to FLL per se. FST computations in POPOOLATION2 were carried out using the same settings as for the population pools (minimum coverage = 10, minimum minor allele count = 2, loci with the 2% highest coverage excluded). FST calculations were repeated after excluding potential outlier loci, i.e. in the 5% tails of the FST distribution (see below). Bootstrapping was performed with 1000 replicates to generate CI’s for the FST estimate.
Outlier SNP detection
We tested for signals of selection using two outlier detection approaches with both population pools and leg pools datasets. In the first approach we extracted loci in the lower and upper tails (0.5% for the population pools and 5% for the leg pools) of the FST distribution, as calculated in POPOOLATION2 (also see Additional file 4). The FST outlier criterion of 5% for the leg pools differed from that for the population pools because it already incorporated the maximum value of FST = 1. We considered the loci with the highest FST values (upper tail) as candidates for divergent selection and the loci with the lowest FST values (lower tail) as candidates for balancing selection, in accordance with the rationale underlying FST-outlier detection tools such as BAYESCAN [62] or LOSITAN [63].
In the second approach to infer signals of selection, we used PCADAPT v. 3.0.4 [57], which employs a PCA to assess population genetic structure prior to outlier identification and is particularly suited to Pool-seq data [57]. PCADAPT was run with 5 replicates for our best K (K = 1 for the leg pools and K = 3 for the population pools) and only SNPs identified across all runs were considered to be candidates under selection.
Further information about the methods used can be found in Additional file 4.
Results
RAD-seq and mapping
Illumina sequencing yielded 9,250,492 reads for the four population pools (average 2,312,623 reads per pool) and 4,235,496 reads for the two leg pools. After filtering, we retained 8,232,334 reads for the population pools (average 2,058,084 reads per pool) and 3,602,671 reads for the two leg pools (see Additional file 5). De novo assembly in STACKS produced 76,168 RAD tags/loci, which we used as reference for mapping. Overall, we could map 6,345,433 reads (68.5%, x̄ = 1,586,358 reads per pool) for the population pools and 2,746,271 reads (64.8%, x̄ = 1,373,136 reads per pool) for the leg pools to our reference (see Additional file 5).
Genome-wide variation and population genetic structure
In total we identified 29,721 SNPs that satisfied our filtering criteria in POPOOLATION2. The number of segregating sites (variable base positions in the genome) per population varied from 7362 to 9912 (x̄ = 8562, Table 2). Genetic diversity estimates, Watterson’s θ and Tajima’s Π, were extremely low for all populations, at θ = 0.0007 and Π = 0.0008 (Table 2). More stringent SNP filtering criteria only slightly increased Watterson’s θ and Tajima’s Π (see Additional file 6).
Average FST values between population pairs were consistent and relatively high, ranging from 0.157 to 0.176 (mean FST across all populations = 0.165, lower 95% CI: 0.164, upper 95% CI: 0.167, Table 3), and even increased under more stringent filtering criteria: 0.219–0.241 (mean FST across all populations = 0.231, see Additional file 7). Excluding outlier loci did not markedly change FST estimates (mean FST across all populations = 0.164, lower 95% CI: 0.163, upper 95% CI: 0.166; see Additional file 7). Furthermore, PCA also supported the population genetic structure inferred by FST and clustered individuals according to their population of origin (Fig. 2).

Population genetic structure of Rediviva longimanus according to principal component analysis (PCA). (a) PCA, performed in PCADAPT, suggested that three principal components explained most of the population genetic structure and revealed a clear separation into four population clusters. (b) Analysing more than three components did not contribute to disentangle further population genetic structure and revealed additional variance within rather than between clusters
We then tested if population genetic differentiation was correlated with population-level differences in mean FLL or rather with geographic distance. The relationship between genetic differentiation (as FST/(1-FST)) and log10 geographic distance was not significant (r2 = 0.21, P > 0.05, Fig. 3a). Genetic differentiation and differences in relative FLL were also not significantly correlated (r2 = 0.34, P > 0.05, Fig. 3b), although there was a positive trend in the relationship. Multiple matrix regression analysis including both geographic distances (log10) and relative FLL was also not significant (r2 = 0.36, P > 0.05).

