
Abstract
Background
Defining species units can be challenging, especially during the earliest stages of speciation, when phylogenetic inference and delimitation methods may be compromised by incomplete lineage sorting (ILS) or secondary gene flow. Integrative approaches to taxonomy, which combine molecular and morphological evidence, have the potential to be valuable in such cases. In this study we investigated the South African scarab beetle genus Pleophylla using data collected from 110 individuals of eight putative morphospecies. The dataset included four molecular markers (cox1, 16S, rrnL, ITS1) and morphometric data based on male genital morphology. We applied a suite of molecular and morphological approaches to species delimitation, and implemented a novel Bayesian approach in the software iBPP, which enables continuous morphological trait and molecular data to be combined.
Results
Traditional morphology-based species assignments were supported quantitatively by morphometric analyses of the male genitalia (eigenshape analysis, CVA, LDA). While the ITS1-based delineation was also broadly congruent with the morphospecies, the cox1 data resulted in over-splitting (GMYC modelling, haplotype networks, PTP, ABGD). In the most extreme case morphospecies shared identical haplotypes, which may be attributable to ILS based on statistical tests performed using the software JML. We found the strongest support for putative morphospecies based on phylogenetic evidence using the combined approach implemented in iBPP. However, support for putative species was sensitive to the use of alternative guide trees and alternative combinations of priors on the population size (θ) and rootage (τ 0 ) parameters, especially when the analysis was based on molecular or morphological data alone.
Conclusions
We demonstrate that continuous morphological trait data can be extremely valuable in assessing competing hypotheses to species delimitation. In particular, we show that the inclusion of morphological data in an integrative Bayesian framework can improve the resolution of inferred species units. However, we also demonstrate that this approach is extremely sensitive to guide tree and prior parameter choice. These parameters should be chosen with caution – if possible – based on independent empirical evidence, or careful sensitivity analyses should be performed to assess the robustness of results. Young species provide exemplars for investigating the mechanisms of speciation and for assessing the performance of tools used to delimit species on the basis of molecular and/or morphological evidence.
Similar content being viewed by others


Background
The identification and delimitation of species is one of the most crucial exercises in the assessment of biodiversity and in understanding the Tree of Life, because species occupy a central role in nearly all disciplines of biology. Species delimitation therefore has broad implications, from biological and ecological conservation, to comparative evolutionary analyses [1–4]. Despite the challenge and importance of defining species units, methods for delimiting species using independent sources of data (e.g., DNA and phenetic data) have only recently been proposed (e.g., [5–15]). Nevertheless, at least since Sneath and Sokal [16], there has been an extensive use of quantitative methods to infer similarity based on morphological traits. Broadly defined as “numerical taxonomy”, or phenetics, these methods have traditionally been used (and criticized) for inferring phylogenetic relationships (e.g., [17, 18]). However, integrative approaches to taxonomy shed new light on the utility of these methods, which have the potential to offer an independent, more reproducible way of inferring species limits [19].
In addition to controversy over the application of different species concepts and their impact for delimiting species [20], delimitation is expected to be especially challenging during the earliest stages of divergence, or speciation, when both molecular and morphological characters exhibit low levels of differentiation [21]. At this stage it can be extremely difficult to detect genetic isolation (i.e., the ultimate outcome of speciation) due to gene flow among populations and incomplete lineage sorting between species [22, 23]. Although molecular data can be useful for the rapid identification and delimitation of species, these processes can compromise the interpretation of the results. Incomplete lineage sorting – shared ancestral polymorphisms between species – can lead to perceived genetic similarity among phenotypically divergent species. Consequently, gene flow and incomplete lineage sorting can result in similar patterns among inferred gene trees [24–26]. To further complicate matters, introgressive hybridization – secondary gene flow between species – can also produce similar patterns among inferred gene trees (e.g., [27–29]).
A suite of new methods have been proposed that can incorporate incomplete lineage sorting in a multilocus framework for the estimation of species trees [30–33] and/or species delimitation [20, 33, 34]. Although these methods rely on the a priori assignment of individuals to pre-defined units (species or populations; [20]), they can be used to test explicit hypotheses of species delimitations. However, studies of recent radiations, or speciation in a young species, will be characterized by uncertain species designations, and are likely to remain challenging.
In contrast to DNA-based taxonomy, common practise for the traditional taxonomic treatment of taxa is an assessment of the organism’s entire morphology. In most groups of insects this includes detailed examination of the copulation organs, which often undergo rapid morphological divergence, driven by sexual selection [35]. However, quantitative data on insect genitalia are rarely obtained for the purposes of integrative taxonomy, and so methods for combining this type of morphological information with molecular data are still underdeveloped [19]. Previously, the only available methods for delimiting species on the basis of morphology were clustering approaches [8, 9, 36, 37]. Unfortunately, these methods quickly loose power when too many species are included, or when dealing with specimens whose closest phylogenetic relatives are unknown [7, 14]. Here we use morphometric and molecular data in an integrative framework, to delimit species in the scarab beetle genus Pleophylla Erichson, 1847. Following the recommendation of Carstens et al. [6], we implemented a suite of methods, including a recently developed approach that incorporates continuous morphological trait data with the multispecies coalescent [14, 34].
Pleophylla is a highly conspicuous genus, found only in isolated parts of the South African escarpment and the East African highlands. The genus belongs to the tribe Sericini (Coleoptera: Scarabaeidae), a highly diverse clade of herbivorous beetles with nearly 4,000 described species. The adults feed polyphagously on a variety of angiosperms, while the larva feed on humus and plant roots in the upper soil layers. Morphological and molecular evidence has shown that the genus belongs to one of the most ancestral-branching lineages of the Sericini, together with its presumptive sister group, Omaloplia, in the eastern Mediterranean [38, 39]. Members of the genus exhibit extreme homogeneity in external morphology, and identification of species usually relies on examination of the male genitalia – a trait used to commonly distinguish between homogenous species of insects [40], including most members of the tribe Sericini [41]. Current taxonomic classification recognises only three valid species ([42]; globalspecies.org/ntaxa/2359831; accessed Dec 13, 2015), however, an extensive survey and taxonomic revision of museum collections has identified 24 distinct morphospecies [43] (Eberle J, Beckett M, Özguel-Siemund A, Frings J, Fabrizi S, Ahrens D. Afromontane forests hide nineteen new species of ancient Pleophylla chafers (Coleoptera: Scarabaeidae): phylogeny and taxonomic revision, in preparation). The aim of our study was to provide a primer for the clarification of the taxonomy of this group, and to explore power and limitations of morphological, molecular and combined approaches to species delimitation in an integrative framework for an apparent “complex” case study.
