
Abstract
Background
During the Pleistocene, shifts of species distributions and their isolation in disjunct refugia led to varied outcomes in how taxa diversified. Some species diverged, others did not. Here, we begin to address another facet of the role of the Pleistocene in generating today’s diversity. We ask which processes contributed to divergence in semi-arid southern Australian birds. We isolated 11 autosomal nuclear loci and one mitochondrial locus from a total of 29 specimens of the sister species pair, Chestnut Quail-thrush Cinclosoma castanotum and Copperback Quail-thrush C. clarum.
Results
A population clustering analysis confirmed the location of the current species boundary as a well-known biogeographical barrier in southern Australia, the Eyrean Barrier. Coalescent-based analyses placed the time of species divergence to the Middle Pleistocene. Gene flow between the species since divergence has been low. The analyses suggest the effective population size of the ancestor was 54 to 178 times smaller than populations since divergence. This contrasts with recent multi-locus studies in some other Australian birds (butcherbirds, ducks) where a lack of phenotypic divergence was accompanied by larger historical population sizes. Post-divergence population size histories of C. clarum and C. castanotum were inferred using the extended Bayesian skyline model. The population size of C. clarum increased substantially during the late Pleistocene and continued to increase through the Last Glacial Maximum and Holocene. The timing of this expansion across its vast range is broadly concordant with that documented in several other Australian birds. In contrast, effective population size of C. castanotum was much more constrained and may reflect its smaller range and more restricted habitat east of the Eyrean Barrier compared with that available to C. clarum to the west.
Conclusions
Our results contribute to awareness of increased population sizes, following significant contractions, as having been important in shaping diversity in Australian arid and semi-arid zones. Further, we improve knowledge of the role of Pleistocene climatic shifts in areas of the planet that were not glaciated at that time but which still experienced that period’s cyclical climatic fluctuations.
Similar content being viewed by others

Background
Understanding the effect that cycles of cooling and warming during the Pleistocene had on the diversification and distribution of fauna and flora is a central theme in evolutionary biology [1]. The impact of the Pleistocene is particularly well understood in the temperate northern hemisphere [1]. It is widely accepted that species underwent range contractions, persisting in one or few refugia during glacial maxima. During warmer interglacials, ranges expanded opening up potentially novel habitats and providing opportunity for secondary contact and gene flow [1]. The effect that these isolation events and shifts in distribution had on the diversification of species varies across taxa, some species having diverged and others remained cohesive [2]. A largely unanswered central question is which processes drove these varying outcomes [2]? When speciation did occur, was it accompanied by increased genetic drift due to large reductions in population size when in contracted refugia, colonisation of newly suitable habitat, or divergent selection? If these processes acted in concert, how might their relative contributions be teased apart?
These questions are especially challenging in understanding the effect of Pleistocene cycles of cooling and warming on diversification in less studied areas such as unglaciated regions of the world, including arid or semi-arid regions (e.g., [3, 4]). Recently, in the context of southern and central Australian temperate, dry, semi-arid and arid woodland habitats, Kearns et al. [5] demonstrated that arid-adapted populations of Grey Butcherbirds Cracticus torquatus underwent range expansions during Pleistocene glacial maxima due to the expansion of the arid zone at that time. They based these conclusions on a robust multi-locus molecular dataset together with environmental niche models for butcherbirds (Cracticus spp.). In other southern Australian semi-arid fauna, the large and vagile Western Grey Kangaroo (Macropus fuliginosus) lost genetic diversity as its range expanded from south western Australia to the east according to analysis of microsatellite data [6].
Two recent studies are particularly pertinent to this issue. One, a multi-species hierarchical approximate Bayesian computation (hABC) approach found that 26 of 32 Australian avian populations showed concordant signatures of population expansion just prior to the Last Glacial Maximum (LGM) (at 35,225 years ago; 95 % quantiles 18,963–67,545) [7]. The 26 populations of birds are ecologically and phylogenetically diverse and inhabit a variety of different biomes. So the response appears to be a general one, across mesic, monsoon tropical, semi-arid and arid environments but some questions remain. Did those 26 population expansions follow significant contractions or were they simply expansions from already large populations into newly available habitat (or a combination across the 26 taxa)? Was speciation or, at least, morphological differentiation a more likely outcome if populations contracted substantially? The timing of this co-expansion is concordant with the expansion of the western populations of the Grey Butcherbird study (25,000 to 30,000 years) [5]. Species distribution modelling in [5] showed that the population expanded from an already large distribution. Other species of butcherbird showed slow but steady increases from before this time (eastern Grey Butcherbird) or maintained stable populations (Silver-backed and Black-backed Butcherbirds (C. argenteus, C. mentalis, respectively)).
A second, earlier study applied hABC to mitochondrial DNA of an assemblage of ten southern Australian semi-arid zone birds, three of which were a subset of species studied in [7]. It focussed on how many divergence events were necessary to explain the data and revealed two to three separate divergence events having affected them during the mid-late Pleistocene; these involved the Nullarbor and/or Eyrean Barriers [8]. Summary statistics of population expansion for individual taxa further revealed varying patterns among these and two further species (or species pairs). The presence and absence of signatures of population expansion varied across species and parts of species ranges [9]. Notably, the analyses highlighted varying degrees of morphological differentiation among the species relative to each other, and suggested different evolutionary forces operating on phenotype in broadly co-distributed species.
Multi-species mitochondrial DNA (mtDNA) studies as discussed above benefit from increased statistical power gained from pooling data into a single analysis [10]. They can accommodate coalescent, mutational and demographic variance associated with individual species and, especially for single loci, high variance of estimated parameters [11]. However, mtDNA can be prone to departures from neutral equilibrium due to effects of selection or migration-drift disequilibrium (either undetected because of confounding processes or untested), which leads to errors in estimates of demographic parameters [12]. These effects may be amplified in species with complex social structures inhabiting heterogeneous environments, as in the White-browed Babbler Pomatostomus superciliosus [13]. Pavlova et al. [14] also showed that in the Eastern Yellow Robin Eopsaltria australis high intraspecific mtDNA divergences can be misleading and unrepresentative of the evolution of the genome and historical demography. It was argued that the mtDNA in that case was under strong environmental selection despite ongoing nuclear gene flow [14, 15]. These issues can only be uncovered by jointly analysing multiple loci.
