
Abstract
Background
Populations that have repeatedly colonized novel environments are useful for studying the role of ecology in adaptive divergence – particularly if some individuals persist in the ancestral habitat. Such “contemporary ancestors” can be used to demonstrate the effects of selection by comparing phenotypic and genetic divergence between the derived population and their extant ancestors. However, evolution and demography in these “contemporary ancestors” can complicate inferences about the source (standing genetic variation, de novo mutation) and pace of adaptive divergence. Marine threespine stickleback (Gasterosteus aculeatus) have colonized freshwater environments along the Pacific coast of North America, but have also persisted in the marine environment. To what extent are marine stickleback good proxies of the ancestral condition?
Results
We sequenced > 5800 variant loci in over 250 marine stickleback from eight locations extending from Alaska to California, and phenotyped them for platedness and body shape. Pairwise FST varied from 0.02 to 0.18. Stickleback were divided into five genetic clusters, with a single cluster comprising stickleback from Washington to Alaska. Plate number, Eda, body shape, and candidate loci showed evidence of being under selection in the marine environment. Comparisons to a freshwater population demonstrated that candidate loci for freshwater adaptation varied depending on the choice of marine populations.
Conclusions
Marine stickleback are structured into phenotypically and genetically distinct populations that have been evolving as freshwater stickleback evolved. This variation complicates their usefulness as proxies of the ancestors of freshwater populations. Lessons from stickleback may be applied to other “contemporary ancestor”-derived population studies.
Similar content being viewed by others


Background
The ecological theory of adaptive divergence predicts that populations diverge phenotypically and genetically if they reside in distinct environments [1, 2], potentially resulting in speciation. Some of the most striking examples of adaptive divergence come from species in which contemporary populations persist in environments putatively occupied by ancestral populations (e.g. [3,4,5,6,7,8,9,10,11]). To the extent that such populations have not undergone evolution, contemporary descendants of populations in ancestral environments can be used as a proxy for the ancestor, with phenotypic and genetic differences between these “contemporary ancestors” and “derived” populations being used to infer the direction, source, and pace of adaptation to derived environments. However, recent changes in the ancestral environment may stimulate evolutionary responses in the contemporary populations that inhabit it, complicating their utility as a proxy.
Standing genetic variation (SGV), defined as the variety of alleles segregating in a population [12, 13], is expected to play an important role in parallel evolution. In particular, SGV permits rapid adaptation compared to de novo mutation, and increases the likelihood that the same beneficial allele will be present in different derived populations [14,15,16]. The role of SGV in adaptive divergence is readily measurable: if an allele fixed in the derived population is present in the contemporary ancestor at low frequencies, it likely contributed to adaptation [13]. However, the inference that an allele present in the contemporary ancestral population resulted in adaptation via SGV requires three assumptions. (1) The subset of individuals that originally colonized the derived environment must have contained the rare adaptive allele at some frequency; otherwise it arose from de novo mutation or subsequent gene flow. (2) The contemporary ancestor has undergone little evolution, including gene flow from the derived population (e.g., [17]). (3) The ancestral population has to have been properly characterized (i.e., population structure and allele frequencies associated with SGV). If there are multiple potential ancestral populations, each with a different pool of SGV, inference about the source and pace of evolution in the derived population will vary depending on which putative ancestral population is investigated (e.g. [12]). These assumptions must be verified to characterize accurately the role of SGV during population divergence.
Threespine stickleback (Gasterosteus aculeatus) provide perhaps the best documented examples of adaptation from SGV. Marine threespine stickleback occur widely in the northern hemisphere, including along the Pacific coast of North America from Alaska south to southcentral California. Across the north Pacific coast, much freshwater habitat formed recently (~ 10,000–20,000 years ago) in association with isostatic rebound following glacial retreat. The subsequent colonization of this habitat by stickleback allows tests of the significance of de novo mutation and SGV for adaptation (e.g. [17,18,19]). For instance, marine stickleback bodies are often covered by > 29 bony lateral plates, but fewer plates (0–10) have evolved in parallel in freshwater populations through selection on a rare marine allele [18]. Despite numerous studies indicating the role of SGV at either a single locus for platedness (Ectodysplasin – hereafter Eda) or for multiple loci with unknown phenotypic effects [20,21,22], assumptions about the appropriateness of considering extant marine sticklebacks as representative of the ancestors of freshwater populations remains untested. Despite evident genetic variation in threespine stickleback among geographic clades [23,24,25,26], marine stickleback on the eastern Pacific are largely assumed to constitute a single population (e.g. [27,28,29,30,31]). This assumption is justified by the absence of barriers to gene flow in the marine environment [27], the migratory capacity of marine stickleback [32], the relative “evolutionary stasis” of marine stickleback inferred from the fossil record [28, 29], and the low marine population structure reported from several local studies ([20, 33], but see [34]). Nevertheless, substantial evidence indicates local adaptation even in highly migratory marine fishes [35,36,37], and indeed in Baltic Sea threespine stickleback [38,39,40]. If Pacific threespine stickleback constitute a single population, their large population size should limit the effects of genetic drift and high gene flow should offset local adaptation [41]. Given these conditions, marine stickleback should not exhibit local differentiation that would generate regional differences in the initial colonists of freshwater lakes and streams. SGV should be the same along the Pacific coast – and all freshwater populations could have evolved from the same initial pool of marine SGV facilitating parallel adaptation. These assumptions require formal testing to elucidate the role of SGV during adaptive divergence.
In this study, we consider phenotypic and genotypic variation of > 200 marine threespine stickleback from eight locations from California to Alaska to test hypotheses about the genetic structure of marine stickleback and its evolutionary consequences. Based on variation in plate phenotypes and genotypes associated with SGV at Eda, three-dimensional body morphology from micro-computed tomography (μCT) scans quantified using geometric morphometrics, and Genotype-by-Sequencing [42], we assess whether marine stickleback constitute a single population. By doing so, we test assumptions about the distribution of SGV in “contemporary ancestors” of freshwater stickleback. Additionally, we test predictions regarding the influence of SGV on the source of adaptation based on genomic sequences for a freshwater population from British Columbia. We specifically test the following null predictions: (1) Marine stickleback populations will not vary in the frequency and content (e.g. private alleles) of SGV. (2) Similarly, marine stickleback will not exhibit genetic population structure, which would otherwise influence the SGV regionally available for selection. (3) Marine populations will exhibit no phenotypic divergence in body shape or platedness [e.g. 18]. (4) If differences in SGV among marine populations occurs, genetic variation will show no evidence of having been shaped by natural selection. (5) If population structure in marine stickleback occurs, geographic proximity to a freshwater population will determine the extent of genetic divergence between marine and freshwater stickleback. (6) Differences in SGV among marine populations, if they occur, will not affect the candidate loci identified as contributing to adaptation in freshwater stickleback.
Methods
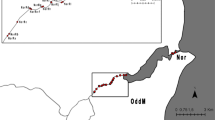
Threespine stickleback (n = 383, Table 1) were collected with minnow traps or seines during the summers of 2010 (Brannen Lake, British Columbia, hereafter BCFW), 2012 (Alaska) and 2013 (all other localities). Sampling locations extended along a 21.8 degree latitudinal spread (Table 1, Fig. 1), from California (south to north, CA01, CA02, CA03), through Oregon (OR01, OR02), the Puget Sound area of Washington (WA01), Vancouver Island (BC01) and Alaska (AK01). Locations varied in terms of benthos, freshwater input, and protection – for instance, CA01 fish were sampled in a slough with freshwater input determined by precipitation, while OR02 were collected near a tidal gate close to the mouth of a river. Other marine species were collected alongside stickleback, such as bay pipefish (Syngnathus leptorhyncus) or smelt (Atherinops/Atherinopsis sp.). Adults were captured in all localities with the exception of WA01, while OR01 contained a range of age classes. Stickleback were euthanized using buffered tricaine methanesulfonate (MS-222) or Eugenol (clove oil) and preserved in 70% ethanol. Fin clips were preserved in 95% ethanol for later sequencing. All collections were conducted in accordance with CCAC guidelines (AUP AC13–0040) and state/provincial/national collection and import permits.

Map of sampling localities. See Table 1 for code designations. Marine sites = triangles, freshwater site = circle
Sex was identified using primers developed by [43] that amplify sex-specific alleles at the idh locus. Alleles were visualized in a 2% agarose gel for 367 individuals.