Methods
Taxon sampling and molecular data collection
A total of 110 individuals of eight putative morphospecies of the genus Pleophylla were collected from eight localities in South Africa (Additional file 1: Table S1-S2; Fig. 1). So far, all known species are endemic to South Africa and represent a limited selection of the morphological diversity of Pleophylla (Eberle J, Beckett M, Özguel-Siemund A, Frings J, Fabrizi S, Ahrens D. Afromontane forests hide nineteen new species of ancient Pleophylla chafers (Coleoptera: Scarabaeidae): phylogeny and taxonomic revision, in preparation). Four of these species have not been described yet, therefore we refer to all putative morphospecies using the same numerical format throughout the text for consistency. Omaloplia nigromarginata and O. ruricola from the putative sister lineage of Pleophylla [38] were included as outgroup taxa. We assessed support for the monophyly of putative morphospecies using standard molecular markers – the nuclear ribosomal rRNA 28S gene, the nuclear internal transcribed spacer 1 (ITS1), and the mitochondrial cytochrome oxidase subunit 1 (cox1) and 16S rRNA (rrnL) genes. Details of DNA extraction, sequencing, alignment and model selection are provided in the Additional file 1.

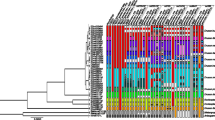
a Maximum likelihood (RAxML) tree of Pleophylla for the combined molecular dataset. Specimens are colored according to morphospecies (Additional file 1: Table S1). Branch length corresponds substitutions per site. Support values for ML and Bayesian posterior probabilities are shown next to branches in grey (RAxML) or indicated below (PhyML/MrBayes). ITS1 GMYC clusters are indicated by an asterisk (*). b Map of South African sampling localities (Additional file 1: Table S2). c Bayesian species tree obtained using *BEAST. Clade posterior probabilities are indicated next to branches. Confidence intervals (grey bars) show the upper limits of the 95 % HPDs obtained using a divergence rate for cox1 of 2 % My-1, and the lower limits obtained using a rate of 4 %. Mean node ages arbitrarily correspond to the mean estimates obtained using a rate of 2 % My-1 (Additional file 1: Table S6). A cloudogram of 10,000 posterior samples shows the uncertainty in the inferred species tree, obtained using the program DensiTree [65]; different colours (blue, red, green) correspond to each consensus topology in the total set of trees
Morphometric analysis
The partial outline of the male’s left paramere (part of the intromittent genital organs, in dorsal view) (Additional file 1: Figure S1) was digitized from images captured on a microscope. The partial outline was extracted from 68 male specimens where the paramere was well preserved. The outlines were resampled as a set of 150 semi-landmarks using tpsDig 2.1 [44]. Standard eigenshape analysis [45, 46] was performed in Eigenshape 2.6, as implemented in morpho-tools [47]. Of the 67 eigenshape axes produced, further analysis was performed on the four eigenshape axes that together explained 75 % of the variation in the samples. Based on these informative eigenshape axes we performed a canonical variate analysis (CVA), grouping the samples according to the morphospecies assignments.
Model-based hierarchical clustering [37, 48] was applied to identify groups of individuals that resemble each other, independent of other evidence or a priori assignments, using the R package mclust 4.4 [36, 37]. The function mclust was used to evaluate the fit of all available clustering models to the morphometric data that explained 75 % (eigenshape axes 1–4) and 95 % (eigenshape axes 1–14) of total paramere shape variance. This method uses expectation maximization (EM) to estimate the maximum likelihood of alternative multivariate mixture models that describe shape variation in the morphometric data [49, 50], and estimates the optimal number of clusters based on the Bayesian Information Criterion (BIC) [51]. All models were evaluated for a predefined number of 1 to 20 clusters and the best-fit result was used for further analyses.
To assess the fit of the a priori morphospecies assignments and the hierarchical clusters found using mclust to the data, we performed a linear discriminant analysis based on the respective specimen groupings and calculated the probability of group membership for each individual. This was done using the R package MASS 7.3.35 [52]. The prior probability that a specimen belonged to a given group was set to be equal for all individuals and groups.
Finally, to investigate the impact of phylogeny on the inferred morphospace, the RAxML tree topology (based on the partitioned combined molecular dataset) was projected onto the paramere morphospace (eigenshape axes 1 and 2) using the function phylomorphospace in the R package phytools [53]. This function estimates the positions of the ancestral nodes using a maximum likelihood approach [53]. In addition, a three-dimensional version of this plot was produced based on eigenshape axes 1, 2 and 3 using the function phylomorphospace3d. The code was modified to make coloration for species group affiliation possible.
Phylogenetic analysis
Phylogenetic analyses of individual and combined markers were performed using likelihood and Bayesian methods. Each analysis was run with the substitution model and partitions selected using PartitionFinder [54] (Additional file 1: Table S3). Unpartitioned maximum likelihood analysis was performed using PhyML 3.0 [55], and partitioned maximum likelihood analysis was performed using RAxML 7.3 [56, 57]. Bayesian phylogenetic analysis was performed using MrBayes 3.1.2 [58]. The default prior on branch lengths implemented in MrBayes can sometimes lead to spuriously large estimates of internal branch lengths [59, 60]. Because the GMYC approach to species delineation is sensitive to estimates of branch lengths, we ran four sets of analyses using an exponential prior on the branch lengths with mean = 0.1 (default), 0.05, 0.01 or 0.005 substitutions/site.