Our focus here is to examine the historical demography of the speciational history of two sister species of southern Australian birds, the Chestnut Quail-thrush Cinclosoma castanotum and the Copperback Quail-thrush C. clarum (sensu [9]). C. clarum and C. castanotum are semi-arid adapted, largely terrestrial species which inhabit fairly dense bushy shrubs and undergrowth of mallee scrub, Acacia scrubs, dry sclerophyll woodland, heath, and native pine [16]. Toon et al.’s [17] exploration of phylogenetic relationships within Cinclosoma using multiple nuclear markers provides biogeographic context for this study. Cinclosoma species diversified out of mesic habitat into semi-arid and arid environments. C. clarum and C. castanotum diverged 2.16 million years ago (mya), while the more arid adapted Cinclosoma species began to diversify 6.08 mya. Semi-arid and arid adapted groups diverged from a common ancestor 13.80 mya.
C. clarum and C. castanotum exemplify a situation where there is both substantial genetic structure (4.38 % net divergence in mtDNA between them [9]) and moderate phenotypic divergence accompanied by differing patterns of sexual dimorphism [9, 18]. When this work commenced, C. clarum and C. castanotum were treated as subspecies of a single species (hypotheses varied regarding location of subspecies’ limits; see [9]). Dolman and Joseph [9] proposed that the level of mtDNA divergence across the Eyrean Barrier coupled with concordantly patterned nuclear DNA (nDNA) [17] and taxonomic divergence warranted recognition of two species. Here, sampling of more individuals and more loci has various advantages. It will: i) enable assessment of population structure from multiple loci without a priori assignment of individual specimens to populations; ii) a more precise test of the model of divergence that is applicable to C. clarum and C. castanotum; iii) test whether gene flow between structured populations has been restricted since divergence; and key to the main objective of this study, iv) examine whether the process of divergence was accompanied by reductions in population size and/or population expansions; and if so v) estimate the timing of population expansion in divergent populations. In using the methodologies herein we also robustly test the species rank of C. clarum and C. castanotum using multiple-step, species delimitation methodology [19–21]: discovery of genetic discontinuities using GENELAND [22]; and coalescent divergence modelling using IMa2 [23] to estimate the level of gene flow between species since divergence. We stress that the present study addresses historical not contemporary demography. Therefore, our use here of 12 multiple loci alleviates the lack of sampling in some gaps such as the small one between the ranges of the two species across the broad region of the Eyrean Barrier and the larger one across much of the range of the subspecies C. clarum fordianum. Sampling in those gaps is of course more relevant to study of contemporary gene flow, and we are addressing this separately in a study that will necessarily use historical DNA from museum specimens.
Methods
Sampling
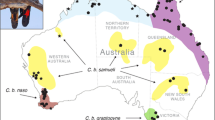
A total of 29 C. castanotum (n = 7) and C. clarum (n = 22) liver tissues were sourced from the Australian National Wildlife Collection, CSIRO National Research Collections; Academy of Natural Sciences at Drexel University, Philadelphia; and South Australian Museum, Adelaide. Details are in Additional file 1: Table S1. Localities of tissue samples used in this study, together with a guide to the general distribution of C. castonotum and C. clarum (localities of all preserved msueum specimens) are presented in Fig. 1.

Map of Australia showing key geographic features, distribution information and sampling localities. Atlas of Living Australia data points for C. clarum and C. castanotum (grey circles) (filtered to represent museum specimens only) and specimens used in this study: C. clarum (hollow circles); C. castanotum (crosses). SB indicates Shark Bay and LEB indicates Lake Eyre Basin
DNA sequencing
Total cellular DNA was extracted according to the salting-out method [24]. DNA sequences from the ND2 gene region of the mtDNA genome were reported in Dolman and Joseph (2015; GenBank numbers KM280401-KM280429). Eleven autosomal nuclear loci were amplified using the PCR primers detailed in Table 1. PCRs were performed in 25 μl reactions containing 1× reaction buffer (Eppendorf), 0.2 mM dNTP, 0.2 μM each primer, 1 Unit HotMaster Taq DNA Polymerase (Eppendorf) which specifically has no requirement for additional MgCl2. Cycling conditions included an initial denaturation of 94 °C for 2 min followed by a touch-down protocol of 34 total cycles of 94 °C denaturation for 30 s, 55 °C to 48 °C annealing for 30 s, extension at 65 °C for 45 s, and a final extension of 65 °C for 5 min. PCR products were purified with Ampure magnetic beads (Agencourt), and sequenced directly in both directions. Autosomal nuclear DNA sequences acquired for this study have been deposited in GenBank (accession numbers KX913359-KX913664). Phases of DNA sequences containing insertions/deletions (indels) were separated manually from the DNA sequence chromatograms as described in Dolman and Moritz [25]. For the remaining sequences with multiple heterozygous sites, phase was determined using the program Phase 2.1.1 [26, 27]. To avoid systematic bias in downstream coalescent analyses, we included all best allele pairs that were consistently resolved at a probability of 0.6 or higher in independent runs [28].
Number of populations and population assignment
Allelic data from 11 autosomal nuclear loci were analysed together with spatial data for each individual using GENELAND [22, 29]. We used GENELAND to estimate the number of populations (k) and their spatial distribution. GENELAND was chosen over other clustering models because of the clear benefits of using a spatial model in cases of diverging subpopulations, potentially subject to admixture [30]. The number of populations, k, was estimated from multiple runs with 10 million iterations and 10,000 thinning and 10 % burn-in generations. Results were produced (with k fixed) from multiple runs of 50 million iterations, 50,000 thinning and 10 % burn-in generations. Runs were repeated 5 times to ensure convergence on a single result. We tested for hierarchical population structure, such that populations identified in the first round of analyses were put into another round of analyses to estimate the number of populations (k) and their distribution.
Tests of assumptions
The isolation with migration model implemented in the program IMa2 assumes that there has been no recombination or gene conversion within the genealogical history of independent loci [31]. We used DNAsp 5.10.01 [32] to perform the four gamete test and to find gene regions excluding recombinant events [33].