Library preparation and analysis
Reduced representation DNA sequencing was used to generate Single Nucleotide Polymorphisms (SNPs) in order to assess population structure and adaptive divergence. Two hundred nanograms total genomic DNA was extracted per fish in January 2016 using Qiagen DNeasy Blood and Tissue kits (n = 265) and digested with EcoRI and MseI restriction enzymes (chosen after in silico digestion [44]). Thirty to thirty-five individuals were included from each marine location, and 15 from BCFW. After digestion-ligation, fish were pooled into groups of nine. Cleanup and size selection were performed simultaneously using SPRI beads (Beckman Coulter), at a bead ratio of 0.8× and 0.61× for left and right-side cleanup, respectively. This left a fragment range between 250 and 600 bp. Pooled samples were divided into three technical replicates to ameliorate stochastic differences during PCR, and were amplified. Replicates were pooled and left-side cleaned using SPRI beads. Pooled samples were quantified using a 2200 TapeStation (Agilent) and Qubit (Thermofisher) dsDNA high sensitivity assay. Equal volumes of each 2 nM pooled sample were pooled to make the final library. Library preparation followed the Illumina protocols for the Illumina NextSeq 500 Mid-Output kits with version 2 chemistry. Two sequencing runs were completed on the Illumina Next Seq 500 using 150 cycles and different final library concentrations - the first at 1.8 pM final concentration, the second at 1.1 pM final concentration. A 20% PhiX spike-in was used for both to compensate for the low diversity nature of the library. Results from the two sequencing runs were merged.
Sequenced reads were cleaned and processed using Stacks v.1.35 [45, 46]. Reads were de-multiplexed and cleaned using process_radtags, rescuing barcodes if the correction of a single sequencing error made them identifiable. GSnap [47] was used to align reads to the stickleback reference genome (Ensembl release 72 [48]), allowing for five mismatches with soft masking disabled. SNP calls were corrected in rxstacks using a bounded SNP model with an upper error rate of 0.1. Stringent filtering criteria were applied to the data, with slightly different filtering criteria used to address different questions. In order to determine population structure, each sampling site was assumed to constitute a distinct population. The filtering criteria for this “marine site” data set included: i) log likelihood threshold > − 60; ii) sequenced in more than 75% of individuals; iii) in 6 of 8 populations; iv) with minimum 4× coverage; v) minimum minor allele frequency of 2%, and vi) FIS > − 0.3. Individuals were included if they retained > 10,000 RAD-loci after cleaning (n = 239, Table 1). After population structure was determined, Adegent-recognized clusters rather than sampling location were used in the second Stacks run. This “marine cluster” data set used the same filtering criteria as above, with the exception that the variant needed to be present in all clusters. Finally, a “marine-freshwater” data set was run, which treated each sampling site as a distinct population and additionally included a freshwater population (see below). In this case the variant needed to be present in all eight marine populations and in the freshwater population. For each data set, all population statistics except for FST were calculated using the populations module of Stacks.
Population genetic structure
Pairwise global and per-locus FST were calculated using the Weir and Cockerham [49] adaptation implemented in hierfstat v0.04–22 [50] in R [51]; pairwise global FST were tested for significance (> 0) using 999 permutations in GenoDive [52]. Discriminant Analysis of Principal Components (DAPC) [53] was used on the “marine site” data to assess population structure in the marine environment using Adegenet v2.0.1 [54], as it has shown to perform better than Structure under a stepping-stone model of dispersal [53]. The optimal number of Principal Components (PCs) to retain was calculated using both xvalDAPC and a-scores, which gave similar answers. The optimal number of clusters was assigned based on the lowest Bayesian Information Criteria (BIC) score using k-means clustering. As several possible k clusters had similarly low BIC scores, analyses were run and compared using 3 to 8 clusters.
An analysis of molecular variance (AMOVA), implemented in poppr v2.3.0 [55, 56] using the Ade4 package [57], was used to determine the proportions of genetic variance among versus within sampling sites or Adegenet-recognized clusters. Missing values were replaced with the average frequency for a locus; ignoring missing values did not alter overall patterns. To explore the possibility of cryptic population structure, each sampling site was further analysed individually using Stacks and Adegenet.
The distance between each sampling site was measured as distance along the coast (km) using Google Maps. Distances were measured to or from the mouth of each bay. Neighbouring localities were separated by 242–479 km, except for BC01-AK01, which were separated by approximately 2500 km of coastline. The location of WA01 in Puget Sound resulted in all locations south of Washington being closer to BC01 than they were to WA01. Genetic distance was calculated using the pairwise global Weir and Cockerham FST measures from hierfstat. Geographic and genetic distance matrices were compared using a Mantel test from the Adegenet package with 999 replications to determine Isolation-by-Distance (IBD).
Population statistics were also estimated for the “marine cluster” data set in Stacks using the optimal Adegenet-identified clusters.
A phylogenetic network was calculated using SNPhylo [58] and visualized using FigTree v.1.4.3 [59]. The “marine sites” data set was used, but SNPhylo additionally filtered loci based on linkage disequilibrium. Individuals were colour-coded according to their recognized genetic cluster. A hierarchical clustering tree was additionally constructed using BayPass v.2.1 [60].
Platedness
Plate variation among populations was assessed using plate number and Eda genotype. Adult stickleback (i.e. fish > 30 mm standard length (SL)) (n = 281, Table 1, Additional file 1: Table S2) were stained in Alizarin red. Plate number, including keel, was counted on both sides of the body and summed. Low-plated individuals without keel (LPNK) were defined as individuals with < 20 anterior plates. Partially-plated keeled (PPK) fish had 21–59 plates, including at least one plate at the caudal keel. Fully-plated keeled (FPK) fish had ≥ 60 plates. Additionally, some low-plated fish had a keel (LPK) and were defined as having < 20 anterior plates plus additional plates at the caudal keel. Partially plated stickleback that lacked a keel (PPNK) had > 20 anterior plates but had no plates at the caudal keel. Individuals were also genotyped at the Stn382 locus [18] as this microsatellite is linked to an indel in intron 1 of the Eda gene, yielding a 218 bp “fully-plated” allele (C) or a 158 bp “low-plated” allele (L) [61]. Genotyping followed the protocol of [43]. Individuals were genotyped as LL (homozygous for the low-plated allele), CL (heterozygous), or CC (homozygous for the fully-plated allele). This approach allowed juveniles (< 30 mm SL) to be included in the analysis (total n = 361), and provided genetic information at a locus with known adaptive significance that was not recovered from sequencing. Hardy-Weinberg equilibrium (HWE) was assessed for Stn382 for each marine site using a goodness-of-fit Chi-squared test.
Morphometrics
Phenotypic variation among populations was further assessed using morphometric analysis. Stickleback > 30 mm SL that had been preserved with relatively little bending (n = 272) were straightened, and spines and fins held flat against the body using plastic wrap. μCT scanning at a resolution of 20 μm was conducted in a standardized fashion for all individuals using a Scanco μCT35 instrument (Scanco AG). Three-dimensional images were generated from the anterior point of the premaxilla to the posterior tip of the pelvic spine, using standardized isosurface thresholds in Amira 5.4 (FEI Visualization Sciences Group). Fifty-five landmarks were plotted on the left side of each fish (Additional file 1: Table S1, Fig. 2) and raw landmark scores were exported to MorphoJ v1.06a [62] for further analyses. A prior study had removed the operculum on the left side of all AK01 stickleback, so landmarks were plotted on their right sides. Data were first transformed to remove differences associated with isometric scaling, rotation and translation using Procrustes superimposition. Residuals from a within-marine site multivariate regression on centroid size were estimated and used in all subsequent analyses. Principal Components Analysis (PCA) determined the major axes of phenotypic variation. Canonical Variate Analysis (CVA) was used to determine Procrustes distances using sex and marine site as categorical variables, although BC01 had only one female and OR01 included one individual of unknown sex. The significance of Procrustes distances among pairwise comparisons of marine site-sex combinations was determined based on 10,000 permutations and a corrected α of 0.0005. Discriminant Function Analysis (DFA) was used to determine the likelihood that individuals could be reassigned to their site of origin, given their phenotypes. For this analysis the effect of sex on reassignment success was not assessed.

Position of 55 landmarks used for the morphometric analysis. See Table S1 for identity of landmarks
Selection on phenotypic variation
Patterns of phenotypic variation among populations may be the result of genetic drift, natural selection, or phenotypic plasticity. One way to rule out neutral evolutionary processes is to compare estimates of phenotypic divergence (PST) with neutral expectations based in part on observed neutral genetic differentiation. Observed phenotypic divergence was estimated as:
where σ2B and σ2W were the between- and within-population components of variance, respectively, for plate count and the first four PCs from the morphometric analysis (as per [63, 64]). Variance components were estimated for all marine sampling sites together (global PST) and pairwise using lme4 [65], with population as a random effect. Genetic divergence at the Stn382 locus for Eda (FSTQ) was estimated using the Weir and Cockerham method in Genepop V4 [66]. Neutral genetic divergence (FST) was estimated in hierfstat using non-genic SNPs identified from our data set using Biomart [67]. Non-genic SNPs may still be linked to loci under selection, so this approach provides a conservative estimate of neutrality.