Regression of genetic differentiation of Rediviva longimanus population pools upon (a) foreleg length (FLL) and (b) geographic distance (log10). Although we failed to detect a significant pattern of isolation by adaptation (IBA) or isolation by distance (IBD) for the R. longimanus populations, there was a positive trend in both
Because non-significant results may arise through lack of statistical power, we estimated the power of our current analyses and the sample size needed to reject the null hypothesis of no IBD and IBA using the R package PWR v. 1.2.2 [64]. This analysis showed that, given our sample size, we needed an effect size of r = 0.90 to detect IBD and IBA. Our observed effects sizes (r) were clearly less than 0.90. Indeed, the statistical power of our analyses, given the observed effect sizes, was found to be low: 15% for IBD and 24% for IBA. Power analysis suggested that 10 or 8 populations would be needed to achieve power (1-β) of 0.80 to detect IBD or IBA, respectively.
Demographic history of Rediviva longimanus
Demographic inference using FASTSIMCOAL2 suggested a bottleneck scenario to best fit our population pools (all four populations) since this scenario’s AIC value was smaller than those for the two alternative demographic models: constant Ne and population expansion (Table 4).
Genetic differentiation by FLL using leg pools
Genetic differentiation between the leg pools (FST = 0.296, lower 95% CI: 0.292, upper 95% CI: 0.299) was nearly twice as high as genetic differentiation among the population pools and dropped only slightly after excluding outlier loci (FST = 0.290, lower 95% CI: 0.291, upper 95% CI: 0.299). Hence, FLL might indeed play a non-negligible role in limiting gene flow (mating) between relatively long and relatively short-legged bees within and between populations.
Outlier SNPs in the population pools and leg pools
We identified 172 RAD-tags in the tails of the FST distribution for the population pools (0.5% threshold) and 652 in the FST tails for the leg pools (5% threshold, see Table 5). PCADAPT analyses detected 326 candidate SNPs in 309 RAD-tags shared across all five runs for the population pools, though only two of these also appeared in the tails of the FST distribution of the same dataset (Fig. 4a, Table 5). For the leg pools, PCADAPT did not identify any statistically significant outlier loci. Moreover, there was little overlap between the empirical FST outliers identified in the leg pools and the population pools (12 outliers overall, Fig. 4b). More detailed information on the outlier analyses can be found in Additional files 8 and 9.

Overall we identified 1119 unique outlier RAD-tag loci. Shown is the overlap between (a) the different methods for the population pools and between (b) the population pools and leg pools datasets. Diagrams were created with http://bioinformatics.psb.ugent.be/webtools/Venn
Discussion
Our RAD-seq analyses of four R. longimanus population pools suggested marked genetic structuring of populations. Genetic differentiation between long-legged and short-legged bees pooled across populations was even more marked than population genetic differentiation, hinting at reduced gene flow based on leg length. All populations exhibited low genetic diversity estimates and seemed to have experienced bottleneck events, which is in accordance with field observations that R. longimanus populations are small and scattered.
Genetic diversity, genetic structure and demographic history of Rediviva longimanus populations
Pronounced population genetic structuring and differentiation were detected between all population pairs of R. longimanus. Even over the relatively short geographic distance of 34 km, populations seemed to be highly differentiated (FST for LI versus LF: 0.163, lower 95% CI: 0.160, upper 95% CI: 0.166). For comparison, genetic differentiation of other ground-nesting wild bee species is often only significant over greater (> 50 km) geographic distances [65, 66].