The Hudson-Kreitman-Aguade (HKA) test [34] was used to test for neutrality across the 12 loci. HKA tests were performed on recombination-free segments of nuclear loci and the entire mitochondrial locus with 10,000 simulations using the computer program HKA (Jody Hey, Rutgers University). Deviations between observed and expected levels of divergence between populations identified using GENELAND, were summed across loci and the probability from Chi-square distribution (22 degrees of freedom) was calculated. The mtDNA locus (ND2) was also subjected to the McDonald Kreitman test [35] across divergent lineages in DnaSP v5.0 [36].
Population divergence model
DNA sequence data from 11 autosomal nuclear loci and one mtDNA locus from populations identified using GENELAND were fitted to the Isolation with Migration model using IMa2 [23]. For two populations the parameters estimated, scaled by the neutral mutation rate (μ, geometric mean across loci of mutation rate per year per locus), were θA, θ1, θ2, effective population diversity for the ancestral population, and two daughter populations since divergence; m1 and m2, directional migration rates; and t, time since divergence. Isolation with migration was implemented with the Infinite Sites model for 11 nuclear loci (recombinant-free segments) and HKY for ND2. The mutation rate for ND2 was given as 2.96 × 10−5 mutations per locus (1019 bp) based on an evolutionary rate of 0.029 per million years for the ND2 gene [37]. Mutation rates were not provided for nuclear loci. We assumed an average generation time of 2 years based on published data [38–40] Several, relatively short preliminary metropolis coupled runs (10 million steps) were required to optimize the number of chains, heating parameters, and maximum parameter limits. Broad parameter limits were subsequently refined. Twenty chains with geometric heating (0.975 for the first heating parameter and 0.84 for the second heating parameter) were found to maintain a sufficient level of swapping between chains (average 74.5 % between chain 0 and chain 1 and 62.9 % between 18 and 19). Seven final M-mode runs were run with different random seeds for 45 million steps, and burn-in of 1,000,000. Swap rates for genealogy: (branch, topology and time since most recent ancestor, mutation rate scalar, HKY Model Kappa parameter (for mtDNA) and effective sample sizes were all adequate. Results from these seven runs confirmed convergence upon parameter distributions. Plots of parameter trends gave no indication of non-random trends. Genealogies were combined from these runs and 300,000 were used in an L-mode run to estimate joint posterior density and the corresponding joint posterior parameter estimates and to conduct model tests to assess which, if any, model appropriately fits the data. To address concerns that non-neutral evolution of mtDNA may affect demographic parameter estimates, analyses were repeated omitting mtDNA, using relative mutation rates for nuclear loci from posteriors of mutation rate scalars from initial runs with mtDNA. IMa2 is reportedly robust to population sub-structuring [41], but to verify the situation with C. c. clarum and C. c. fordianum, analyses were also repeated omitting the two specimens potentially representing C. c. fordianum according to mtDNA. The scaled mutation rates so recovered for nDNA loci were thought to be reasonable estimates based on published avian nuclear mutation rates of between 0.34 × 10−9 and 1.20 × 10−9 [42]. All but one nuclear mutation rate (minimum = 0.18 × 10−9) were within the published range (maximum = 0.97 × 10−9; average = 0.56 × 10−9).
Timing of population expansion post-divergence
Bayesian skyline plots [43] can estimate the timing of the most recent population expansion. Using a single locus, moving backwards in time beyond the initial bottleneck, information on polymorphisms is lost, and any further population fluctuations are unlikely to be detectable [44, 45]. Using multiple loci to estimate Bayesian skyline plots has the potential to “see past” at least one population expansion event. This of course depends on the number of loci sampled, and the size of the most recent bottleneck. Population size histories of C. clarum and C. castanotum were inferred using the extended Bayesian skyline plot (EBSP) [45] on 12 and 11 loci respectively, in BEAST v1.7.5 [46]. Substitution models, clock models and trees for each partition (locus) were unlinked. Substitution models were chosen to maintain consistency with IMa2 analyses, i.e. simplest model for nuclear loci (HKY) and HKY for ND2, and a strict molecular clock was used. The evolutionary rate for ND2 was fixed at 0.029 substitutions per million years [37]. The nuclear locus Mame-AL08 was fixed at 0.547 × 10−9 substitutions as this was the closest mutation rate to the average across all 11 loci of calculated from mutation rate scalars in IMa2 (average = 0.560 × 10−9). All other nuclear loci were estimated using normal distribution priors around starting estimates of rates calculated from estimates from mutation scalar in the IMa2 analyses. Two individuals representing putative C. clarum fordianum according to mtDNA were not included in the EBSP analysis of C. clarum as this population structure may result in false signals of population declines [47]. BEAST was run for 500 million generations, sampled every 50,000 generations with a 10 % burnin. Effective sample sizes of posterior parameter estimates were monitored to be at least above 100 and stationarity of parameter estimates were checked across multiple runs.
Results
DNA sequencing
The length of DNA sequence data for 11 nuclear loci for 29 individuals (average = 25 individuals per locus) ranged from 187 to 997 bp (average = 527 bp) (Table 1). Eight of 11 loci have indels which could be aligned unambiguously. The first position of each indel was recoded as a segregating character for use in IMa2; otherwise all gaps following the first position were coded as missing data.
Number of populations and population assignment
Phase resolved bi-allelic data were used to estimate the number of populations and to assign individuals to those populations. The peak of the posterior distribution in GENELAND for the number of clusters was k = 2. With k then fixed to k = 2, several runs were found to all converge upon the same population assignment. Population assignment is concordant with mtDNA differentiation at the Eyrean Barrier and therefore concordant with current species delimitation of C. clarum and C. castanotum [9]. All individuals were assigned to their respective populations either side of the Eyrean Barrier with a probability of 1.0, except for specimen ANWC B33368 (Australian National Wildlife Collection, CSIRO) from Tamala Station, south of Shark Bay, which was assigned to C. clarum with a probability of 0.99. Posterior densities for k = 1 to 4 clusters and maps of posterior probability to belong to cluster 1 or 2 are presented in Additional file 2: Figure S1. Hierarchical analyses within these two populations revealed no further clustering of populations. The high level of mtDNA divergence of two specimens (assigned to C. clarum) from C. clarum (one from Tamala Station, Western Australia, and one from Shed Tank, South Australia), suggest that these specimens, at least according to mtDNA, could represent the subspecies C. clarum fordianum, or gene flow from it, or incomplete sorting of mtDNA within the two recognized subspecies of C. clarum. As n = 2 and geographic sampling is sparse, there may be insufficient power to detect a second genetic discontinuity between C. clarum clarum and C. c. fordianum using GENELAND. We included these two specimens as C. clarum in coalescent analyses of divergence, supported by available mtDNA evidence suggesting that the C. clarum clarum - C. clarum fordianum subspecies level divergence is indeed more recent than the C. castanotum - C. clarum species level divergence. However, analyses of divergence were also repeated in three independent runs with these two specimens removed and results changed very little as presented in Table 3.