Bayesian species tree estimation
The multispecies coalescent was implemented in *BEAST 1.75 [31, 61, 62] to co-estimate the species tree, individual (cox1 and ITS1) gene trees and divergence times. The less informative ribosomal markers were excluded because analysis in *BEAST that included rrnL and 28S failed to converge, despite extensive efforts to improve convergence diagnostics. Putative morphospecies were used to define taxonomic units a priori – all 14 female individuals, for which there was ambiguity regarding a priori species assignment, were excluded from the analyses (Additional file 1: Table S1). The mean substitution rate of cox1 was fixed, clock model parameters were unlinked across genes, and the rate of ITS1 was estimated relative to cox1. Estimates for the substitution rate of cox1 among insect species vary substantially across different studies, and are dependent on a large number of variables [63, 64]. We therefore applied a range of mean branch rates, in five independent sets of analyses (2, 2.5, 3, 3.5 or 4 % My−1). The resulting posterior sample of species trees was additionally visualized with DensiTree [65]. Further details of all phylogenetic analyses, including prior parameter and chain settings, are provided in the Additional file 1.
Distinguishing incomplete lineage sorting from hybridization
To assess whether low genetic variation observed among morphospecies could be attributed to incomplete lineage sorting, we used the posterior predictive checking approach developed by Joly et al. [66] and implemented in the software JML [67]. This approach uses simulated datasets of gene trees and sequence alignments generated under a coalescent model that assumes no migration (or hybridisation) for a given species tree. The proportion of simulated datasets for which the minimum pairwise distance is lower than the observed, can be interpreted as the posterior probability (P) that the model is correct. A small P value therefore suggests that a model that assumes no hybridization does not fit the data well (e.g., the observed minimum genetic distances are lower than expected). To account for uncertainty, simulations were performed for individual partitions using 10,000 trees from the posterior distribution of species tree output by *BEAST, which include estimates of population size and branch lengths. Further details of the simulations are provided in the Additional file 1.
DNA-based species delimitation
For single marker species delimitation (cox1 and ITS1) we used four widely implemented approaches: statistical parsimony analysis [68], automated barcode gap detection (ABGD) [69], the generalized mixed Yule-coalescent (GMYC) model [12, 70, 71], and the Poisson tree processes (PTP) model [72]. Outgroup species (Omaloplia) and specimens with duplicate haplotypes were pruned from the dataset (or tree) prior to analysis, otherwise some methods have been shown to produce false positives [73].
Haplotype networks for each individual marker were generated using statistical parsimony analysis [68] implemented in TCS 1.2 [42]. Statistical parsimony analysis partitions the data into networks of closely related haplotypes connected by changes that are non-homoplastic with a 95 % probability; if applied to mtDNA, the inferred networks have been found to be largely congruent with Linnaean species [74]. The GMYC model [12, 70, 71] was used to estimate species boundaries with the trees obtained from MrBayes and RAxML using in the R package splits [70], with single and multiple threshold options. This method is based on the phylogenetic species concept and identifies species clusters by recognising the apparent increase in the branching rate from interspecific diversification to population-level coalescence, and defining the threshold based on an ultrametric tree. Trees were converted to ultrametric using PATHd8 [75] and the penalized likelihood method implemented in r8s 1.7 [76], with the optimal smoothing parameter selected using the cross-validation procedure. The age of the ingroup was assigned an arbitrary age of 1, and the resultant trees were fully resolved using TreeEdit 1.0 [77] using an arbitrary branch length of 4 x 10−6. Finally, we estimated uncertainty in the number of GMYC species clusters based on the Akaike Information Criterion (AIC), using the method outlined in [78]. This approach uses a modified AIC score, corrected for sample size (AICc), to assess the relative support for alternative (single and multiple threshold) models, versus the maximum likelihood model, and the null model (no change in the branching rate). Akaike weights (the relative support for each model) are assigned to each model based on the AICc scores. Model-averaged estimates of the number of GMYC species are obtained from the models withinδAICc = 2. The phylogenetic species concept also underlies the Poisson tree processes (PTP) model for species delimitation [72]. However, in contrast to the GMYC approach, the PTP infers speciation events based on a shift in the number of substitutions at internal nodes. We employed the maximum likelihood variant of PTP using the RAxML trees. For the ABGD approach we used the online version (last modified on 10/29/2015 and accessed on 01/23/2016, http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html, [69]). This method is based on the assumption that divergence among organisms belonging to the same species will be less than the divergence observed among organisms of different species. The first significant gap in the distribution of sequence distances beyond intraspecific sequence divergence can thus be used to infer operational taxonomic units (OTU) that may be related to species (e.g., [79]). ABGD analyses were performed on matrices of pairwise sequence divergence, calculated for each marker using MEGA (v6.06, [80]). Distances were corrected using the best fitting substitution models. Prior maximum divergence of intraspecific diversity was set to 0.01, which has previously been demonstrated to recover species accurately [69].
Finally, the results of competing approaches to species delimitation were compared using the “entities counts” (i.e., inferred species counts) and the match ratio = 2*Nmatch/(Ni + Nmorph), where Nmatch is the number of species with exact matches (i.e., all specimens of a given morphospecies – and only those – belong to a single GMYC entity) and Ni and Nmorph are the number of inferred molecular operational taxonomic units (MOTUs) and morphospecies, respectively [73]. If there is complete congruence between the MOTU entities and the morphospecies the match ratio = 1, otherwise the ratio will be < 1.