Selection was inferred based on two methods. The first assessed the association between PST-FST and FSTQ-FST using Mantel tests. This measure is based on the expectation that phenotypic or QTL divergence will be uncorrelated with neutral genetic divergence – by extension implicating selection to explain such patterns. The second test involved Whitlock and Guillaume’s [68] method using the R-code from [69]. In brief, the expected between-population variance component for a neutral phenotype was estimated by using observed non-genic FST and observed within-population variance component for the phenotype:
As per [69], the distribution of neutral σ2B was estimated by generating a χ2 distribution with six degrees of freedom (one less the number of sampling sites excluding Washington), and multiplying a randomly drawn value from this distribution by σ2B. From this new distribution expected neutral σ2B were drawn 10,000 times and used to create a distribution of neutral PST-FST. The observed PST-FST was then compared to this distribution and the quantile of the neutral distribution that lay beyond the observed value was used as the probability of the observed outcome in the absence of selection, p. Under the expectation of no selection, p is > 0; selection is evidenced if p = 0. FSTQ-FST values was also compared to the neutral PST-FST, as per [40].
To quantify the relation between phenotype and Stn382 genotype, a generalized linear model (GLM) was fit to the data in R, using the glm routine, as per [40], with plate number as the dependent variable, genotype as a fixed effect, and using a log-link function with a quasi-Poisson error distribution. Furthermore, a Mantel test was used to estimate the correlation between pairwise FSTQ and PST measures.
Selection on genetic variation in the ocean
Under the assumption that marine stickleback populations have a shared history, the covariance matrix of population allele frequencies (Ω) was estimated in BayPass [60] using the “marine site” data. From this a hierarchical clustering tree [60] was generated, assuming no gene flow. A covariate-free genome scan was then performed to identify outlier loci putatively under selection, using per-locus measures of differentiation (XtX). The simulate.baypass function was used to estimate the posterior predictive distribution of XtX using a pseudo-observed data set (POD) [60]. Any loci in the “marine site” data set with XtX values above the POD-estimated threshold were scored as outlier loci potentially under selection. Genic outliers were identified using BioMart [67].
Marine-freshwater genetic divergence
To assess the extent to which the choice of putative “contemporary ancestor” affected inference about adaptation to fresh water, pairwise FST was estimated for each marine-freshwater pair using hierfstat. In BayPass, covariance matrices, PODs and XtX thresholds were estimated for each marine-freshwater comparison. Outlier loci were examined to determine if the same outliers were being consistently recovered irrespective of the origin of the marine fish.
Results
Sex
A total of 205 males and 162 females were sampled. Sex bias was particularly striking in CA01 (26 M, 9 F), BC01 (47 M, 4 F), and AK01 (8 M, 23 F).
Sequencing results
Over 192 million reads passed initial filters (Additional file 1: Table S3) in the “marine sites” data set. Two hundred eighty-two RAD-loci were excluded due to excess heterozygosity. Filtering minor alleles at a threshold of 2% reduced the number of retained loci by 32%. After filtering, between 230,010 (AK01) and 426,018 (OR01) loci were retained for each site sampled, generating between 1877 (AK01) and 5204 (OR01) SNPs (Table 2), for a total of 6655 variant loci.
Standing genetic variation
All marine samples exhibited SGV, ranging from an average of 0.82% (AK01) to 1.22% (OR01) of the total SNPs genotyped in a given population; however, the pool of SGV varied from California to Alaska (Table 2). Stickleback from each marine location contained multiple private alleles (alleles found only at that location) (Fig. 3) and were polymorphic for a portion of the variant loci (loci that were polymorphic in at least one marine site). Polymorphism among variant loci varied from 57% (AK01) to 80% (OR01). For variant loci, the average frequency of the major allele (present in > 50% of all sequenced stickleback) ranged from 88% (OR02) to 93% (AK01) (Additional file 1: Figure S1), suggesting that the frequencies of SGV also differed among locations. Heterozygosity ranged from 0.10 (AK01) to 0.17 (OR02) for variant loci. Population-level average FIS over all variant loci varied from 0.032 (CA01) to 0.064 (OR01) (Table 2). Although FIS was close to 0 for most loci, it approached 1 for a few loci (Additional file 1: Figure S2).

The distribution of private allele frequencies per putative marine population. a CA01, b CA02, c CA03, d OR01, e OR02, f WA01, g BC01, h AK01. See Table 1 for label meanings
Population genetic structure
All pairwise comparisons of global FST significantly exceeded 0 (p < 0.001), and ranged from 0.020 to 0.181 (Table 3). Pairwise FST between the northern marine groups (WA01, BC01, and AK01) were all small (< 0.05), although other comparisons showed moderate (between 0.05–0.15), and three showed great (between 0.15–0.25) differentiation.
Significant population genetic structure was detected. The best supported number of clusters from the eight marine locations sampled was five (BIC = 1379, Fig. 4, Additional file 1: Table S4). The five clusters were, from south to north, CA01, CA02, CA03-OR01, OR02, and WA01-BC01-AK01. The CA03-OR01 cluster also contained seven individuals from OR02 and a single individual from AK01; otherwise individuals clustered with others from their sampling locality. The possibility of a single genetic cluster was as well-supported as ten genetic clusters (BIC = 1392). Three to six clusters had BIC values that differed little from the best-supported model. Altering the number of putative clusters revealed different population structures (Additional file 1: Table S4), with WA01 and AK01 continuing to cluster until k = 13. Cryptic population structure was not evidenced for any sampling locality (Additional file 1: Table S5). Even when WA01, BC01, and AK01 were included in a single analysis, k = 1 was the best supported cluster (BIC = 436.9). However, k = 2 (BIC = 438.2) and k = 3 (BIC = 440.2) still separated individuals by locality.

Adegenet-identified clusters for k = 5. Inset shows hypothetical range of each cluster. Note that the cluster identified as CA03, OR01 contains one AK01 and seven OR02 individuals. See Table 1 for label meanings
The genetic variance partitioned between clusters by AMOVA was low (19%) compared to within clusters (81%). When clusters were not considered, 33% of variation occurred between marine sites. A Mantel test of pairwise geographic distances and pairwise Weir and Cockerham FST was non-significant when all sites were included (r = − 0.2, p = 0.8). This was largely driven by the extreme distance between the WA01, BC01, and AK01 sites. If AK01 was excluded from the analysis the association between geographic and genetic distance was weakly significant (r = 0.5, p = 0.02) (Additional file 1: Figure S3).
A total of 4299 variant loci were sequenced in the “marine clusters” data set, with 2441 (CA01) to 3869 (CA03-OR01) SNPs per cluster. The CA01 sample included the most private alleles (n = 80), but the cluster of WA01-BC01-AK01, which individually had few private alleles (1 to 25), now had 47 (Additional file 1: Figure S4, Additional file 1: Table S6 and S7). The proportion of variant loci that were polymorphic within a cluster varied from 57% (CA01) to 90% (CA03-OR01). Observed heterozygosity for variant loci varied from 13% (CA01) to 17% (OR02). FIS was lowest in southern California and highest in CA03-OR01 (Additional file 1: Table S7).
The phylogenetic network largely agreed with Adegenet assignment (Fig. 5). The network revealed greater intermixing of groups than did Adegenet, but WA01, BC01, and AK01 still largely clustered together and comprised a separate lineage from most southern stickleback. CA01 and CA02 constituted distinct lineages. Most OR02 individuals appeared to be derived from the CA03-OR01 clade, with 75% bootstrapping confidence. Similarly, the hierarchical clustering method grouped WA01-BC01-AK01 together, but placed OR02 as basal to all groups (Fig. 5).

Platedness
Fish sampled from each site differed in the frequencies of plate morphs (Additional file 1: Table S2). FPK morphs comprised 100% of samples from BC01 and OR01. Four LPNK individuals were sampled from AK01, with the rest being FPK. All other sites were at least trimorphic for LPNK, PPK, and FPK. California in particular had high frequencies of LPNK stickleback, comprising 77% of samples. Five individuals from OR02 and CA01 exhibited the rare LPK morph, and a single individual from OR02 was a PPNK morph.