Marked genetic differentiation over a small geographic distance might be a general feature of Rediviva bees and potentially of other flying insects in the South African winter rainfall area due to the region’s harsh climatic conditions [39]; the peak flowering season (August–September) is often cold and wet [67], which forces bees to forage under often unfavourable weather conditions. This might reduce bees’ foraging ranges and gene flow, resulting in increased genetic differentiation between populations [36, 39, 68]. In addition, Rediviva bees built their nests in the ground [25] and might have special nesting requirements. This might make the landscape appear fragmented to Rediviva bees and further reduce gene flow among their populations.
Low Ne could also account for the high population differentiation detected. A possibility is that bees are blown around by the harsh conditions of the Succulent Karoo, leading to high natal dispersal (i.e. near-panmixia), which, when coupled with very low Ne, could generate a pattern of consistently high population genetic differentiation independent of geography. Though plausible, we consider this scenario unlikely as it cannot easily account for the consistent relationship between foreleg length and local Diascia floral spur length observed in R. longimanus (Hollens-Kuhr et al., unpublished observations) and other Rediviva spp. [28, 29].
Low Ne is, however, in agreement with personal observations of the rarity of the species (Kuhlmann, Hollens-Kuhr), and could also explain our low genetic diversity estimates for R. longimanus. Genetic diversity, measured as Watterson’s θ and Tajima’s Π, were at least one order of magnitude lower than estimates in other Pool-seq studies of non-threatened species [69, 70], though comparable to genetic diversity estimates for small island populations [71]. Our demographic analyses of R. longimanus, suggesting genetic bottlenecks, would also fit the low genetic diversity estimates inferred. During dry years, flower production can be very poor in the Succulent Karoo biome and most Rediviva species are likely to experience a marked reduction in population size, with some populations collapsing completely (Pauw, Kuhlmann, unpublished observations). Populations might thus frequently go through genetic bottlenecks, which would result in reduced genetic diversity, and which is probably only slowly restored once conditions become more favourable. The small, highly structured and potentially genetically depauperate R. longimanus populations are of conservation concern.
Pooling of individuals for genetic analysis might also have inflated our estimates of genetic differentiation, as suggested by Anderson et al. [72], who found that pools comprising few individuals might result in an artificial surplus of fixed loci. However, Anderson et al.’s conclusions were based on a very extreme example involving only six individuals per pool while our lowest pooled size (for population AP) contained 11 individuals. Thus, although Pool-seq might lead to biased allele frequency estimates and erroreous population genetic inferences when pool sizes and sequencing coverage are insufficient [72, 73], ours were likely adequate to ensure robust inference. First, our pool sizes were usually of the appropriate order of magnitude for accurate allele frequency estimates (≥ 25 individuals with a coverage of ≥20-30x), as suggested by Ferretti et al. [54]. Second, we followed highly stringent criteria in building our reference RAD-tags and in SNP calling (quality score ≥ 20, minimum coverage ≥ 10, minor allele frequency ≥ 2) to ensure accurate allele frequency estimation and reduce sequencing errors.
Population genetic structure of R. longimanus, genetic drift and selection
We did not detect an association between population genetic differentiation in R. longimanus and geographic distance or variation in FLL, probably because we analysed only a relative small number of populations and thus lacked statistical power. Rediviva longimanus populations are small, thus the issue of statistical power is difficult to resolve. Reciprocal transplant experiment would help to assess if differences in FLL are locally adaptive [74]. But they are also difficult to implement on endemic and rare species that are of conservation concern, such as R. longimanus.
However, it is likely that genetic drift plays an important role in determining R. longimanus population genetic structure. First, populations are small in size (Kuhlmann, Hollens-Kuhr, unpublished observations), suggesting they are highly vulnerable to the effects of genetic drift. Moreover, we inferred population bottlenecks as the most likely demographic scenario for all populations studied. We also found low genetic diversity for all populations. Furthermore, populations seem to be significantly structured (average FST = 0.165), suggesting limited genetic exchange between populations, which would exacerbate the effects of drift.