Total-evidence species delimitation
We assessed support for the a priori morphospecies assignments using a total-evidence-based Bayesian approach, implemented in the programs iBPP 2.1.2 [14] and BPP 3.0, [20, 34]. Briefly, this method uses a multispecies coalescent model to assess competing hypotheses of species delimitations, allowing for conflict between gene and species trees. The results are conditioned on a user specified guide tree and depend on estimates of the species divergence times (τ) and population sizes (θ). Individuals are assigned to independent populations and alternative delimitation hypotheses are proposed by collapsing one or more internals nodes in the guide tree. In the original implementation, the likelihood calculation is based on molecular data [34], while iBPP includes an extension of the model that allows continuous trait data to be included in the likelihood calculation [14]. This latter approach therefore enables both molecular and morphological data to be combined in the assessment of a priori species assignments.
It has been demonstrated that the results of this method can be sensitive to both prior parameter and guide tree choice [81]. For example, for high values of θ the model tends to (over-) split species, and for low values of θ the model tends to lump species together. To assess the robustness of our results, we compared the results obtained under variable combinations of the specified priors on the root age (τ 0 ) and the population mutation rate (θ) (Table 1). To assess the influence of the guide tree, we compared the results obtained using three alternative input trees: (a) the topology estimated using *BEAST, (b) the topology estimated from the concatenated DNA matrix using RAxML/MrBayes, (c) a modified version of the *BEAST topology based on morphological similarity among species (Additional file 1: Figure S2). All combinations of prior parameter (Table 1) and guide tree choices were performed in iBPP (a) without data, to evaluate the impact of the priors, and using the following three datasets: (b) molecular data only, (c) morphometric data only, and (d) molecular and morphometric data. The analysis sometimes got stuck in a single species model, resulting in poor overall convergence, and so all analyses were repeated 10 times with different random seeds to ensure stability of the results.
In an additional set of analyses, we implemented unguided species delimitation using the program BPP [20]. This method accounts for uncertainty in the guide tree, by proposing changes to the species tree topology using nearest-neighbour interchange (NNI), as well as proposing changes to species assignments. Morphometric data cannot be analysed in BPP, so this analysis was performed for the molecular dataset only. The analyses were performed using the above combinations of priors and initial guide tree choices.
To explore the impact of distinct single-marker genotypes within the same morphospecies, in combination with the morphological trait data, we also analysed an additional guide tree with guided and unguided BPP, in which sp10 was specified as two species entities (This split received strong support in several single marker delimitations, see results).
Results
Phylogenetic analysis and the monophyly of morphospecies
Phylogenetic analysis of independent and combined datasets using different approaches and parameter choices (PhyML, RAxML, and MrBayes) produced overall similar topologies (Fig. 1, Additional file 1: Figures S3-S5). Changing the branch length prior implemented in MrBayes had no impact on the inferred topology but had a large impact on tree length (the sum of branch lengths) (Additional file 1: Table S4). Analysis of different datasets (mitochondrial, nuclear or combined) mainly differed in their degree of tree resolution, and the level of support for the monophyly of individual morphospecies and/or interspecific relationships. There is remarkably low interspecific molecular variation observed across the entire genus. The trees produced using the ribosomal markers (rrnL and 28S) were poorly resolved. The cox1 data provided better resolution and supported the monophyly of two out of eight putative morphospecies. ITS1 provided the best resolution and supported the monophyly of all but two morphospecies (sp01 and sp02) (Additional file 1: Figure S3-S5).
The topology obtained using the combined dataset that included all four markers was identical to the ITS1 gene tree (Fig. 1), but support values for most nodes were greater than those obtained using individual genes. In the combined analyses of all four markers, the monophyly of all putative morphospecies was strongly supported with the aforementioned exception. Morphospecies sp01 and sp02 were never recovered as monophyletic, although these groups occupied distinct areas of the morphospace in the morphometric analysis of the genitalia (Fig. 2, Additional file 1: Figure S1, Additional file 2).

Plot of the 2D phylomorphospace using the RAxML tree topology (based on the partitioned combined molecular dataset) projected onto the paramere morphospace explained by eigenshape axes 1 and 2. Colors refer to Fig. 1
The Bayesian species tree estimated using *BEAST for the combined cox1 and ITS1 dataset resulted in strong support for the interspecific relationships estimated using the cox1 data, rather than the ITS1 data. Although the species tree topology differed to that obtained using alternative phylogenetic methods (PhyML, RAxML and MrBayes), the individual gene trees (for cox1 and ITS1) obtained using *BEAST were not different. The age of the most recent common ancestor of the sampled members of the genus, was estimated to be 2.64 – 35.97 and 3.69 – 17.88 Mya, based on the 95 % highest posterior density intervals for the slowest and fastest cox1 substitution rates (2 and 4 % My−1), respectively (Additional file 1: Table S5). The use of a higher cox1 substitution rate produced younger and, unexpectedly, more precise posterior age estimates. The ages for the two youngest divergence events (sp01 + sp02 and sp06 + sp10) were estimated to be no older than 0.17 Mya and 0.65 Mya, respectively (Additional file 1: Table S6).
Evidence of hybridization was assessed using the posterior predictive checking approach as implemented in the software JML (Joly, 2012), based on the minimum pairwise sequence distances among morphospecies for each marker partition (cox1 [P1 vs. P2 vs. P3], and ITS1), and the resulting posterior probability (P) of observing these distances under the multispecies coalescent model assuming no hybridization (Additional file 1: Table S7). In all cases the observed pairwise distances between individuals of all morphospecies were not lower than expected at the 5 % level (P > 0.05), given the null model (the coalescent with no migration or hybridization) across all partitions (cox1 P1 and P2, P > 0.1; P3, P > 0.05; ITS1, P > 0.2). The distances observed between individuals of the two species pairs that could not be resolved using cox1 (sp06 + sp10) or both cox1 and ITS1 (sp01 + sp02) were not lower than expected for either marker (i.e., sp06 + sp10, P > 0.2; sp01 + sp02, P > 0.6). The tests performed suggest that incomplete lineage sorting is sufficient to explain the observed genetic variation (although mitochondrial partition P3 produced anomalous results for sp09 and sp11, see Additional file 1: Table S7).