Tests of assumptions
Recombination-free alignments of nuclear loci range from 207 to 832 bp (average = 369 bp) (Table 1). These recombination-free alignments were used in all downstream analyses. The Hudson-Kreitman-Aguade (HKA) test [34] used to test for neutrality across the 12 loci showed no significant deviation from neutrality: sum of deviations = 10.892; d.f. = 22; Chi Square P = 0.976.
The McDonald-Kreitman test [35] on the coding ND2 gene showed evidence for purifying selection in divergent populations of C. castanotum and C. clarum (synonymous mutations: 37 fixed and 32 polymorphic; non- synonymous mutations:1 fixed and 9 polymorphic; P = 0.015; neutrality index = 10.406). While neutral mutations are still subject to genetic drift, deleterious mutations are removed. This will not affect downstream analyses except for lowering the overall mutation rate of ND2, possibly underestimating the timing of demographic events.
DNA diversity
Recombination-free DNA sequence data revealed low to moderate levels of variation at the 11 nuclear loci. The number of polymorphic sites ranges from 2 to 11 (average = 6) and number of haplotypes ranges from 3 to 13 (average = 7) (Table 2). Average nucleotide diversity across all 11 loci is 0.236 % for C. clarum and 0.182 % for C. castanotum (Table 2). The average number of distinct haplotypes across all 11 nuclear loci is 6 for C. clarum and 3 for C. castanotum and average haplotype diversity is 0.492 and 0.493, respectively for C. clarum and C. castanotum (Table 2).
Population divergence model
Seven final M-mode runs of ca. 60 million steps each converged upon comparable peak parameter estimates (Fig. 2). Table 3 shows highest posterior parameter estimates and the corresponding 95 % upper and lower posterior density bounds for each parameter. Marginal posterior density distributions for each parameter are presented on a demographic scale in Table 4 and Fig. 2. The tree files for these seven runs were combined for an L-mode run. In the L-mode run based on 300,000 genealogies, the divergence model that attained the highest likelihood was the full model which allows migration estimates greater than zero and all population size parameter estimates to vary. However, models based on i) a coalescent migration rate of zero from C. clarum to C. castanotum or ii) equal migration rates could not be rejected (LLR =0.0 and 1.805, respectively; Table 3). Joint posterior estimates from the L-mode run for each of these three models are provided in Table 3. Effective population size (Ne) of the ancestral population is substantially reduced (2287 according to joint posterior estimates for the full model) compared with the two daughter populations (C. castanotum = 123,031 and C. clarum = 406,598). The timing of divergence is during the Middle Pleistocene according to the highest posterior estimate at 0.814 mya and 95 % lower and upper posterior density bounds, 0.514–1.180 mya. Migration since divergence (2 Nm) from west to east, i.e., C. clarum to C. castanotum, is not significantly different from zero. In contrast, migration since divergence from east to west (C. castanotum to C. clarum) is significantly greater than zero, but is nonetheless low (2 Nm <1.0; highest posterior estimate = 0.238; and 95 % lower and upper posterior density bounds = 0.020, 0.820). Repeating analyses without mtDNA produced essentially the same results. Note that the mutation rates for nuclear loci in these analyses were based on relative mutation rates compared with mtDNA in analyses including the full dataset. The importance lies in that the relative demographic parameter estimates are robust to omitting the mtDNA. Repeating analyses without the two specimens more suggestive genetically of being C. c. fordianum or affected by gene flow from it produced very low deviation from all peak parameter estimates.

Multi-locus coalescent-based analyses of population divergence (IMa2). Marginal posterior density distributions from IMa2 analyses – seven independent MCMC analyses (M-mode) of DNA sequence data from one mitochondrial locus and 11 nuclear loci for the divergence of C. clarum and C. castanotum from their common ancestor. On a demographic scale, parameters are (a) time since divergence in years; b effective population size, Ne; and c effective number of migrants per generation, 2 Nm
Timing of population expansion post-divergence
Signatures of population expansion are evident in both C. clarum and C. castanotum. C. castanotum began to increase in population size around 25 thousand years ago (kya) (HPD of ca. 35-0 kya), and C. clarum began to increase in population size around 75 ka (HPD of ca. 75-25 kya) (see Fig. 3).

Extended Bayesian skyline plots (EBSP). Estimates of effective population size (Ne) over time (in millions of years) for (a) C. castanotum and (b) C. clarum based on combined nuclear and mitochondrial loci, using EBSP in BEAST v 1.7.5. The median posterior estimates are presented with the shaded area indicating the 95 % HPD. Effective population size, Ne is scaled to demographic scale as per IMa2 results and presented on a log10 scale. At time 0, Ne is comparable to current Ne in Table 3. *Indicates comparable Ne on EBSP plots with different scales
Discussion
We used multi-locus DNA sequence data to assess historical population structure within sister species of quail-thrush, Cinclosoma clarum and C. castanotum, from southern Australia. The genetic data defined spatial location of population clusters and individuals were assigned to these clusters. A critical result of our analyses, which we expand below, was that population structure was concordant with recently revised species limits [9] and congruent with the Eyrean Barrier being a major historical barrier in these species. The Lake Eyre Basin/Eyrean Barrier has long been hypothesized as a major phylogeographic break in birds [48–50] and indeed in quail-thrush, Cinclosoma spp. [18, 51]. This theory, based originally on morphology, has been supported by molecular data in other Australian birds, such as the Australian ringneck parrots (Barnardius zonarius) [52], the splendid fairy-wren (Malurus splendens) [53] the White-eared Honeyeater (Nesoptilotis leucotis) [8, 9] and White-browed Babbler [13].