Yet FLL might also have a non-negligible effect on genetic differentiation and result in reduced gene flow, as suggested by the high FST estimate between our leg pools. Though our leg pools dataset comprised individuals from different populations as well as with different leg lengths, the effect of population of origin per se on the FST estimate was probably slight. This is because we found all populations to exhibit a consistent, marked FST independent of geography and because we incorporated individuals from multiple – and often the same – populations into both the long and the short leg pools.
How variation in FLL per se translates into increased genetic differentiation, potentially because of reduced gene flow and limited mating between long and short legged morphs, is unknown. Sexual selection acting on FLL seems unlikely since variation in FLL is only displayed by females and, in bees, males are usually not the choosy sex but rather undertake scramble competition for mates [75]. Partial reproductive isolation is more likely due to habitat preference. It has already been shown that long legged Rediviva bees preferentially use long spurred flowers and vice versa [28, 34]. Long-legged bees might prefer localities with mainly long spurred plants while short-legged bees might preferentially occur at localities where short-spurred plants dominate, since bees will be more successful in extraction oil and hence provisioning offspring when they occur in localities with host plants possessing spur lengths that fit their FLL. Partial reproductive isolation due to habitat preference may then arise if mating takes place in the appropriate localities of daughters and their sons [7]. However, it is unknown where mating occurs in R. longimanus. Nevertheless, examples where local adaptation to a host plant increases genetic differentiation and may finally lead to reproductive isolation have been documented for several other insects, in particular phytophagous insects, e.g. the walking stick insect Timema cristinae [4, 9, 76], the leaf beetle Neochlamisus bebbianae [5] or the apple maggot fly Rhagoletis pomonella, [19, 20]; see also [77] for a more complete list.
Candidate genes under selection
Overall, we detected 1119 outlier loci, though there was little consistency in outlier loci identified by the different methods and datasets. Our pooled RAD-seq approach analysed only a small proportion of the R. longimanus genome and thus likely missed important genes underpinning local adaptation. Whole genome sequencing rather than our RAD-seq approach would be more powerful to address the genetics of local adaptation and to identify candidate loci underlying FLL, which we assume to be locally adaptive and experience strong selection.
We note, though, that selection may not always favour bees with the longest legs but rather may favour those with the best fitting legs. Simulations suggest that bees with legs much longer than the floral spurs of their hosts are unable to successfully collect oil [6]. Selection, even if strong, may then maintain multiple alleles, namely for both long and for short legs, in the same population.
Conclusions
We found pronounced genetic differentiation among R. longimanus populations and low genetic diversity, likely because of low Ne and limited dispersal, compounded by recent bottleneck events. Genetic drift seemed to be important in structuring R. longimanus populations, but FLL might also reduce gene flow, as indicated by high genetic differentiation between our leg pools. Future studies including additional populations are required to test if neutral evolutionary processes such as genetic drift and migration or host plant adaptation are more important in structuring R. longimanus populations and whether FLL is associated with reduced gene flow and reproductive isolation. Nevertheless, our study is a first step to understand better the population genomics of an important pollinator in the Succulent Karoo biodiversity hotspot.
References
Hartl DL, Clark AG. Principles of population Genetics. 4th ed. Sunderland, Massachusetts: SinauerAssociates; 2007.
Palstra FPP, Ruzzante DEE. Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol. 2008;17:3428–47.
Egan SP, Ragland GJ, Assour L, Powell THQ, Hood GR, Emrich S, et al. Experimental evidence of genome-wide impact of ecological selection during early stages of speciation-with-gene-flow. Ecol Lett. 2015;18:817–25.
Nosil P, Egan SP, Funk DJ, Article O. Heterogeneous genomic differentiation between walking-stick ecotypes: “isolation by adaptation” and multiple roles for divergent selection. Evolution (N Y). 2007;62:316–36.
Egan SP, Nosil P, Funk DJ. Selection and genomic differentiation during ecological speciation: isolating the contributions of host association via a comparative genome scan of Neochlamisus bebbianae leaf beetles. Evolution (N Y). 2008;62:1162–81.