Molecular tree- and character-based species delimitation
We investigated DNA-based species delimitation and associated uncertainty using (i) statistical parsimony, (ii) the GMYC model, (iii) the PTP model, and (iv) ABGD approach. The analyses using the rrnL and 28S data did not provide support for any of the putative morphospecies (results not shown). Of the 13 resulting cox1 networks, three matched exclusively a single putative morphospecies. ITS1 networks provided a closer correspondence to the morphospecies. Of the 9 ITS1 networks, four matched exclusively a single putative morphospecies: sp09, sp11, sp12 and spX2. Individuals of morphospecies sp06 shared two networks, and individuals of morphospecies sp10 shared two networks. Individuals of morphospecies sp01 and sp02 shared a single network. Together these results suggest that there is a higher degree of incomplete lineage sorting among cox1 than ITS1, and that species sp01 and sp02 cannot be distinguished on the basis of the molecular markers used here.
The GMYC results obtained using cox1 were very sensitive to the input tree, but there were no obvious differences in the GMYC output that could be attributable to the trees generated using MrBayes versus RAxML, or PATHd8 versus r8s (Additional file 1: Table S8). Bayesian trees with longer branch lengths tended to result in more GMYC entities (species clusters + singletons), but not ubiquitously. Consequently, the cox1 trees produced very variable results. In most cases several (up to 8) models contributed to a majority of the Akaike weight (>0.5), suggesting that no single model best represented the data. Accounting for uncertainty in model selection resulted in the number of entities ranging between 3.00 (σ2 = 0) and 16.54 (σ2 = 0.89), depending on the input tree; these GMYC units were widely incongruent with the a priori morphospecies assignments (further details therefore not shown here). There was less variation in the GMYC results obtained using the ITS1 trees – the single threshold models were always preferred to the multiple threshold models. In the majority of cases only one single threshold model was found within δAICc = 2, suggesting that the preferred model provided an appreciably better fit to the data than the alternatives. The ITS1 data resulted in a minimum of 8 (σ2 = 0) and a maximum of 10.99 (σ2 = 4.05) entities, depending on the input tree. In 8 out of 10 cases, the preferred model resulted in eight entities, corresponding to morphospecies sp01 + sp02, sp06, sp09, sp11, sp12, spX2, and two clusters of morphospecies sp10.
In general, congruence between the inferred MOTUs and the morphospecies was more dependent on marker choice than species delimitation method (Table 2, Additional file 2: Table S9). For cox1 the number of MOTUs ranged from 7 (PTP) to 13 (GMYC), while the analyses based on ITS1 resulted in 8 (GMYC, PTP, ABGD), 9 (TCS) and 10 (GMYC) entities. The PTP and ABGD analyses largely confirmed the results of the GMYC model for the ITS1 data; five of the eight MOTUs were fully congruent with the morphospecies (sp11, spx2, sp9, sp6, sp12). Finally, the match ratios obtained for cox1 were consistently lower (0.27-0.42) than those obtained using ITS1 (0.47-0.63) (Table 2).
Morphometric evidence for species delimitation
We first assessed quantitative support for the eight putative morphospecies assignments among Pleophylla based on an open shape outline of the left paramere of the male genitalia, using (i) standard eigenshape analysis, (ii) canonical variate analysis (CVA), (iii) hierarchical clustering, and (iv) linear discriminant analysis. The first four eigenshape axes represented 75 % of the cumulative variation of the outline shape (Additional file 1: Table S10, Figure S6). Eigenaxis 1, 2, 3 and 4 represented 51.5 %, 15.6 %, 6.8 % and 6.0 % of the variation, respectively. The first 14 eigenshape axes account for 95 % of the cumulative variation. The plots of the 2D and 3D phylomorphospace (Fig. 2, Additional file 5) showed clear separation between all but one of the morphospecies, with no intermediate states between the morphospecies. The only exception was sp12, which overlapped in morphospace with sp02. CVA on eigenshape axes 1–4 (Additional file 1: Figure S1) revealed a clear distinction between five of the eight morphospecies (sp01, sp02, sp06, sp10 and sp11), with the exception of those for which only one or two specimens were available for analysis (sp09, sp12 and spX2). This was in contrast to the DNA-based tree topology and species delimitation, where specimens of two species pairs (sp01 + sp02, and sp06 + sp10) could not be distinguished based on the analysis of cox1 and/or ITS1.
Hierarchical model-based cluster analysis [37] can identify unique morphological clusters of individuals without requiring a priori species assignments (e.g., [8]). The results of this analysis were extremely sensitive to the model choice (Fig. 3). Different mixture models favoured strikingly different numbers of clusters (e.g., 9, 7, 5, and 3 clusters were found for eigenshape axes 1–4 under different models) (BIC, Fig. 3a). The best model obtained for eigenshape axes 1–4 (the ellipsoidal, equal shape model; VEV) resulted in 3 clusters, but only morphospecies sp11 and sp10 (with the exception of one individual) were recovered as independent unique clusters. The best-fit model obtained for eigenshape axes 1–14 (the diagonal, varying volume, equal shape model; VEI) resulted in 12 clusters (Fig. 3b), with all morphospecies recovered in more than one group, with the exception of the singletons and sp6; the latter was recovered together with individuals of sp9 and spX2.

Species estimates of hierarchical clustering and confidence evaluation of a priori defined morphospecies and morphoclusters by LDA. Columns show results for 75 % and 95 % of total variation in the morphometric data. a, b: Choice of the best fitting cluster model by BIC. Reassignment probabilities to the clusters from hierarchical clustering with individuals ordered by (c, d) clusters and (e, f) by a priori defined morphospecies, and (g, h) reassignment probabilities to a priori defined morphospecies. Bars below plots C-H indicate prior group assignment for LDA, bars above plots E and F indicate affiliation to a priori defined morphospecies. Individuals in plots E-H are ordered identically
Linear discriminant analysis (LDA) with respect to the a priori defined morphospecies recovered one of the eight species (sp11, 100 % of individuals) based on eigenshape axes 1–4 (Fig. 3, Additional file 1: Table S11). Two of the eight morphospecies were recovered with the LDA based on eigenshape axes 1–14 (sp10, sp11, 100 % of individuals; the remaining morphospecies were recovered for 50–92 % of individuals). LDA with respect to groups identified by the model-based cluster analysis recovered all three clusters correctly based on eigenshape axes 1–4 (Fig. 3, Additional file 1: Table S12). Finally, LDA on clusters from the second analysis based on eigenshape axes 1–14 recovered all but two of the groups for 100 % of individuals.