Juvenile and adult plate morphs could be estimated using Stn382 genotypes (Fig. 6, Additional file 1: Table S2). Only 2 of 50 WA01 individuals were heterozygous CL; the remainder were CC. Among juvenile OR01 there was a single LL, 11 CL, and 17 CC individuals. Furthermore, although all OR01 adults were FPK, six of these were CL heterozygotes. All polymorphic populations were in HWE for Stn382, except for AK01 (observed 24 CC, 0 CL, 3 LL, expected 21.3 CC, 5.3 CL, 0.3 LL; Chi-Squared test: 1 d.f., p = 0) (Additional file 1: Table S2).

The frequency of different Eda genotypes using the Stn382 marker, for each sampling site. “The North” refers to samples from Washington, British Columbia, and Alaska. CC = homozygous for the fully-plated allele. LL = homozygous for the low-plated allele. CL = heterozygous
Morphometrics
Phenotypes varied extensively among sites. The first eight Principal Components explained 71% of all phenotypic variance (Additional file 1: Figure S5). The first two Canonical Variates (CVs) explained 57% of the variation among combinations of site and sex, and the first four CVs explained 77% (Additional file 1: Table S8). CV1, after accounting for differences in centroid size, revealed that BC01 fish had narrow, streamlined bodies with dorsal and ventral landmarks both shifting inward relative to the consensus fish (Figs. 7 and 8). Californian fish were grouped close together on CV1 and had squatter, less streamlined bodies with dorsal and ventral landmarks shifted away from one another relative to the consensus. AK01, OR01 and OR02 had intermediate phenotypes between BC01 and California. CV2 showed a gradual transition from CA01 to BC01, but AK01 was clearly distinct from all other sites along this axis. AK01 showed substantial dorsolateral and anterior-posterior constriction of the body relative to all other sites (Figs. 7 and 8).

a Canonical Variate (CV) 1 vs CV2, and b CV3 vs CV4 for body shape. See Table 1 for label meanings

Wireframes of stickleback oriented (left) left laterally, showing the head and anterior tip of the pelvic spine situated left to right, and (right) superiorly. a CV1 for a BC01-type body shape; b CV1 for a CA01-type body shape; c CV2 for an AK01-type body shape; d CV2 for a BC01-type body shape. Light blue wireframe shows the consensus morphology, while dark blue shows the conformational change
The sexes differed morphologically at all sites except AK01 (Additional file 1: Table S9), but the sexes still largely grouped together according to sampling location. CA02 and OR02 were exceptions, with males from both sites clustering with OR01 males. Similarly, CA01 and CA02 females had morphologies that were not significantly distinct.
The DFA revealed that most fish could be classified according to marine site (based on Procrustes distance, p < 0.001 for all pairwise comparisons), with a single fish misclassified (Additional file 1: Table S10). Cross-validation misclassified an average of 2.8 fish per pairwise comparison (n = 59 total misclassifications), but this varied from 0 to 7 (CA02 – OR02), 8 (CA03 – OR02), 9 (CA01 – CA02), and 10 (CA02 – CA03). Only three fish were misclassified when comparing sites from within an Adegenet-recognized cluster. Thus, most misclassifications occurred among, rather than within, genetic clusters.
PST-FST and FSTQ-FST comparisons
PST was estimated as 0.46 for platedness, 0.60 for PC1, 0.29 for PC2, 0.23 for PC3, and 0.09 for PC4. FSTQ was 0.60. Plate PST and FSTQ greatly exceeded the range of the neutral PST-FST distribution (p = 0 for both), as did PST for PC1 (p = 0). PST for PC2 was marginally significant but within the tail of the neutral distribution (p = 0.002), while PST for PC3 (p = 0.02) and PC4 (p = 0.7) were well within the neutral distributions (Fig. 9).

The expected neutral distribution of PST contrasted with the observed values of PST-FST for a platedness, including FSTQ-FST for the Eda allele, and principal components for body shape, including b PC1, c PC2, d PC3, and e PC4
Of all of the Mantel tests between PST, FST, FSTQ, and distance, only one was significant and two were marginally significant with corrected α = 0.05/18 = 0.0027: PC1 PST correlated positively with geographic distance, plate count PST correlated positively with FSTQ, and, surprisingly, PC1 PST correlated positively with FSTQ (Table 4, Additional file 1: Figure S6 and S7). The relation between plate PST and FSTQ was substantiated with a generalized linear model which showed a decrease in plate number with number of L alleles (null deviance = 4813 with 277 d.f., residual deviance = 349 with 275 d.f., p < 0.0001, Hosmer and Lemeshow goodness of fit text: χ2 = − 5 × 10− 26, d.f. = 8, p = 1).
Selection on genetic variation in the ocean
102 of 6655 loci from the “marine sites” data set were flagged as outliers, using an XtX threshold of 15.52 (Additional file 1: Figure S8). Although variant loci were sequenced across all 21 chromosomes and an additional 65 scaffolds, outliers were only detected on 16 chromosomes and two scaffolds. Of these, 8 of 36 (22%) variant loci on scaffold 37 were outliers, followed by 13 of 178 (7%) on linkage group (LG) XXI, 11 of 184 (6%) on LGXIX, and 18 of 498 (4%) on LGIV. Of these 102 outliers, 20 were located within 16 genes (Table 5), although none of these genes have been previously studied in stickleback.
Genetic differentiation and outlier analysis for marine-freshwater comparisons
The “marine-freshwater” data set identified 132,415 loci, of which 1912 were variant. As expected, marine-freshwater divergence was high and in all but one instance was > 0.25 (“very great” differentiation). The lowest pairwise FST values involved the Oregon sites (FST(BCFW, OR01) = 0.27, FST(BCFW,OR02) = 0.18) (Additional file 1: Table S11). The maximum difference in pairwise per-locus FST estimates for a single locus was 0.991, and the average difference was 0.19. 314 (17%) SNPs had a minimum FST estimate of little genetic differentiation (< 0.05) in at least one marine-freshwater contrast, but great genetic differentiation (> 0.25) in another.
97 of 1912 loci were identified as outliers in at least one marine-freshwater comparison, using XtX thresholds of 4.9 (CA03 – FW) to 6.0 (CA01 – FW). Seventeen outlier loci resided in 13 genes (Table 6). Only 3 of 97 loci were flagged as outliers in all eight marine-freshwater comparisons, while nearly a third (31%) were flagged in only a single comparison. Five of the genic loci were outliers in seven comparisons (tub, S100P, and three novel genes), and five were outliers in only one (including a different S100P locus).
Surprisingly, 12 of the loci flagged as outliers in the “marine sites” data set were also flagged as outliers in at least one marine-freshwater comparison (Table 6), including a novel gene on LGII (n = 7 comparisons), and csnk1g2a (n = 3 comparisons). The gene OVGP1 was also flagged as containing outlier loci in both analyses, but different loci were flagged in each case.
Discussion
Marine stickleback exhibit between-population genetic variation
The significance of SGV for parallel evolution depends on its occurrence in the ancestral populations. Marine stickleback harbour SGV, and it differs between populations. Differences in gene expression between two marine BC populations suggested this possibility [10], but here it has been quantified across an extensive latitudinal range. The extent of SGV, 0.8–1.2% of all sequenced loci, is intermediate to that reported from other studies [34, 70]. Nucleotide diversity varied from 0.0016 to 0.0027, consistent with results from Alaska (0.0022 and 0.0025 [20]) and slightly lower than that reported from Oregon (0.003–0.0036 [34]). All marine locations harboured some degree of private alleles, even after ignoring minor alleles at < 2% frequency, suggesting that not just frequencies of SGV but also content of SGV can differ from site to site. Furthermore, the best-known example of SGV, Eda, was present at varying frequencies between populations and is likely under selection in the marine environment. Such variation in the content and frequency of SGV, in turn, led to compelling evidence for population genetic structure. Marine threespine stickleback showed substantial population genetic structure along the Pacific coast of North America. Although FST values (average FST = 0.088) were generally lower than those reported for marine-freshwater divergence (e.g. [20, 34, 71]), they were higher than those reported for other marine stickleback populations along the North American Pacific coast (two Alaskan populations: FST = 0.0076 [20]; three Oregonian populations: FST = 0.007 [34]). However, they align with studies from Europe [39, 40, 72].
Five genetic clusters were identified for the eight sampled localities, although structuring was hierarchical, with some clusters more genetically diverged than others. The most widespread cluster occupied > 2700 km of coastline from Washington to Alaska. Marine stickleback from this genetic cluster have been well characterized, with genetic divergence reported to be low between populations separated by up to 1000 km [20, 33, 73]. Such low structuring between proximate marine populations has led to a basic assumption in stickleback literature that marine stickleback exhibit little genetic or phenotypic diversity globally (e.g. [27,28,29, 64, 74]) – an assumption supported by the iconic image of distinct freshwater stickleback forms radiating from a single fully-plated marine stickleback type (e.g. [75]). In contrast, results from a broad range suggest that such generalizations should be restricted to the northern genetic cluster – and even it contains morphological and genetic differentiation that could be adaptively significant.