Chapman MA, Hiscock SJ, Filatov DA. The genomic bases of morphological divergence and reproductive isolation driven by ecological speciation in Senecio (Asteraceae). J Evol Biol. 2015;29:98–113.
Nosil P. Ecological speciation. 1st ed. USA: Oxford University Press; 2012.
Simon J-C, D’Alençon E, Guy E, Jacquin-Joly E, Jaquiéry J, Nouhaud P, et al. Genomics of adaptation to host-plants in herbivorous insects. Brief Funct Genomics. 2015;14:413–23.
Nosil P. Divergent host plant adaptation and reproductive isolation between ecotypes of Timema cristinae walking sticks. Am Nat. 2007;169:151–62.
Heidel-Fischer HM, Vogel H. Molecular mechanisms of insect adaptation to plant secondary compounds. Curr Opin Insect Sci. 2015:8:8–14.
Thorp RW. The collection of pollen by bees. In: Dafn A, Hesse M, Pacini E, editors. Pollen Pollinat. Vienna, Austria, Austria: Springer; 2000.
Jaquiéry J, Stoeckel S, Nouhaud P, Mieuzet L, Mahéo F, Legeai F, et al. Genome scans reveal candidate regions involved in the adaptation to host plant in the pea aphid complex. Mol Ecol. 2012;21:5251–64.
Hoang K, Matzkin LM, Bono JM. Transcriptional variation associated with cactus host plant adaptation in Drosophila mettleri populations. Mol Ecol. 2015;24:5186–99.
Rundle HD, Nosil P. Ecological speciation. Ecol Lett. 2005;8:336–52.
Cockerham CC. Analyses of gene frequencies. Genetics. 1973;74:679–700.
Nosil P, Funk DJ, Oritz-Barrientos D. Divergent selection and heterogeneous genomic divergence. Mol Ecol. 2009;18:375–402.
Wu C. The genic view of the process of speciation. J Evol Biol. 2001;14:851–65.
Feder JL, Egan SP, Nosil P. The genomics of speciation-with-gene-flow. Trends Genet Elsevier Ltd. 2012;28:342–50.
Michel AP, Sim S, Powell THQ, Taylor MS, Nosil P, Feder JL. Widespread genomic divergence during sympatric speciation. 2010;21:9724–9.
Filchak KE, Roethele JB, Feder JL. Natural selection and sympatric divergence in the apple maggot Rhagoletis pomonella. Nature. 2000;407:739–42.
Whitehead VBB, Whitehead VBB, Steiner KEEKE. Oil-collecting bees of the winter rainfall area of South Africa (Melittidae, Rediviva). Ann South African Museum. 2001;108:143–277.
Whitehead VB, Steiner KE, Eardley CD. Oil collecting bees mostly of the summer rainfall area of southern Africa (Hymenoptera: Melittidae: Rediviva). J Kansas Entomol Soc. 2008;81:122–41.
Vogel S. The Diascia flower and its bee - an oil-based symbiosis in southern Africa. Acta Bot Neerl. 1984;33:509–18.
Steiner KE, Whitehead VB. The association between oil-producing flowers and oil-collecting bees in the Drakensberg of southern Africa. Monogr Syst Bot Missouri Bot Gard. 1988;25:259–77.
Kuhlmann M. Nest architecture and use of floral oil in the oil-collecting south African solitary bee Rediviva intermixta (Cockerell) (Hymenoptera: Apoidea: Melittidae). J Nat Hist. 2014;48:2633–44.
Pauw A. Floral syndromes accurately predict pollination by a specialized oil-collecting bee (Rediviva peringueyi, Melittidae) in a guild of south African orchids (Coryciinae). Am J Bot. 2006;93:917–26.
Kahnt B, Montgomery GA, Murray E, Kuhlmann M, Pauw A, Michez D, et al. Playing with extremes: origins and evolution of exaggerated female forelegs in south African Rediviva bees. Mol Phylogenet Evol Elsevier. 2017;115:95–105.