Bayesian species delimitation
The total-evidence approach to Bayesian species delimitation [14, 34] provided strong support for the a priori defined morphospecies, however, for independent data types (molecular versus morphometric), the results were sensitive to the priors on the root age (τ 0 ) and population size (θ) parameters (Fig. 4, Additional file 3: Table S13). Broadly, posterior probabilities (i.e., support for species delimitations) increased in the integrative analyses that combined molecular and morphological trait data (Fig. 4). While results were sensitive to both the choice of τ 0 and θ, the choice of θ seemed to be more influential. The most consistent pattern that emerged is that low values of θ sometimes lead to low support for species delimitations. Species remained relatively well supported with high prior values of τ 0 . When the model was run under the prior (e.g., without data), with exception of the deepest divergences (sp09), the model did not result in any support (P > 0.95) for the a priori species assignments. This indicates that although the results were sensitive to the priors, the data contained informative signal.

Mean posterior probabilities of Bayesian species delimitations from 10 repeated runs with commonly used priors. Means inferred under 9 different θ and τ prior combinations are color-coded in 3x3 boxes on each putative speciation split of the guide trees. The arrows in the legend point to the direction of more conservative prior choices. Columns from left to right: 3 alternative guide tree topologies from *BEAST, ML and MrBayes analyses, and a modified *BEAST topology based on morphological similarity of the species; rows: analyses using (a) no data (prior only), (b) molecular data, (c) morphometric trait data, and (d) both data sources. The colours of the large 3x3 inset boxes indicate the number of repeat-analyses that were stuck in the one species model. Gamma distribution densities of θ and τ priors 1–3 are depicted in the bottom left corner. Dashed lines indicate the respective distribution means
Based on morphometric data alone, the divergence between sp01 + sp02 and sp12 in tree A was strongly supported (P > 0.95), however, the combination with low θ values reduced support at these nodes (Fig. 4). The analysis based on molecular data alone provided overall support for the a priori morphospecies assignments. Exceptions occur for all nodes given low values of θ with all data sets. For example, sp01 and sp02 were strongly supported in analyses with higher values of θ (P > 0.95), while there was low support for this divergence in analyses with the lowest value of θ (P < 0.32). The delimitation between sp02 and sp12 (tree C) was the only split that consistently received low support under all θ prior values and with all data sets.
As expected, the results were also sensitive to the guide tree choice. For example, when sp02 and sp12 were specified as belonging to separate groups of species, they were always strongly supported with high posterior probabilities (tree A, B). However, when the guide topology was modified to accommodate the observed high morphological similarity between sp02 and sp12 (guide tree C), they were almost never recovered as independent species (Fig. 4). Interestingly, none of the a priori defined species gained high support for all prior combinations across all guide trees, even using the integrative total evidence approach (Fig. 4d).
The unguided analyses (molecular data only) that applied nearest neighbor interchanges (NNI) to the initial guide tree topologies largely confirmed the results of the guided (iBPP) analyses. While the initial guide tree and the choice of the τ 0 prior did not alter the results, the choice of the θ prior had strong influence on the posterior probabilities of the speciation splits. All a priori defined morphospecies were well supported under θ 1 and θ 2 (Table 1) however, under the narrow and small θ 3 prior, in particular sp1 and sp2, but also sp1, sp10, sp12, sp2, and sp6, were lumped into one species (Additional file 4: Table S14 and Additional file 5: Table S15).
In the final set of analyses, in which sp10 was specified as two separate entities, corresponding to two distinct genotypes (Additional file 1: Figure S7), this split was not supported based on the analysis of the morphometric data alone, as expected. However, this split received strong support based on the analysis of both the molecular only and combined datasets (Additional file 1: Figure S7).
Discussion
Congruence between single DNA markers and morphometric evidence
Using a wide range of morphometric and phylogenetic tools, we tested for congruence between morphological, molecular, and integrative approaches (i.e., iterative sensu [19]) to species delimitation in the chafer beetle genus Pleophylla. Morphometric analysis (eigenshape analysis) of the left paramere of the male genitalia, as well as subsequent CVA and LDA provided quantitative support for the majority species assignments based on morphology. In contrast, model based hierarchical clustering showed much less congruence with the morphospecies (Fig. 3e, f), indicating that this approach may not be suitable for delimitation at the level of species.
Molecular-based species delimitation resulted in a wide range of support for morphospecies, based on the analysis of standard markers used among beetles (e.g., [82–85]), from zero (28S and rrnL) to moderate or high (cox1 and ITS1). The ribosomal markers were insufficiently informative to support any of the putative morphospecies (Additional file 1: Figure S3-S5; Table S8), due to the remarkably low interspecific molecular variation observed across the entire genus. This is less surprising for the slowly evolving 28S rRNA marker, but rrnL has previously provided reasonable resolution at the species level among scarabs (e.g., [82]). The mitochondrial gene cox1 and the nuclear region ITS1 were more informative, while the latter provided the best resolution. There was overall congruence between the morphospecies and the ITS1 MOTUs (GMYC, ABGD, PTP), despite the fact that ITS1 had fewer haplotypes than cox1 (23 versus 53) and a lower relative substitution rate (Additional file 1: Table S5). A wide range of tree building methods, parameters and tree linearization approaches did not improve the results of the GMYC model using cox1. In particular, there were three putative morphospecies that were difficult to distinguish on the basis of molecular data alone (sp01 vs 02; sp6 vs sp10; sp02 vs sp12). At one extreme, individuals belonging to a single morphospecies (sp10) were assigned to two MOTUs on the basis of two distinct ITS1 genotypes. The genotypes had a total of 31 segregating sites, including one 2-base-deletion, two 4-base-deletions, and one 2-base-insertion, indicating that a single mutation is unlikely to the cause of the molecular variation, although this pattern was not recovered by any other marker. At the other extreme, individuals belonging to two distinct morphospecies were assigned to a single MOTU and shared identical cox1 and ITS1 haplotypes (sp01, sp02), which may be attributed to introgressive hybridisation or incomplete lineage sorting.