Our data suggest that C. clarum and C. castanotum diverged across the Eyrean Barrier during the Middle Pleistocene. Inference from stratigraphic sequences within the distribution of C. castanotum suggests that their divergence likely coincided with a reduction of woodland and tall shrub cover during the Pleistocene [54]. This trend towards aridity in southern Australia was progressive and step-wise [55] and led to the presence of dry-land vegetation in south-eastern Australia at a site in western Victoria at 1.2 mya [56].
The divergence time estimated between C. clarum and C. castanotum in this study can be viewed in context of Cinclosoma species’ divergences within mesic, semi-arid and arid environments across Australia and New Guinea generally [17]. A species tree estimated in *BEAST [57] using eight nuclear markers (common to this study) and a slightly lower evolutionary rate (0.0207 mutations per million year versus an ND2 rate of 0.029 mutations per million year in this study) inferred that semi-arid and arid species of Cinclosoma diverged from the ancestor of the eastern Australian mesic species C. punctatum 17.67 mya. Semi-arid species (C. clarum and C. castanotum) and the remaining four arid species diverged from each other 13.8 mya. The divergence between the semi-arid species (C. clarum and C. castanotum) was inferred as 2.1 mya, earlier than the estimate from this study. This difference may be reconciled by i) *BEAST not accounting for gene flow between populations, or ii) limitations of the methods. Gene flow estimates in the current study were low, and, if anything, a species tree discounting gene flow would appear shallower than one with positive levels of gene flow. The answer is most likely to be in the limitations of the methods, e.g., number of populations that can be studied, differences in models of nucleotide evolution. Interestingly, a comparison between a Poisson random field (PRF) method and *BEAST divergence time estimates across the Isthmus of Panama for 22 bird sister pairs reported that *BEAST almost consistently estimated deeper divergence times [58]. The continued improvement of these and related methods will facilitate improved parameter estimates in the future [59].
Next, estimates of rates of migration since divergence also clarified the history of speciation between C. clarum and C. castanotum. The model based on a coalescent migration rate of zero from C. clarum to C. castanotum could not be rejected. The effective number of migrants per generation since divergence in the reverse direction was significantly greater than zero, but low (2 Nm = 0.238). This level of migration since divergence (2 Nm <1) is not enough to maintain cohesion of a single species of quail-thrush and is consistent with current species delimitation of C. clarum and C. castanotum. The similarly low migration rate estimates from analyses repeated without the mtDNA locus are noteworthy in light of evidence from elsewhere of mtDNA divergence despite nuclear gene flow in the Eastern Yellow Robin Eopsaltria australis [14, 15]. In that case mtDNA divergence was driven by positive and purifying selection on the mitochondrial genome, but here we confirm results are genome wide and more likely reflect demographic processes.
In using the methodologies herein we have robustly tested the species rank recently accorded to C. clarum and C. castanotum [9] using multiple-step, species delimitation methodology [19–21]. A population clustering method (GENELAND) using multi-locus allelic data was used to identify distinct populations without prior assignment of individuals to populations. A coalescent-based ‘isolation with migration’ model was used to confirm that migration since divergence has been low enough to prevent breakdown of species barriers and therefore maintain distinct species.
Bayesian inference of population size history (EBSP) suggested that a significant population expansion event in C. clarum began before the LGM. C. clarum began to expand ca. 50 kya and continued to expand through the Holocene until the present. The signature of population increase in C. castanotum was more recent, around 20 kya, and not as substantial. It is only evident in the mean and highest posterior density and not clear according to lowest posterior density. This may be a power-affect due to fewer individuals sampled in this species. More substantial population expansion in C. clarum compared with C. castanotum according to EBSP is consistent with earlier results from mtDNA summary statistics (Ramos and Onsin’s R 2 and Fu’s Fs) suggesting statistically significant expansion to the west of the Eyrean Barrier in C. clarum, but not to the east of the Eyrean Barrier in C. castanotum [9]. Notably, the timing of population expansion in both species are broadly concordant with the timing in butcherbirds [5] where estimates for western grey butcherbirds (between 30 and 25 ka), eastern grey butcherbirds (from 50 ka) and 26 of 32 Australian avian populations which showed signatures of population expansion just prior to the LGM, ca 35 ka (19–68 ka).
Similar geographical patterns of greater expansion west of the Eyrean Barrier than to its east have been observed in ringneck parrots Barnardius spp. [47] and Variegated Fairy-wrens Malurus lamberti [60] though these patterns were not rigorously dated. This pattern may reflect an interaction between habitat (these are all birds of mallee woodlands) and, simply, the far greater area of that habitat that is available to populations west of the Eyrean Barrier (Fig. 1). The asymmetric pattern of gene flow we observed (greater from C. castanotum into C. clarum than the reciprocal) may reflect fine scale habitat availability in the region of the southern Flinders Ranges-Eyre Peninsula. It warrants closer attention through finer scale sampling of the populations of C. clarum fordianum in that region than was possible in this study. We are pursuing that work from museum specimen-based genomic data.
A key objective of this study was to examine the historical demography that accompanied speciation of these two quail-thrush. Notably, we found that the effective population size of the ancestor of C. clarum and C. castanotum was substantially reduced compared with current population sizes. The joint posterior estimate of the ancestral effective population size was 2287, which is 54 times (C. castanotum) to 178 times (C. clarum) lower than joint posterior estimates of current population. Current effective population sizes on a demographic scale (123,031 for C. castanotum and 406,598 for C. clarum) seem large compared to these species’ current conservation status of vulnerable, near threatened or rare (depending on state/territory) (Atlas of Living Australia accessed 4 January 2016). There are several assumptions used in the analyses which may contribute to this discrepancy: i) effective population sizes are assumed to be constant in each time period (pre- and post-divergence); and ii) potential error in mutation rate estimates used for ND2 (mtDNA) and/or generation time. Estimates may reflect effective population sizes prior to more recent population declines due to habitat loss, fragmentation and degradation post-European settlement [61–63]. Results from EBSP demographic analyses suggest the violation of the assumption of constant effective population size in each time period (at least in C. clarum in the time period since divergence). Notwithstanding these problems of model-fitting, there is a consistent and clear signal of a large reduction in population size in the speciation history of these birds, whether it is at the time of population splitting or post-divergence, followed by significant population expansion.