The southern genetic clusters were sequentially separated by a few hundred kilometres, well within the migratory ability of marine stickleback [76,77,78]. IBD was evident only after removing AK01 from the dataset, suggesting that limited migration could explain patterns of divergence between the southern genetic clusters. However, IBD needs to be interpreted with caution, as geographic distance was correlated with latitude, and latitudinal variation can be associated with environmental clines [79]. Whatever the causes that shape genetic variation between stickleback populations, the distribution of SGV among different marine populations affects inference about the source and pace of selection in the freshwater environment, and complicates attempts to uncover loci that are under selection in derived populations.
Marine stickleback exhibit between-population phenotypic variation
Marine stickleback are generally considered to be fully-plated (e.g. [27]), yet the Eda genotype for low-platedness has an ancient marine origin [18]. The low-plated allele has been hypothesized to exist in the marine environment as SGV only when transported from the freshwater environment [17] or when masked by marine modifying alleles [18]. If the low-plated allele exists at low frequencies in the ocean, behaviours that facilitate the movement of low-plated marine stickleback into fresh water could also account for the consistent colonization of rare low-plated stickleback in lakes and streams [80]. We found substantial variation in the frequency of the low-plated allele, to the point that it was the major allele in some Californian and Oregonian populations, but was absent from BC01. This finding is consistent with other records of low-plated marine stickleback in California ([81], but see [18, 82]) and high frequencies of low-platedness in European marine stickleback [61, 74, 83,84,85]. However, it contrasts with other Pacific and Atlantic North American studies that focussed on northern sites [86,87,88,89,90,91].
The observed SGV at Eda could affect the rate at which adaptation to lakes occurred in the past. Indeed, it may explain why reduced plate size has evolved in some freshwater populations, rather than reduced plate number, as an alternative strategy that may have been required in the absence of SGV at Eda [92, 93]. Such variation at Eda is a particularly striking reminder that the function of full-platedness in marine stickleback remains unknown (for an example of its possible use, demonstrated in freshwater populations, see [94]).
Marine stickleback body shape also varied extensively along the coast. Californian populations tended to have squatter body shapes that appeared to be less streamlined than their northern counterparts. The functional significance of these differences requires testing – but it is interesting that the streamlined fish were from a single genetic cluster, while the squat Californian fish exhibited significant population structuring between neighbouring localities. This potentially indicates extensive migration along the northern coast that is not mirrored in the south. Jamniczky et al. [95] reported considerable morphological divergence between neighbouring sampling sites in British Columbia – groups presumably with little to no genetic divergence, implicating plasticity as a driver of morphological variation. Morris et al. [79] similarly reported variation in vertebral number and standard length with latitude. However, two related analyses suggest that selection may also play a role in shaping phenotypic diversity.
Based on DeFaveri and Merilä’s [40] method, Pacific coast stickleback exhibited selection for platedness similar to that observed for Baltic Sea stickleback. There was also suggestive evidence for selection on PC1 of body shape, which largely corresponded to CV1 – more streamlined northern fish, more squat southern fish. The role of plasticity in affecting these results remains to be determined. PC1 was marginally associated with Eda genotype. To our knowledge, this is the first study to demonstrate pleiotropic or linked effects of Eda on body shape in the marine environment (for marine-freshwater or freshwater-only evidence for pleiotropy, see [96]; for other forms of phenotypes associated with Eda see [80, 97,98,99,100,101]), which could result in low-platedness being an indirect target of selection in some marine habitats. It is possible, for instance, that a pleiotropic relationship exists between body shape, Eda, and thermal tolerance – a possibility suggested by the relationship between low plate frequency and latitude in anadromous populations of Europe [83]. This is one of several possible explanations that requires formal testing.
Although Eda is the best-characterized example of SGV, the outlier analysis revealed other potential candidate genes for selection in the marine environment. Extensive differentiation was evidenced at some loci. Local adaptation despite gene flow has been found in other marine fish populations [35, 102,103,104], including European marine stickleback [72]; but the extent to which stickleback south of Washington exhibited population structure was unanticipated.
Smaller bodies and distinct body shapes tend to evolve in freshwater stickleback populations [105, 106], often with significant correlations between morphology and freshwater biotic and abiotic factors [107]. Given the morphological variation among marine stickleback, elucidating whether freshwater morphology is the result of plasticity, SGV, or de novo mutation requires informed decisions about what constitutes the marine ancestor.
Inferring the source and pace of adaptation in freshwater stickleback
The choice of marine stickleback affected inference concerning the source and pace of adaptation in one freshwater population from Vancouver Island. Few outlier loci were consistently recovered in all marine-freshwater comparisons; had only a single marine population been used in this study, the outliers reported would differ depending on which marine population had been chosen. Many studies involve comparisons between geographically proximate marine and freshwater stickleback pairs (e.g. [21, 22]), presumably to account for the possibility of population structure in the marine environment. Yet FST was the lowest when the freshwater population was paired with a geographically distant population from northern Oregon, a finding that is difficult to reconcile with the assumption that the nearest marine population is the most suitable ancestral type. The occurrence of Japanese mtDNA haplotypes in Haida Gwaii lake populations that are not present in Haida Gwaii marine populations [108, 109] suggests that this may not be an isolated incident. Similarly, several studies have noted, but not explained, the fact that northern marine stickleback are genetically more similar to southern than northern freshwater stickleback [17, 21, 22].
Nearly one third of outlier loci were only outliers for a single marine-freshwater comparison. Thus, one’s choice of marine population could produce spurious inferences about the loci under selection, or miss true candidate genes. Clearly more information is needed going forward about the relationship between marine and freshwater stickleback.
Inferring the role of SGV during adaptive divergence in ancestral-derived comparisons requires three conditions to be met, for which we have provided varying evidence. The first, that colonists had to contain the same variants as present in the ancestral population, could not be tested here. Second, the contemporary ancestor has to have been relatively evolutionarily static. Both phenotypic and genetic evidence demonstrates that this condition does not hold for marine stickleback. Contemporary marine populations have diverged phenotypically in a manner beyond neutral evolutionary expectations, and genetically in a way partially explicable by selection. This means that contemporary marine threespine stickleback populations are genetically and phenotypically distinct from their own ancestors – and it was these ancestors that also originally colonized lakes and streams along the coast. Thus the term “contemporary ancestor” is a misnomer, as contemporary marine threespine stickleback populations do not reflect the ancestral condition. Interpretations of freshwater stickleback evolution need to be tempered by marine stickleback evolutionary history.
Third, the ancestral population has to have been properly characterized. Eastern Pacific stickleback exhibit some population genetic structure, although consistent with other reports [20, 33] stickleback north of Oregon constitute a single genetic population. This means that along much of the coast freshwater environments were likely colonized by distinct marine stickleback populations, which differed in SGV frequency and content. Furthermore, it is likely that marine stickleback have exhibited range contractions and expansions along the southern and northern coasts throughout their evolutionary history, most recently in the north after the last glacial retreat [110]. This means that there is no a priori reason to expect that a marine population currently proximate to freshwater populations are descendants of the ancestors of those freshwater populations. Population structuring and evolutionary history thus changes our understanding of the ancestral condition of marine stickleback, and requires that we carefully consider the use of contemporary marine populations when addressing evolutionary questions.
Conclusions
Studies that compare marine and freshwater stickleback may need to adjust their methodologies in light of marine stickleback variation. The typical image in textbooks is of a single marine stickleback form from which numerous freshwater forms radiate [75]. Our data suggests that there is phenotypic and genetic variation in marine threespine stickleback which likely impacts freshwater stickleback diversification – to assume a single marine form is no longer tenable. Furthermore, assuming ancestral status for the marine population most geographically proximate to the freshwater population of interest is problematic, unless it can be directly demonstrated (e.g. [111]). So where does this leave the comparative method? One possibility would be to reconstruct the genotype of the ancestor to all eastern Pacific marine stickleback – but this would ignore the important role that local variation has played in freshwater stickleback evolution. Another possibility could be to conduct larger-scale geographic sampling than has heretofore been done, of both marine and freshwater forms, in order to determine a more thorough evolutionary history of this species. Then, having taken evolutionary relationships into account, comparisons can be made using better-justified “contemporary ancestors”.