Steiner KE, Whitehead VB. Oil flowers and oil bees: further evidence for pollinator adaptation. Evolution (N Y). 1991;45:1493–501.
Steiner KE, Whitehead VB. Pollinator adaptation to oil-secreting flowers-Rediviva and Diascia. Evolution (N Y). 1990;44:1701–7.
Steiner KE, Whitehead VB. Oil secretion and the pollination of Colpias mollis (Scrophulariaceae). Plant Syst Evol. 2002;235:53–66.
Kuhlmann M, Hollens H. Morphology of oil-collecting pilosity of female Rediviva bees (Hymenoptera: Apoidea: Melittidae) reflects host plant use. J Nat Hist. 2015;49:561–73.
Waterman RJ, Bidartondo MI, Stofberg J, Combs JK, Gebauer G, Savolainen V, et al. The effects of above- and belowground mutualisms on orchid speciation and coexistence. Am Nat. 2011;177:E54–68.
Pauw A, Kahnt B, Kuhlmann M, Michez D, Montgomery GA, Murray E, et al. Long-legged bees make adaptive leaps: linking adaptation to coevolution in a plant – pollinator network. Proc R Soc B. 2017;284.
Hollens H, van der Niet T, Cozien R, Kuhlmann M. A spur-ious inference : pollination is not more specialized in long- spurred than in spurless species in Diascia - Rediviva mutualisms. Flora Morphol Distrib Funct Ecol Plants. 2016;232:73–82.
Whitehead VBB, Steiner KEE. Two new species of oil-collecting bees of the genus Rediviva from the summer rainfall region of South Africa (Hymenoptera, Apoidea, Melittidae). Ann South African Museum. 1992;102:143–64 Available from: http://biostor.org/reference/110298.
Linder HP, Johnson SD, Kuhlmann M, Matthee CA, Nyffeler R, Swartz ER. Biotic diversity in the southern African winter-rainfall region. Curr Opin Environ Sustain. 2010;2:109–16.
Procheş Ş, Cowling RM, du Preez DR. Patterns of geophyte diversity and storage organ size in the winter-rainfall region of southern Africa. Divers Distrib. 2005;11:101–9.
Pauw A. Collapse of a pollination web in small conservation areas. Ecology. 2007;88:1759–69.
Kahnt B, Soro A, Kuhlmann M, Gerth M, Paxton RJ. Insights into the biodiversity of the succulent Karoo hotspot of South Africa: the population genetics of a rare and endemic halictid bee, Patellapis doleritica. Conserv Genet. 2014;15:1491–502.
Gustincich S, Manfioletti G, Del Sal G, Schneider C, Carninci P. A fast method for high-quality genomic DNA extraction from whole human blood. BioTechniques. 1991;11:298–300.
Hebert PDN, Cywinska A, Ball SL, DeWaard JR. Biological identifications through DNA barcodes. Proc R Soc B Biol Sci. 2003;270:313–21.
Cane JH. Estimation of bee size using Intertegular span (Apoidea). J Kansas Entomol Soc. 1987;60:145–7.
Buckley J, Holub EB, Koch MA, Vergeer P, Mable BK. Restriction associated DNA-genotyping at multiple spatial scales in Arabidopsis lyrata reveals signatures of pathogen-mediated selection. BMC Genomics. 2018;19:496.
Wang H, Zhao S, Mao K, Dong Q, Liang B, Li C, et al. Mapping QTLs for water-use efficiency reveals the potential candidate genes involved in regulating the trait in apple under drought stress. BMC Plant Biol. 2018;18:136.
Lecaudey LA, Schliewen UK, Osinov AG, Taylor EB, Bernatchez L, Weiss SJ. Inferring phylogenetic structure, hybridization and divergence times within Salmoninae (Teleostei: Salmonidae) using RAD-sequencing. Mol Phylogenet Evol. 2018;124:82–99.