Available data suggest a dynamic history of vegetation assemblages inhabited by these birds in semi-arid southern Australia, one that is consistent with our findings. Studies in south-eastern Australia suggest a progression from species-rich sclerophyll in the Early Pleistocene, to reduced tree-cover in the Middle Pleistocene, to herb fields and chenopod shrubs dominating the Late Pleistocene to initial Holocene [54, 64, 65]. By the early Holocene, tall trees and shrubs returned to form the current semi-arid adapted woodland as broadly evident today [54]. Following divergence, EBSP analyses suggest that these species had (or continued to have) restricted effective population sizes in the Late Pleistocene, probably due to the lack of tall trees and shrubs and dominance of herbs and chenopod shrubs at that time. Population expansion most likely coincided with the very recent increase in numbers of tall trees and shrubs transitioning into the semi-arid adapted woodland evident today.
A comparable multi-locus study of population demography associated with divergence across southern Australia in birds, albeit of an estuarine and aquatic species, is that between south-west and south-east populations of the Chestnut Teal (Anas castanea) [66]. This divergence was more recent at 0.21 mya, was subject to higher numbers of effective migrants per generation since divergence (2 Nm = 16 from southeast into southwest and 2 Nm = 1.0 from southwest into southeast), and notably was not associated with reduction in population size in the history of divergence according to IMa2. In the Chestnut Teal, the southwest population may have contracted since divergence, while the southeast population remained stable or expanded from an effective population size of 400,000. These demographic factors (more recent divergence, higher gene flow and higher ancestral population size) may well explain the basis for the lack of phenotypic divergence in the Chestnut Teal compared with moderate differentiation between C. clarum and C. castanotum. Further, roles of Z-linked loci and sexual selection in the divergence of the Chestnut Teal from its close relative the Grey Teal A. gracilis are now apparent in that case [67]. Clearly, the roles of natural and sexual selection need to be explored in the moderate differentiation between C. clarum and C. castanotum. More multi-locus data sets from species that have undergone phenotypic divergence, like these quail-thrush, such as the Blue Bonnet and Naretha Bluebonnet (Northiella haematogaster and N. narethae, respectively) and those that have remained relatively more stable phenotypically, despite substantial time since divergence, such as the White-eared Honeyeater (Nesoptilotis leucotis) and the Scarlet Robin Petroica boodang [8, 9] are required to more fully partition which processes drove these varying outcomes of Pleistocene divergence across southern Australia. We also draw attention to the different patterns of sexual dimorphism in C. castanotum and C. clarum [9] and so the worth of exploring a role of sexual selection in their differentiation. This study indicates that drastic reductions in population size associated with isolation and expansion into newly available habitat could have been among the key drivers or, at least, correlates of corresponding phenotypic divergence across southern semi-arid Australia.
Conclusions
Multi-locus data are important for understanding the processes involved in divergence and for improving understanding of why some species differentiate and maintain integrity as separate species, while others subjected to apparently the same climatic regime continue to be a single cohesive species. Here we posed the question, when speciation does occur, is it accompanied by increased genetic drift due to large reductions in population size when in contracted refugia, colonisation of newly suitable habitat, or divergent selection? While further research is required to unravel the relative roles of natural and sexual selection in this case, differing patterns of sexual dimorphism in the two species suggests a role for sexual selection operating in concert with variation in effective population size. Certainly, a role for drift is consistent with our findings. As it becomes more common to obtain data from a larger number of loci through methods such as exon capture and RAD sequencing approaches (e.g. [68, 69]), as well as whole genome approaches to estimate effective population size over 10 mya [70], the power to obtain demographic parameters associated with divergence will improve. Integration of this with species’ ecology, such as the mallee habitats of the species studied here, and palaeoenvironmental history of different continents and regions will likely also be important in explaining patterns of concordance and discordance among species. For example, numerous bird species in Australia now appear to have undergone expansion prior to the LGM [7]. Yet large post-LGM declines in population size are not apparent in the quail-thrush studied here and the Grey Butcherbird Cracticus torquatus [5], all sedentary birds of semi-arid woodlands and shrublands. That such a decline has been reported in the Budgerigar Melopsittacus undulatus, a small nomadic parrot of similar habitats in inland Australia, may reflect its very different ecology tied to free-standing water. Alternatively, it may be related to the increased power of the whole genome analysis from which that decline was inferred in that species [70].
Abbreviations
- bp:
-
Base pairs
- EBSP:
-
Extended Bayesian Skyline Plot
- hABC:
-
Hierarchical Approximate Bayesian Computation
- HKA:
-
Hudson-Kreitman-Aguade
- HKY:
-
Hasegawa, Kishino and Yano
- HPD:
-
Highest Posterior Density
- indels:
-
Insertions or deletions
- kya:
-
Thousand years ago
- LGM:
-
Last Glacial Maximum
- mtDNA:
-
Mitochondrial DNA
- mya:
-
Million years ago
- nDNA:
-
Nuclear DNA
- RAD:
-
Restriction site Associated DNA markers
References
Hewitt GM. Genetic consequences of climatic oscillations in the quaternary. Philos Trans: Biol Sci. 2004;359(1442):183–95.
Knowles LL, Richards CL. Importance of genetic drift during Pleistocene divergence as revealed by analyses of genomic variation. Mol Ecol. 2005;14(13):4023–32.
Lessa EP, Cook JA, Patton JL. Genetic footprints of demographic expansion in North America, but not Amazonia, during the Late Quaternary. Proc Natl Acad Sci. 2003;100(18):10331–4.
Soltis DE, Morris AB, McLachlan JS, Manos PS, Soltis PS. Comparative phylogeography of unglaciated eastern North America. Mol Ecol. 2006;15(14):4261–93.
Kearns AM, Joseph L, Toon A, Cook LG. Australia’s arid-adapted butcherbirds experienced range expansions during Pleistocene glacial maxima. Nat Commun. 2014;5:3994.
Neaves LE, Zenger KR, Prince RIT, Eldridge MDB, Cooper DW. Landscape discontinuities influence gene flow and genetic structure in a large, vagile Australian mammal, Macropus fuliginosus. Mol Ecol. 2009;18(16):3363–78.
Chan YL, Schanzenbach D, Hickerson MJ. Detecting concerted demographic response across community assemblages using hierarchical approximate Bayesian computation. Mol Biol Evol. 2014;31(9):2501–15.