“Contemporary ancestors” are used in a number of systems [3,4,5,6,7,8,9,10,11] for addressing evolutionary questions. They are particularly useful for determining the role of SGV during evolution, and for identifying the alleles involved in adaptation to new environments. Clearly, care must be exercised in characterizing these proxies of the ancestral form, as unaccounted population structure and current evolution can lead to spurious interpretations of adaptation. Whether the lessons from stickleback apply to other species with smaller geographic distributions or more limited opportunities for gene flow waits to be seen.
Abbreviations
- AK:
-
Alaska
- AMOVA:
-
Analysis of molecular variance
- BC:
-
British Columbia
- BIC:
-
Bayesian Information Criteria
- C:
-
Fully plated Eda allele
- CA:
-
California
- CV:
-
Canonical variate
- CVA:
-
Canonical variate analysis
- DAPC:
-
Discriminant Analysis of Principal Components
- DFA:
-
Discriminant Function Analysis
- Eda :
-
Ectodysplasin
- F:
-
Female
- FPK:
-
Fully plated with keel
- FSTQ :
-
Genetic divergence at Eda allele
- FW:
-
Freshwater
- GLM:
-
Generalized linear model
- HWE:
-
Hardy-Weinberg equilibrium
- IBD:
-
Isolation-by-Distance
- km:
-
Kilometer
- L:
-
Low plated Eda allele
- LG:
-
Linkage group
- LPK:
-
Low plated with keel
- LPNK:
-
Low plated without keel
- M:
-
Male
- OR:
-
Oregon
- PC:
-
Principal Component
- PCA:
-
Principal Components Analysis
- POD:
-
Pseudo-observed data set
- PPK:
-
Partially plated with keel
- PPNK:
-
Partially plated without keel
- PST :
-
Estimate of phenotypic divergence
- SGV:
-
Standing genetic variation
- SL:
-
Standard length
- WA:
-
Washington
- μCT:
-
Micro-computed tomography
- μm:
-
Micrometer
- σ2 B :
-
Between-population component of phenotypic variance
- σ2 W :
-
Within-population component of phenotypic variance
References
Bernatchez L. Ecological theory of adaptive radiation: an empirical assessment from coregonine fishes (Salmoniformes). In: Hendry AP, Stearns SC, editors. Evolution illuminated: salmon and their relatives. Oxford: Oxford University Press; 2004. p. 175–207.
Rundle HD, Nosil P. Ecological speciation. Ecol Lett. 2005;8:336–52.
Berry RJ, Jakobson ME, Peters J. The house mice of the Faroe Islands: a study in microdifferentiation. J Zool. 1978;185:73–92.
Losos JB, Creer DA, Glossip D, Goellner R, Hampton A, Roberts G, Haskell N, Taylor P, Ettling J. Evolutionary implications of phenotypic plasticity in the hindlimb of the lizard Anolis sagrei. Evolution. 2000;54:301–5.
Hendry A. Adaptive divergence and the evolution of reproductive isolation in the wild: an empirical demonstration using introduced sockeye salmon. Genetica. 2001;1:515–34.
Des Roches S, Robertson JM, Harmon LJ, Rosenblum EB. Ecological release in White Sands lizards. Ecol Evol. 2011;1:571–8.
Domingues VS, Poh Y-P, Peterson BK, Pennings PS, Jensen JD, Hoekstra HE. Evidence of adaptation from ancestral variation in young populations of beach mice. Evolution. 2012;66:3209–23.
Cahill JA, Green RE, Fulton TL, Stiller M, Jay F, Ovsyanikov N, Salamzade R, St. John J, Stirling I, Slatkin M, Shapiro B. Genomic evidence for island population conversion resolves conflicting theories of polar bear evolution. PLoS Genet. 2013;9:e1003345.
Yavno S, Fox MG. Morphological change and phenotypic plasticity in native and non-native pumpkinseed sunfish in response to sustained water velocities. J Evol Biol. 2013;26:2383–95.
Morris MRJ, Richard R, Leder EH, Barrett RDH, Aubin-Horth N, Rogers SM. Gene expression plasticity evolves in response to colonization of freshwater lakes in threespine stickleback. Mol Ecol. 2014;23:3226–40.
Dennenmoser S, Vamosi SM, Nolte AW, Rogers SM. Pool-seq reveals adaptive genomic divergence under high gene flow between freshwater and brackish-water ecotypes of prickly sculpin (Cottus asper). Mol Ecol. 2017;26:25–42.
Przeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evolution. 2005;59:2312–23.
Barrett RD, Schluter D. Adaptation from standing genetic variation. Trends Ecol Evolut. 2008;23:38–44.
Elmer KR, Meyer A. Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends Ecol Evolut. 2011;26:298–306.
Hendry AP. Key questions in the genetics and genomics of eco-evolutionary dynamics. Heredity. 2013;111:456–66.
Peichel CL, Marques DA. The genetic and molecular architecture of phenotypic diversity in sticklebacks. Phil Trans R Soc B. 2017;372:20150486.
Schluter D, Conte GL. Genetics and ecological speciation. Proc Natl Acad Sci. 2009;106:9955–62.
Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G Jr, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science. 2005;307:1928–33.
Chan YF, Marks ME, Jones FC, Villarreal G Jr, Shapiro MD, Brady SD, Southwick AM, Absher DM, Grimwood J, Schmutz J, Myers RM, Petrov D, Jónsson B, Schluter D, Bell MA, Kingsley DM. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010;327:302–5.
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010;6:e1000862.
Jones FC, Chan YF, Schmutz J, Grimwood J, Brady SD, Southwick AM, Absher DM, Myers RM, Reimchen TE, Deagle BE, Schluter D, Kingsley DM. A genome-wide SNP genotyping array reveals patterns of global and repeated species-pair divergence in sticklebacks. Curr Biol. 2012;22:83–90.
Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, Birney E, Searle S, Schmutz J, Grimwood J, Dickson MC, Myers RM, Miller CT, Summers BR, Knecht AK, Brady SD, Zhang H, Pollen AA, Howes T, Amemiya C. Broad institute genome sequencing platform and whole genome assembly team, lander ES, Di Palma F, Lindblad-Toh K, Kingsley DM. The genomic basis of adaptive evolution in threespine stickleback. Nature. 2012;484:55–61.
Haglund TR, Buth DG, Lawson R. Allozyme variation and phylogenetic relationships of Asian, north American, and European populations of the threespine stickleback, Gasterosteus aculeatus. Copeia. 1992;1992:432–43.
Ortí G, Bell MA, Reimchen TE, Meyer A. Global survey of mitochondrial DNA sequences in the threespine stickleback: evidence for recent migrations. Evolution. 1994;48:608–22.
Mäkinen HS, Merilä J. Mitochondrial DNA phylogeography of the three-spined stickleback (Gasterosteus aculeatus) in Europe - evidence for multiple glacial refugia. Mol Phylogenet Evol. 2008;46:167–82.
Lescak EA, Marcotte RW, Kenney LA, von Hippel FA, Cresko WA, Sherbick ML, Colgren JJ, López JA. Admixture of ancient mitochondrial lineages in three-spined stickleback populations from the North Pacific. J Biogeogr. 2015;42:532–9.
Bell MA. Evolution of phenotypic diversity in Gasterosteus aculeatus superspecies on the Pacific coast of North America. Syst Zool. 1976;25:211–27.
Bell MA. Paleobiology and evolution of threespine stickleback. In: Bell MA, Foster SA, editors. The evolutionary biology of the threespine stickleback. NY: Oxford University Press; 1994. p. 438–71.
Foster SA, Baker JA, Bell MA. The case for conserving threespine stickleback populations. Fisheries. 2003;28:10–8.
Spoljaric MA, Reimchen TE. 10 000 years later: evolution of body shape in Haida Gwaii three-spined stickleback. J Fish Biol. 2007;70:1484–503.
Baker JA, Heins DC, Foster SA, King RW. An overview of life-history variation in female threespine stickleback. Behaviour. 2008;145:579–602.
Williams DD, Delbeek JC. Biology of the threespine stickleback, Gasterosteus aculeatus, and the blackspotted stickleback, G. wheatlandi, during their marine pelagic phase in the bay of Fundy, Canada. Environ Biol Fish. 1989;24:33–41.
Withler RE, McPhail JD. Genetic variability in freshwater and anadromous sticklebacks (Gasterosteus aculeatus) of southern British Columbia. Can J Zool. 1985;63:528–33.
Catchen J, Bassham S, Wilson T, Currey M, O’Brien C, Yeates Q, Cresko WA. The population structure and recent colonization history of Oregon threespine stickleback determined using restriction-site associated DNA-sequencing. Mol Ecol. 2013;22:2864–83.