Theodorou P, Radzevičiūtė R, Kahnt B, Soro A, Grosse I, Paxton RJ. Genome-wide single nucleotide polymorphism scan suggests adaptation to urbanization in an important pollinator, the red-tailed bumblebee (Bombus lapidarius L.). Proc R Soc B Biol Sci. 2018;285:20172806.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376.
Miller MR, Dunham JP, Amores A, Cresko W a, Johnson E a. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17:240–8.
Andrews S. FastQC: a quality control tool for high throughput sequence data. [Internet]. 2010. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–40.
Kofler R, Pandey RV, Schlötterer C. PoPoolation2 : identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics. 2011;27:3435–6.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Ferretti L, Ramos-Onsins SE, Pérez-Enciso M. Population genomics from pool sequencing. Mol Ecol. 2013;22:5561–76.
Kofler R, Orozco-terWengel P, de Maio N, Pandey RV, Nolte V, Futschik A, et al. Popoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One. 2011;6:e15925.
Efron B, Tibshirani R. An introduction to the Bootstrap. New York. London: Chapman and Hall; 1993.
Luu K, Bazin E. Blum MGB. Pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour. 2017;17:67–77.
Wang IJ. Examining the full effects of landscape heterogeneity on spatial genetic variation: a multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution (N Y). 2013;67:3403–11.
Goslee SC, Urban DL. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw. 2007;22:1–19.
Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013;9:e1003905.
Boitard S, Kofler R, Françoise P, Robelin D, Schlötterer C, Futschik A. Pool-hmm: a Python program for estimating the allele frequency spectrum and detecting selective sweeps from next generation sequencing of pooled samples. Mol Ecol Resour. 2013;13:337–40.
Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–93.
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart GLOSITAN. A workbench to detect molecular adaptation based on a F st -outlier method. BMC Bioinformatics. 2008;9:1–5.
Stephane C. pwr: Basic Functions for Power Analysis. 2018.
Soro A, Field J, Bridge C, Cardinal SC, Paxton RJ. Genetic differentiation across the social transition in a socially polymorphic sweat bee, Halictus rubicundus. Mol Ecol. 2010;19:3351–63.
Davis ES, Murray TE, Fitzpatrick ÚNA, Brown MJF, Paxton RJ. Landscape effects on extremely fragmented populations of a rare solitary bee, Colletes floralis. Mol Ecol. 2010;19:4922–35.
Mucina L, Jürgens N, Le Roux A, Rutherford MC, Schmiedel U, Esler KJ, Powrie LW, et al. Succulent Karoo biome. In: Mucina L, Rutherford MC, editors. Veg South Africa, Lesotho Swasil. Pretoria: SANBI; 2006. p. 220–99.
Kuhlmann M. Patterns of diversity, endemism and distribution of bees (Insecta: Hymenoptera: Anthophila) in southern Africa. South African J Bot. 2009;75:726–38.
Guo B, Lu D, Liao WB, Merilä J. Genome-wide scan for adaptive differentiation along altitudinal gradient in the Andrew’s toad Bufo andrewsi. Mol Ecol. 2016;25:3884–900.
Guo B, DeFaveri J, Sotelo G, Nair A, Merilä J, Li Z, et al. Population genomic evidence for adaptive differentiation in the Baltic Sea herring. Mol Ecol. 2016;25:2833–52.
Funk WC, Lovich RE, Hohenlohe PA, Hofman CA, Morrison SA, Sillett TS, et al. Adaptive divergence despite strong genetic drift: genomic analysis of the evolutionary mechanisms causing genetic differentiation in the island fox (Urocyon littoralis). Mol Ecol. 2016:1–19.
Anderson E, Skaug HJ, Barshis DJ. Next-generation sequencing for molecular ecology: a cavaet regarding pooled samples. Mol Ecol. 2014;23:502–12.
Futschik A, Schlötterer C. The next generation of molecular markers from massively parallel sequencing of pooled DNA samples. Genetics. 2010;186:207–18.