Dolman G, Joseph L. A species assemblage approach to comparative phylogeography of birds in southern Australia. Ecol Evol. 2012;2(2):354–69.
Dolman G, Joseph L. Evolutionary history of birds across southern Australia: structure, history and taxonomic implications of mitochondrial DNA diversity in an ecologically diverse suite of species. Emu. 2015;115(1):35–48.
Beaumont MA, Rannala B. The Bayesian revolution in genetics. Nat Rev Genet. 2004;5(4):251–61.
Beaumont MA. Approximate Bayesian computation in evolution and ecology. In: Futuyma DJ, Shafer HB, Simberloff D, editors. Annual review of ecology, evolution, and systematics, Vol 41. Palo Alto: Annual Reviews; 2010. p. 379–406.
Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13(4):729–44.
Norman JA, Blackmore CJ, Rourke M, Christidis L. Effects of mitochondrial DNA rate variation on reconstruction of Pleistocene demographic history in a social avian species, Pomatostomus superciliosus. PLoS One. 2014;9(9):e106267.
Pavlova A, Amos JN, Joseph L, Loynes K, Austin JJ, Keogh JS, Stone GN, Nicholls JA, Sunnucks P. Perched at the mito-nuclear crossroads: divergent mitochondrial lineages correlate with environment in the face of ongoing nuclear gene flow in and Australian bird. Evolution. 2013;67(12):3412–28.
Morales HE, Pavlova A, Joseph L, Sunnucks P. Positive and purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol Ecol. 2015;24(11):2820–37.
Johnstone R, Storr G. Handbook of western Australian birds, vol. II – passerines (blue-winged pitta to goldfinch). Perth: Western Australian Museum; 2004.
Toon A, Austin JJ, Dolman G, Pedler L, Joseph L. Evolution of arid zone birds in Australia: leapfrog distribution patterns and mesic-arid connections in quail-thrush (Cinclosoma, Cinclosomatidae). Mol Phylogenet Evol. 2012;62(1):286–95.
Schodde R, Mason I. Directory of Australian birds: passerines. Melbourne: CSIRO Publishing; 1999.
Carstens BC, Pelletier TA, Reid NM, Satler JD. How to fail at species delimitation. Mol Ecol. 2013;22(17):4369–83.
Fujita MK, Leaché AD, Burbrink FT, McGuire JA, Moritz C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol Evol. 2012;27(9):480–8.
Leaché AD, Fujita MK. Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Proc R Soc Lond B Biol Sci. 2010;277:3071–77. doi: 10.1098/rspb.2010.0662.
Guillot G, Mortier F, Estoup A. GENELAND: a computer package for landscape genetics. Mol Ecol Notes. 2005;5:703–11.
Hey J. Isolation with migration models for more than two populations. Mol Biol Evol. 2010;27(4):905–20.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Dolman G, Moritz C. A multi-locus perspective on refugial isolation and divergence in rainforest skinks (Carlia). Evolution. 2006;60(3):573–82.
Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73(5):1162–9.
Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–89.
Garrick R, Sunnucks P, Dyer R. Nuclear gene phylogeography using PHASE: dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation. BMC Evol Biol. 2010;10(1):1–17.
Guillot G, Estoup A, Mortier F, Cosson JF. A spatial statistic model for landscape genetics. Genetics. 2005;170:1261–80.
Francois O, Durand E. The state of the field: Spatially explicit Bayesian clustering models in population genetics. Mol Ecol Resour. 2010;10(5):773–84.
Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167(2):747–60.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Hudson RR, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA-sequences. Genetics. 1985;111(1):147–64.
Hudson RR, Kreitman M, Aguadé M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116(1):153–9.
McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351(6328):652–4.
Egea R, Casillas S, Barbadilla A, Egea R, Casillas S, Barbadilla A. Standard and generalized McDonald-Kreitman test: a website to detect selection by comparing different classes of DNA sites. Nucleic Acids Research 36 (Web Server). 2008:W157-W162.
Lerner Heather RL, Meyer M, James Helen F, Hofreiter M, Fleischer Robert C. Multilocus resolution of phylogeny and timescale in the extant adaptive radiation of Hawaiian honeycreepers. Curr Biol. 2011;21(21):1838–44.
Higgins PJ. Handbook of Australian, New Zealand and Antarctic birds. Melbourne: Oxford University Press; 1999.
Higgins PJ, Peter JM. Handbook of Australian, New Zealand and Antarctic birds. Melbourne: Oxford University Press; 2002.
Higgins PJ, Peter JM, Steele WK. Handbook of Australian, New Zealand and Antarctic birds. Melbourne: Oxford University Press; 2001.
Sousa VC, Grelaud A, Hey J. On the nonidentifiability of migration time estimates in isolation with migration models. Mol Ecol. 2011;20(19):3956–62.
Ellegren H. The evolutionary genomics of birds. In: Futuyma DJ, editor. Annual review of ecology, evolution, and systematics, Vol 44. 2013. p. 239–59.
Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22(5):1185–92.
Grant WS. Problems and cautions with sequence mismatch analysis and Bayesian skyline plots to infer historical demography. J Hered. 2015;106(4):333–46.
Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol Biol. 2008;8(1):289.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Heller R, Chikhi L, Siegismund HR. The confounding effect of population structure on Bayesian skyline plot inferences of demographic history. PLoS One. 2013;8(5):e62992.
Ford JR. Speciation in Australian birds adapted to arid habitats. Emu. 1974;74(3):161–8.
Keast JA. Bird speciation on the Australian continent. Bull Mus Comp Zool. 1961;123:303–495.
Schodde R. Origin, adaptation and evolution of birds in arid Australia. In: Barker WR, Greenslade PJM, editors. Evolution of the flora and fauna of arid Australia. Adelaide: Peacock Publications; 1982. p. 191–224.
Ford J. Geographical variation in Cinclosoma castanotum and its historical significance. Emu. 1981;81(4):185–92.
Joseph L, Wilke T. Lack of phylogeographic structure in three widespread Australian birds reinforces emerging challenges in Australian historical biogeography. J Biogeogr. 2007;34(4):612–24.
Kearns AM, Joseph L, Edwards SV, Double MC. Inferring the phylogeography and evolutionary history of the splendid fairy-wren Malurus splendens from mitochondrial DNA and spectrophotometry. J Avian Biol. 2009;40(1):7–17.