Hemmer-Hansen J, Therkildsen NO, Pujolar JM. Population genomics of marine fishes: next-generation prospects and challenges. Biol Bull. 2014;227:117–32.
Carvalho GR, Hauser L, Martinsohn J, Naish K. Fish, genes and genomes: contributions to ecology, evolution and management. J Fish Biol. 2016;89:2471–8.
Machado LF, Damasceno JD, Bertoncini AA, Tosta VC, Farro APC, Hostim-Silva M, Oliveira C. Population genetic structure and demographic history of the spadefish, Chaetodipterus faber (Ephippidae) from southwestern Atlantic. J Exp Mar Biol Ecol. 2017;487:45–52.
DeFaveri J, Jonsson PR, Merilä J. Heterogeneous genomic differentiation in marine threespine sticklebacks: adaptation along an environmental gradient. Evolution. 2013;67(9):2530–46.
DeFaveri J, Shikano T, Shimada Y, Merilä J. High degree of genetic differentiation in marine three-spined sticklebacks (Gasterosteus aculeatus). Mol Ecol. 2013;22:4811–28.
DeFaveri J, Merilä J. Evidence for adaptive phenotypic differentiation in Baltic Sea sticklebacks. J Evol Biol. 2013;26:1700–15.
Bernatchez L. On the maintenance of genetic variation and adaptation to environmental change: considerations from population genomics in fish. J Fish Biol. 2016;89:2519–56.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379.
Peichel CL, Ross JA, Matson CK, Dickson M, Grimwood J, Schmutz J, Myers RM, Mori S, Schluter D, Kingsley DM. The master sex-determination locus in threespine sticklebacks is on a nascent Y chromosome. Curr Biol. 2004;14:1416–24.
Lepais O, Weir JT. SimRAD: an R package for simulation-based prediction of the number of loci expected in RADseq and similar genotyping by sequencing approaches. Mol Ecol Resourc. 2014;14:1314–21.
Catchen J, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping loci de novo from short-read sequences. G3. 2011;1:171–82.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–40.
Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–75.
Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–55.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70.
Goudet J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Resour. 2005;5:184–6.
Core Team R. R: a language and environment for statistical computing. R foundation for statistical computing. Vienna; 2016. https://www.r-project.org/
Meirmans PG, Van Tienderen PH. GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Resour. 2004;4:792–4.
Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94.
Jombart T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–5.
Kamvar ZN, Tabima JF, Grünwald NJ. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. 2014;2:e281.
Kamvar ZN, Brooks JC, Grünwald NJ. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front Genet. 2015;6:208.
Dray S, Dufour AB. The ade-4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22:1–20.
Lee T-H, Guo H, Wang X, Kim C, Paterson AH. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics. 2014;15:162.
Rambaut A. FigTree. 2007. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 9 Oct 2017.
Gautier M. Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics. 2015;201:1555–79.
Lucek K, Haesler MP, Sivasundar A. When phenotypes do not match genotypes – unexpected phenotypic diversity and potential environmental constraints in Icelandic stickleback. J Hered. 2012;103:579–84.
Klingenberg CP. MorphoJ: an integrated software package for geometric morphometrics. Mol Ecol Resourc. 2011;11:353–7.
Kaeuffer R, Peichel CL, Bolnick DI, Hendry AP. Parallel and nonparallel aspects of ecological, phenotypic, and genetic divergence across replicate population pairs of lake and stream stickleback. Evolution. 2012;66:402–18.
Ravinet M, Prodöhl PA, Harrod C. Parallel and nonparallel ecological, morphological and genetic divergence in lake-stream stickleback from a single catchment. J Evol Biol. 2013;26:186–204.
Bates D, Maechler M, Bolker B, Walker S. Fitting linear mixed-effect models using lme4. J Stat Softw. 2015;67:1–48.
Rousset F. Genepop ‘007: a complete reimplementation of the Genepop software for windows and Linux. Mol Ecol Resourc. 2008;8:103–6.
Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G, Bardou P, Beck T, Blake A, Bonierbale M, Brookes AJ, Bucci G, Buetti I, Burge S, Cabau C, Carlson JW, Chelela C, Chrysostomou C, Cittaro D, Collin O, Cordova R, Cutts RJ, Dassi E, Di Genova A, Djari A, Esposito A, Estrella H, Eyras E, Fernandez-Banet J, Forbes S, Free RC, Fujisawa T, Gadaleta E, Garcia-Manteiga JM, Goodstein D, Gray K, Guerra-Assunção JA, Haggarty B, Han DJ, Han BW, Harris T, Harshbarger J, Hastings RK, Hayes RD, Hoede C, Hu S, Hu ZL, Hutchins L, Kan Z, Kawaji H, Keliet A, Kerhornou A, Kim S, Kinsella R, Klopp C, Kong L, Lawson D, Lazarevic D, Lee JH, Letellier T, Li CY, Lio P, Liu CJ, Luo J, Maass A, Mariette J, Maurel T, Merella S, Mohamed AM, Moreews F, Nabihoudine I, Ndegwa N, Noirot C, Perez-Llamas C, Primig M, Quattrone A, Quesneville H, Rambaldi D, Reecy J, Riba M, Rosanoff S, Saddiq AA, Salas E, Sallou O, Shepherd R, Simon R, Sperling L, Spooner W, Staines DM, Steinbach D, Stone K, Stupka E, Teague JW, Dayem Ullah AZ, Wang J, Ware D, Wong-Erasmus M, Youens-Clark K, Zadissa A, Zhang SJ, Kasprzyk A. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43:W589–98.
Whitlock MC, Guillaume F. Testing for spatially divergent selection: comparing QST to FST. Genetics. 2009;183:1055–63.
Lind MI, Ingvarsson PK, Johansson H, Hall D, Johansson F. Gene flow and selection on phenotypic plasticity in an island system of Rana temporaria. Evolution. 2011;65:684–97.
Feulner PGD, Chain FJJ, Panchal M, Eizaguirre C, Kalbe M, Lenz TL, Mundry M, Samonte IE, Stoll M, Milinski M, Reusch TBH, Bornberg-Bauer E. Genome-wide patterns of standing genetic variation in a marine population of three-spined sticklebacks. Mol Ecol. 2013;22:635–49.
Mäkinen HS, Cano JM, Merilä J. Genetic relationships among marine and freshwater populations of the European three-spined stickleback (Gasterosteus aculeatus) revealed by microsatellites. Mol Ecol. 2006;15:1519–34.
Lind EE, Grahn M. Directional genetic selection by pulp mill effluent on multiple natural populations of three-spined stickleback (Gasterosteus aculeatus). Ecotoxicology. 2011;20:503–12.
Taylor EB, McPhail JD. Evolutionary history of an adaptive radiation in species pairs of threespine sticklebacks (Gasterosteus): insights from mitochondrial DNA. Biol J Linnean Soc. 1999;66:271–91.
Raeymaekers JAM, Maes GE, Audenaert E, Volckaert FAM. Detecting Holocene divergence in the anadromous-freshwater three-spined stickleback (Gasterosteus aculeatus) system. Mol Ecol. 2005;14:1001–14.
Bell MA, Foster SA. Introduction to the evolutionary biology of the threespine stickleback. In: Bell MA, Foster SA, editors. The evolutionary biology of the threespine stickleback. NY: Oxford University Press; 1994. p. 438–71.
Jones DH, John AWG. A three-spined stickleback, Gasterosteus aculeatus L. from the North Atlantic. J Fish Biol. 1978;13:231–6.
Quinn TP, Light TJ. Occurrence of threespine sticklebacks (Gasterosteus aculeatus) in the open North Pacific Ocean: migration or drift? Can J Zool. 1989;67:2850–2.
Cowen RK, Chiarella LA, Gomez CJ, Bell MA. Offshore distribution, size, age, and lateral plate variation of late larval/early juvenile sticklebacks (Gasterosteus) off the Atlantic coast of New Jersey and New York. Can J Fish Aquat Sci. 1991;48:1679–84.
Morris MRJ, Petrovitch E, Bowles E, Jamniczky HA, Rogers SM. Exploring Jordan’s rule in Pacific three-spined stickleback Gasterosteus aculeatus. J Fish Biol. 2017;91:645–63.
Barrett RD, Vines TH, Bystriansky JS, Schulte PM. Should I stay or should I go? The Ectodysplasin locus is associated with behavioural differences in threespine stickleback. Biol Lett. 2009;5:788–91.
Bell MA. Low-plate morph of the threespine stickleback breeding in salt water. Copeia. 1979;1979:529–33.
Snyder RJ, Dingle H. Adaptive, genetically based differences in life history between estuary and freshwater threespine stickleback (Gasterosteus aculeatus L.). Can J Zool. 1989;67:2448–54.