Distinguishing between secondary gene flow and incomplete lineage sorting is difficult because both processes produce similar phylogenetic patterns [66]. JML analyses [67] indicated that incomplete lineage sorting may be sufficient to explain the observed level genetic variation across species with independent data partitions and species – with the exception of the fast evolving cox1 third codon (Additional file 1: Table S5; sp09 and sp11), the monophyly of these species was otherwise well supported. The basic substitution model implemented in JML may not be sufficient to account for hidden substitutions at this site and may underestimate the genetic distance for this partition (Additional file 1: Table S5, S7). Overall, the JML results provide support for an incomplete lineage sorting scenario, however, this test cannot be treated as definitive against secondary gene flow. The method implemented in JML can only be used to detect hybridization events for sequences that have a coalescence time younger than the speciation event [66], and this approach can result in false negatives [86].
Bayesian species delimitation using an integrative taxonomy framework
In concordance with our results from the Bayesian species delimitation, Solís-Lemus et al. [14] have shown that the integration of morphological evidence together with molecular data may greatly enhance the discriminative power of species delimitation models. However, it has also been shown that errors and uncertainties in upstream analyses (e.g., guide tree inference, individual-species assignment) and prior parameter choice may impact the accuracy of results [81, 87, 88]. Here, we assessed the impact of a wide range of parameter combinations, including prior parameter and guide tree choice.
Leaché and Fujita [81] previously demonstrated the significant impact of using randomly generated guide tree topologies. Rannala [89] questioned the practicality of exhaustive guide tree manipulation, with respect to the increased computation time associated with popular phylogenetic inference methods. In addition, a random set of guide trees will include some unreasonable or unlikely topologies, which can lead to inaccurate delimitations (e.g., over-splitting; [88]). Here, we limited our guide tree choice to three options, justified on the basis of evidence of independent molecular and morphometric evidence, in order to further evaluate incongruences between both data sources (Additional file 1: Figure S2). The use of alternative guide trees had a large impact on the results. For example, the use of guide tree C (based on morphological similarity) allowed us to identify support for a putative species pair (sp02/sp12), which was otherwise not identified using alternative molecular based approaches, including the unguided (NNI) approach in BPP (Additional file 4: Table S14 and Additional file 5: Table S15). In an additional set of experiments, we used a fourth guide tree topology based on the support for a putative case of cryptic diversity obtained using alternative single-marker delimitation approaches (sp10, Additional file 2: Table S9; Additional file 1: Figure S2, S7). This experiment, however, cannot provide definitive support for these species entities, because the units were inferred on the basis of non-independent evidence. Manual inspection of the alignments for sp10 revealed 2 ITS1 genotypes with 43 segregating sites represented by sp10a and sp10b. This is a very strong signal compared to a total of 44 segregating sites in both mitochondrial markers, which did not exhibit any diverging signal between sp10a and sp10b. Only a single site was polymorphic for sp10b in 2 of the 4 sp10b specimens. However, these analyses serve to demonstrate that the results obtained using this model can be extremely sensitive to the signal present in single molecular markers, even in presence of data that provide strong evidence for morphological similarity (Additional file 1: Figure S7).
The use of alternative prior combinations for the population size (θ) and root age (τ) priors each had a large impact on the results. These analyses indicate that these parameters must either be chosen using extreme caution (using independent empirical evidence) or multiple analyses should be performed to assess the robustness of species delimitations to these parameters, such as the analyses performed here. We found that phylogenetically younger species (sp01, sp02, sp06, sp10, sp12) and analyses that employed less data (e.g., single versus combined traits) were typically more sensitive to the results. It has also been demonstrated that strong variation in mutation rate and population size among populations or species can also decrease the accuracy of alternative coalescent-based delimitation models [90].
The inclusion of more individuals (and/or data) can lead to more accurate and precise parameter estimates [91], but increased taxon sampling is sometimes not possible due to the natural rarity of some species [73]. The development of better approaches to account for this uncertainty may be important, because in reality many biodiversity studies will be subject to limited taxon sampling. Further research using empirical and simulated data are required to fully assess the impact of guide tree, prior parameter choice, model violation and taxon sampling. Here, we demonstrate that the inclusion of morphological data can lead to more robust estimates of species delimitations. The results obtained using the combined dataset are less sensitive to prior parameter choice, than the analysis based on molecular or morphological dataset alone (Fig. 4; Additional file 3: Table S13, Additional file 4: Table S14 and Additional file 5: Table S15). Overall, nearly all morphospecies received strong support based on the analysis of the combined dataset (Fig. 4d). All sequence-based inference methods, including tree inference using concatenated data or coalescent-based approaches such as *BEAST and BPP, may be impacted by putative incomplete lineage sorting or introgression. An integrative approach to taxonomy enables all available evidence to be utilized and may be particularly useful for delimitating very young species, which will always be difficult to distinguish on the basis of molecular or morphological data alone.
Conclusions
The earliest stages of speciation will be the point at which it will be the hardest to establish a boundary between population and species level divergence. However, such cases (and their solution) are the “holy grail” of taxonomy and provide an exemplar for investigating the intermediate stages of the “Darwinian continuum” from varieties to species [92] and inevitably create problems for the definition of species. Integrative or multiple strategies may be necessary in such cases where conflicts are most likely to exist [6, 19]. Together with previous studies [7, 14] we have confirmed that morphology can be a highly informative trait within an integrative approach, such as iBPP, to species delimitation.