Cupper ML. Last glacial to Holocene evolution of semi-arid rangelands in southeastern Australia. The Holocene. 2005;15(4):541–53.
McLaren S, Wallace MW. Plio-Pleistocene climate change and the onset of aridity in southeastern Australia. Glob Planet Chang. 2010;71(1–2):55–72.
Wagstaff BE, Kershaw AP, O’Sullivan PB, Harle KJ, Edwards J. An early to middle Pleistocene palynological record from the volcanic crater of Pejark Marsh, western plains of Victoria, southeastern Australia. Quat Int. 2001;83–85:211–32.
Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27(3):570–80.
Amei A, Smith BT. Robust estimates of divergence times and selection with a Poisson random field model: a case study of comparative phylogeographic data. Genetics. 2014;196(1):225–33.
Carling MD, Brumfield RT. Integrating phylogenetic and population genetic analyses of multiple loci to test species divergence hypotheses in Passerina buntings. Genetics. 2008;178(1):363–77.
McLean AJ, Toon A, Schmidt DJ, Joseph L, Hughes JM. Speciation in chestnut-shouldered fairy-wrens (Malurus spp.) and rapid phenotypic divergence in variegated fairy-wrens (Malurus lamberti): a multilocus approach. Mol Phylogenet Evol. 2012;63(3):668–78.
Bennett AF, Ford LA. Land use, habitat change and the conservation of birds in fragmented rural environments: a landscape perspective from the Northern Plains, Victoria, Australia. Pac Conserv Biol. 1997;3(3):244–61.
Saunders DA. Australian developments in conservation evaluation changes in the avifauna of a region, district and remnant as a result of fragmentation of native vegetation: the wheatbelt of Western Australia. A case study. Biol Conserv. 1989;50(1):99–135.
Szabo JK, Vesk PA, Baxter PWJ, Possingham HP. Paying the extinction debt: woodland birds in the Mount Lofty Ranges, South Australia. Emu. 2011;111(1):59–70.
Luly JG. Holocene palaeoenvironments near lake Tyrrell, semi-arid northwestern Victoria, Australia. J Biogeogr. 1993;20(6):587–98.
Sniderman JMK, Jordan GJ, Cowling RM. Fossil evidence for a hyperdiverse sclerophyll flora under a non–Mediterranean-type climate. Proc Natl Acad Sci. 2013;110(9):3423–8.
Dhami KK, Joseph L, Roshier DA, Heinsohn R, Peters JL. Multilocus phylogeography of Australian teals (Anas spp.): a case study of the relationship between vagility and genetic structure. J Avian Biol. 2013;44(2):169–78.
Dhami K, Joseph L, Roshier DA, Peters JL. Recent speciation and elevated Z-chromosome differentiation between a sexually dichromatic and monochromatic pair of Australian teals (Anatidae: Anas). J Avian Biol. 2016;47:92–102.
Bragg JG, Potter S, Bi K, Moritz C. Exon capture phylogenomics: efficacy across scales of divergence. Mol Ecol Resour. 2016;16:1059–68. doi:10.1111/1755-0998.12449.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17(2):240–8.
Nadachowska-Brzyska K, Li C, Smeds L, Zhang G, Ellegren H. Temporal dynamics of avian populations during Pleistocene revealed by whole-genome sequences. Curr Biol. 2015;25(10):1375–80.
Lee JY, Edwards SV. Divergence across Australia’s Carpentarian Barrier: statistical phylogeography of the red-backed fairy wren (Malurus melanocephalus). Evolution. 2008;62(12):3117–34.
Backstroem N, Fagerberg S, Ellegren H. Genomics of natural bird populations: a gene-based set of reference markers evenly spread across the avian genome. Mol Ecol. 2008;17(4):964–80.
Sorenson MD, Ast JC, Dimcheff DE, Yuri T, Mindell DP. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol Phylogenet Evol. 1999;12(2):105–14.
Acknowledgements
We thank the Maralinga-Tjarutja People and Community and the Tjuntjuntjara People and Community for permission to collect on their lands. We thank Steve Donnellan, South Australian Museum, and Andy Austin, the University of Adelaide, for laboratory space and facilities, and Vicki Thomson for discussion. For tissue samples and help with field work, we thank Alex Drew, Robert Palmer, Gilbert Pfitzner and Ian Mason (Australian National Wildlife Collection, Canberra); Nate Rice (Academy of Natural Sciences, Philadelphia) and collection staff at the South Australian Museum and Western Australian Museum. Lynn Pedler assisted with field work. We thank eResearch SA for their support and time on High-performance computing facilities.
Funding
CSIRO Office of the Chief Executive funded a Postdoctoral Fellowship and laboratory costs for Gaynor Dolman when the laboratory work was performed.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the Dryad Repository: doi:10.5061/dryad.v033k and GenBank accession numbers: KM280401-KM280429 (mtDNA) and KX913359-KX913664 (autosomal nDNA).
Authors’ contributions
LJ and GD conceived the study. GD conducted the molecular laboratory work and analysed the data. GD wrote the manuscript with comments from LJ. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
In general tissues used in this study were from previously collected and registered museum specimens. Ethics permits were required for collecting in the Great Victoria Desert to supplement existing collections: i) CSIRO Human Nutrition project number 713-08/08; and CSIRO Sustainable Ecosystems Animal Ethics Committee permit 07-04. Scientific collecting permits for work in the Great Victoria Desert to supplement existing collections were granted by the South Australian Department of Environment, Water and Natural Resources and the Western Australian Department of Parks and Wildlife.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Museum registration and location details of specimens used in this study are listed. (PDF 187 kb)
Additional file 2: Figure S1.
Identification of genetic discontinuities using GENELAND: analyses of allelic data from 11 autosomal nuclear loci reveal two distinct genetic clusters either side of the Eyrean Barrier. Posterior densities for k = 1 to 4 clusters and maps of posterior probability to belong to cluster 1 or 2. (PDF 173 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dolman, G., Joseph, L. Multi-locus sequence data illuminate demographic drivers of Pleistocene speciation in semi-arid southern Australian birds (Cinclosoma spp.). BMC Evol Biol 16, 226 (2016). https://doi.org/10.1186/s12862-016-0798-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-016-0798-6