Kawecki TJ, Ebert D. Conceptual issues in local adaptation. Ecol Lett. 2004;7:1225–41.
Bonduriansky R. The evolution of male mate choice in insects: a synthesis of ideas and evidence. Biol Rev. 2001;76:305–39.
Riesch R, Muschick M, Lindtke D, Villoutreix R, Comeault AA, Farkas TE, et al. Transitions between phases of genomic differentiation during stick-insect speciation. Nat Ecol Evol. 2017;1:1–13.
Matsubayashi KW, Ohshima I, Nosil P. Ecological speciation in phytophagous insects. Entomol Exp Appl. 2009;134:1–27.
Acknowledgements
We are very grateful to Baocheng Guo, Sarah Flanagan, Chrystelle Delord, Renaud Vitalis, Simon Boitard, Anne Royer, Luca Ferretti and Keurcien Luu for technical advice during data analyses. We also thank Martin Husemann, Robin Moritz, Michael Lattorff and Patricia Landaverde for intellectual input and especially the anonymous referees for very helpful and constructive comments on a former version of the manuscript. Furthermore, we are indebted to CapeNature and the Department of Environment and Nature Conservation for permission to collect bees in Western Cape Province and Northern Cape Province respectively.
Funding
We acknowledge the Studienstiftung des deutschen Volkes for funding BK and the German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig for funding sequencing services. Moreover, we acknowledge the financial support of the Open Access Publication Fund of the Martin-Luther-University Halle-Wittenberg.
Availability of data and materials
The dataset (RAD-seq raw data) supporting the conclusions of this article is available in the NCBI Sequence Read Archive database via the Accession Number SRP127322 (http://www.ncbi.nlm.nih.gov/Traces/sra).
Author information
Authors and Affiliations
Contributions
BK designed the project, generated sequencing data, conducted data analyses and wrote the manuscript. PT assisted with data analyses and provided substantial intellectual input to the study. AS and RJP were involved in project design and data interpretation. HH, MK and AP sampled specimens and provided ecological data about the study organism and habitat. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Pairwise test for significant differences in relative FLL between Rediviva longimanus population pools. (XLSX 10 kb)
Additional file 2:
Picture showing the differences in FLL between population pool LC (Keiskie Mountains) and LF (Farm Papkuilsfontain). (PDF 180 kb)
Additional file 3:
Locations and number of samples per location for the two Rediviva longimanus leg pools. Mean FLL and standard deviations for the two leg pools are also indicated. (XLSX 10 kb)
Additional file 4:
Additional methods. (DOCX 110 kb)
Additional file 5:
Summary statistics for the number of reads sequenced, reads retained after filtering and reads successfully mapped to the consensus reference sequence for the Rediviva longimanus population pools and leg pools. (XLSX 10 kb)
Additional file 6:
Genetic diversity estimates for the Rediviva longimanus population pools including only SNPs with a minor allele count of 6, minimum coverage of 20 and maximum coverage ≤ 98%. (XLSX 11 kb)
Additional file 7:
Pairwise FST for the Rediviva longimanus population pools after excluding loci potentially under selection (above diagonal) and FST (below diagonal) under more stringent SNP filtering criteria (minimum count of the minor allele = 6, minimum coverage = 20, maximum coverage ≤ 98%). (XLSX 12 kb)
Additional file 8:
Additional results for the outlier analysis, in particular for outlier annotation and GO enrichment. (DOCX 325 kb)
Additional file 9:
BLAST and GO annotation of all 136 outliers identified for the Rediviva longimanus population and leg pool data. The data set and methods with which the outliers were identified are also given. (XLSX 61 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kahnt, B., Theodorou, P., Soro, A. et al. Small and genetically highly structured populations in a long-legged bee, Rediviva longimanus, as inferred by pooled RAD-seq. BMC Evol Biol 18, 196 (2018). https://doi.org/10.1186/s12862-018-1313-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-018-1313-z