Münzing J. The evolution of variation and distributional patterns in European populations of the three-spined stickleback, Gasterosteus aculeatus. Evolution. 1963;17:320.
Klepaker T. Lateral plate polymorphism in marine and estuarine populations of the threespine stickleback (Gasterosteus aculeatus) along the coast of Norway. Copeia. 1996;1996:832–8.
Ferchaud A-L, Hansen MM. The impact of selection, gene flow and demographic history on heterogeneous genomic divergence: three-spine sticklebacks in divergent environments. Mol Ecol. 2016;25:238–59.
Hagen DW, Gilbertson LG. Geographic variation and environmental selection in Gasterosteus aculeatus L. in the Pacific northwest, America. Evolution. 1972;26:32–51.
Hagen DW, Moodie GEE. Polymorphism for plate morphs in Gasterosteus aculeatus on the east coast of Canada and an hypothesis for their global distribution. Can J Zool. 1981;60:1032–42.
Cresko WA, Amores A, Wilson C, Murphy J, Currey M, Phillips P, Bell MA, Kimmel CB, Postlethwait JH. Parallel genetic basis for repeated evolution of armor loss in Alaskan threespine stickleback populations. Proc Natl Acad Sci. 2004;101:6050–5.
Karve AD, von Hippel FA, Bell MA. Isolation between sympatric anadromous and resident threespine stickleback species in mud Lake, Alaska. Environ Biol Fish. 2008;81:287–96.
Kitano J, Bolnick DI, Beauchamp DA, Mazur MM, Mori S, Nakano T, Peichel CL. Reverse evolution of armor plates in the threespine stickleback. Curr Biol. 2008;18:769–74.
Bell MA, Gangavalli AK, Bewick A, Aguirre WE. Frequency of Ectodysplasin alleles and limited introgression between sympatric threespine stickleback populations. Environ Biol Fish. 2010;89:189.
Leinonen T, McCairns RJS, Herczeg G, Merilä J. Multiple evolutionary pathways to decreased lateral plate coverage in freshwater threespine sticklebacks. Evolution. 2012;66:3866–75.
Wiig E, Reseland JE, Østbye K, Haugen HJ, Vøllestad LA. Variation in lateral plate quality in threespine stickleback from fresh, brackish and marine water: a micro-computed tomography study. PLoS One. 2016;11:e0164578.
Reimchen TE. Predator handling failures of lateral plate morphs in Gasterosteus aculeatus: functional implications for the ancestral plate condition. Behaviour. 2000;137:1081–96.
Jamniczky HA, Barry TN, Rogers SM. Eco-evo-devo in the study of adaptive divergence: examples from threespine stickleback (Gasterosteus aculeatus). Integr Comp Biol. 2015;55:166–78.
Albert AYK, Sawaya S, Vines TH, Knecht AK, Miller CT, Summers BR, Balabhadra S, Kingsley DM, Schluter D. The genetics of adaptive shape shift in stickleback: pleiotropy and effect size. Evolution. 2008;62:76–85.
Barrett RD, Rogers SM, Schluter D. Environment specific pleiotropy facilitates divergence at the Ectodysplasin locus in threespine stickleback. Evolution. 2009;63:2831–7.
Rogers SM, Tamkee P, Summers B, Balabahadra B, Marks M, Kingsley DM, Schluter D. Genetic signature of adaptive peak shift in threespine stickleback. Evolution. 2012;66(8):2439–50.
Mills MG, Greenwood AK, Peichel CL. Pleiotropic effects of a single gene on skeletal development and sensory system patterning in sticklebacks. EvoDevo. 2014;5:5.
Greenwood AK, Mills MG, Wark AR, Archambeault SL, Peichel CL. Evolution of schooling behavior in threespine sticklebacks is shaped by the Eda gene. Genetics. 2016;203:677–81.
Robertson S, Bradley JE, MacColl AD. Eda haplotypes in three-spined stickleback are associated with variation in immune gene expression. Sci Rep. 2017;7:42677.
Knutsen H, Jorde PE, André C, Stenseth NC. Fine-scaled geographical population structuring in a highly mobile marine species: the Atlantic cod. Mol Ecol. 2003;12:385–94.
Knutsen H, Olsen EM, Jorde PE, Espeland SH, André C, Stenseth NC. Are low but statistically significant levels of genetic differentiation in marine fishes ‘biologically meaningful’? A case study of coastal Atlantic cod. Mol Ecol. 2011;20:768–83.
Saha A, Johansen T, Hedeholm R, Nielsen EE, Westgaard JI, Hauser L, Planque B, Cadrin SX, Boje J. Geographic extent of introgression in Sebastes mentella and its effect on genetic population structure. Evol Appl. 2017;10:77–90.
McGuigan K, Nishimura N, Currey M, Hurwit D, Cresko WA. Cryptic genetic variation and body size evolution in threespine stickleback. Evolution. 2011;65:1203–11.
Bowles E, Johnston RA, Vanderzwan SL, Rogers SM. Genetic basis for body size variation between an anadromous and two derived lacustrine populations of threespine stickleback Gasterosteus aculeatus in Southwest Alaska. Curr Zool. 2016;62:71–8.
Magalhaes IS, D’Agostino D, Hohenlohe PA, MacColl ADC. The ecology of an adaptive radiation of three-spined stickleback from north Uist, Scotland. Mol Ecol. 2016;25:4319–36.
Deagle BE, Reimchen TE, Levin DB. Origins of endemic stickleback from the queen Charlotte Islands: mitochondrial and morphological evidence. Can J Zool. 1996;74:1045–56.
Johnson LS, Taylor EB. The distribution of divergent mitochondrial DNA lineages of threespine stickleback (Gasterosteus aculeatus) in the northeastern Pacific basin: post-glacial dispersal and lake accessibility. J Biogeogr. 2004;31:1073–83.
Jacobs DK, Haney TA, Louie KD. Genes, diversity, and geologic process on the Pacific coast. Annu Rev Earth Planet Sci. 2004;32:601–52.
Lescak EA, Bassham SL, Catchen J, Gelmond O, Sherbick ML, von Hippel FA, Cresko WA. Evolution of stickleback in 50 years on earthquake-uplifted islands. PNAS. 2015;112:E7204–12.
Acknowledgements
The authors wish to thank: Friends of the Arcata Marsh, Elkhorn Slough Foundation, South Slough National Estuarine Research Reserve, Bamfield Marine Sciences Centre, and the National Park Service in King Salmon, Alaska for their assistance in locating and capturing marine stickleback; T. Gaskill, L. Basch, K. Wasson, D. Homer, D. Morris, S. Vanderzwan, T. Barry, A. Pistore, R. Kaufman, E. Ellefson, V. Heather, B. Hallgrimsson and the Hallgrimsson lab. This research was enabled in part by support provided by WestGrid (www.westgrid.ca) and Compute Canada Calcul Canada (www.computecanada.ca).
Funding
NSERC Discovery Grants (RT735287 and 418249–2012), Vanier, Killam, and Alberta Innovates Technology Futures funded this paper. No funding body was involved in the design, collection, analysis, or interpretation of the data for this study, or writing of this manuscript.
Availability of data and materials
https://doi.org/10.5061/dryad.450c2v7.
Author’s contributions
MRJM collected stickleback, collected and interpreted data, and wrote the manuscript. EB collected stickleback from Alaska, assisted with genomics data collection and analysis, and contributed to the manuscript. BA assisted with genomics data collection and statistical analysis, and contributed to the manuscript. HAJ provided the μCT scanner, assisted with morphological analysis, and contributed to the manuscript. SMR funded and oversaw each stage of the project. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Animal ethics was approved by the University of Calgary Life and Environmental Science Animal Care Committee, and followed the guidelines for the Canadian Council of Animal Care (CCAC). Animal Use Protocol: AUP AC13–0040.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Additional tables and figures. This document contains information supporting the main text, including: the list of landmarks used for 3D morphometrics, population-specific details on sex and platedness, additional information pertaining to methodology, the distributions of major allele frequencies and FIS per population, isolation-by-distance analyses, details regarding Adegenet-recognized clusters, CVA and DFA morphometrics results, additional Mantel tests, global pairwise FST values for each marine-freshwater comparison, and results pertaining to the outlier analysis. (DOCX 1008 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Morris, M.R.J., Bowles, E., Allen, B.E. et al. Contemporary ancestor? Adaptive divergence from standing genetic variation in Pacific marine threespine stickleback. BMC Evol Biol 18, 113 (2018). https://doi.org/10.1186/s12862-018-1228-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-018-1228-8