Complex cases of species delimitation, such as those among Pleophylla species, demonstrate the sensitivity of delimitation approaches to prior parameter choices and are thus useful for investigating the performance of new methodologies. We have highlighted the importance of examining the effect of prior choice on species delimitation results in BPP and iBPP, especially if highly informative prior distributions (α >1) are used. Previously, specifying a high θ and a low τ 0 value was intended to constitute a conservative prior combination that should not lead to over-splitting [81]. However, we found that this combination actually led to higher support for more splits, which was attributable to the strong influence of the θ parameter. For a conservative estimate of species delimitations, we recommend using a low value of θ to avoid species over-splitting.
The incongruence between trait- or gene-based species delimitations (Fig. 5, Additional file 1: Figure S8) may have multiple independent causes. First, sampling issues and the ability to capture statistically significant entities may be problematic, particularly for trait-based inference [7] (see also above). For example, trait-based clustering algorithms quickly loose power when including too many or too poorly sampled species, or when variation is distributed over too many dimensions, resulting in more noise [14, 93]. These problems may also pose a challenge for combined approaches to species delimitation, however their impacts have not been fully explored. Second, the incongruence among independent methods, employed for the analysis of different data types (molecules versus discrete or continuous morphological traits), may be attributed to the use of competing species concepts [94, 95]. Model based clustering applied on morphological traits is simply based on the morphological species concept; tree-based species inference methods (e.g., GMYC, PTP) are based on the phylogenetic species concept in [88, 96], which rely on the assumption of reciprocal monophyly across gene trees. The assumption of monophyly among independent markers may be problematic because this assumption is known to be violated for closely related species. de Queiroz redefined the criteria inherent to most species concepts [21, 97, 98] that species represent independent metapopulation lineages through time. Instead, in the generalized lineage concept (GLC) the criteria used to demarcate species (e.g., morphological differences, monophyly or reproductive isolation) are treated as attributes that accumulate during the process of lineage diversification [98]. This concept has been broadly adopted by coalescent-based approaches to species delimitation [6, 7, 10, 20, 34, 99–103], which model the lineage diversification process using multiple markers to delimit species (e.g., [104]). Several studies have delimited species successfully using these approaches [5, 81, 94, 95, 105, 106].

Overview of the results from the different species delimitation methods and data. Inferred entities that were fully congruent with the a priori morphospecies assignments are indicated by the bold circumscribed coloured squares, incongruent units remain white; sub-splitting within morphospecies is indicated by horizontal dashes. Additional sub-splitting within morphospecies that share overlapping MOTUs are circumscribed by a narrow line. Uncertain delimitations are indicated by thin lines between a priori morphospecies
BPP (and iBPP) treat species as hypotheses in a probabilistic framework, using objective tests to delineate independent evolutionary lineages (i.e., species), therefore satisfying numerous species concepts [95]. Caution should always be taken when interpreting the results of a single dataset [6, 7], however, an integrative model-based approach to detecting species is likely to have more utility and could result in more robust species delimitations, especially when divergence varies across different phenotypic, genetic or ecological parameters [7].
Finally, based on the outcome of the integrative BPP analysis (Figs. 4 and 5), which was broadly congruent with the single trait evidence, we conclude that in our Pleophylla data set sp1, sp6, sp9, sp10, sp11, and spX2 are valid species, while sp2 and sp12 very likely belong to the same taxon. The results of alternative molecular delimitation methods provided support for potential cryptic species (sp10, Additional file 1: Figure S8). However, this signal comes from one of the four markers only (which we demonstrated can overwhelm the signal of other data in the BPP/iBPP analyses, Additional file 1: Figure S7) and this result is not corroborated by morphological or geographical evidence (the two MOTUs occur in the same location). Therefore, at this stage we do not consider these as two separate species. (These conclusions will be further developed by formal taxonomic treatment, type material and taxonomic revision that will be presented in a separate upcoming study; Eberle J, Beckett M, Özguel-Siemund A, Frings J, Fabrizi S, Ahrens D. Afromontane forests hide nineteen new species of ancient Pleophylla chafers (Coleoptera: Scarabaeidae): phylogeny and taxonomic revision, in preparation). Additional information about the structure of a population or species complex, based on much broader individual, geographical and DNA sequence sampling would very likely have improved our case study. However, natural rarity (linked with the time constraints of most biodiversity studies) will always have an impact on the number of available samples and may strongly bias the results [107].
Simulations have suggested that the number of loci required for robust Bayesian species delimitation may be large [10]. Here, we demonstrate that the signal from a single marker can influence the outcome of a fully integrative analysis, even given the inclusion of morphology. These results further underlay the necessity for upgrading the globally successful Barcoding initiatives to include a broader range of universal markers (e.g., [108]). Despite numerous disadvantages [109, 110], this approach would help to overcome some of the major challenges to accurate species delimitation [111]. Future directions in integrative taxonomy will need to further address these issues, including integrative study design and the interpretation of frequently incongruent results. In addition the development of new tools for integrating disparate types of specimen-based data in taxonomic studies offer an exciting opportunity to free taxonomy from subjectivity.
Availability of supporting data
Voucher specimens have been deposited in the Zoological Research Museum A. Koenig (Bonn). All molecular sequences generated for this study were deposited in GenBank (Additional file 1: Table S1). Sequence alignments, program input files and phylogenetic trees were deposited on Zenodo (doi:10.1186/s12862-016-0659-3) [112]. The perl script used for running (i)BPP with multiple prior combinations, along with all input files, is available at https://github.com/eberlejonas/BPPmulti.git.
References
Agapow PM, Bininda-Emonds OR, Crandall KA, Gittleman JL, Mace GM, Marshall JC, Marshall JC, Purvis A. The impact of species concept on biodiversity studies. Q Rev Biol. 2004;79(2):161–79.
Daugherty CH, Cree A, Hay JM, Thompson MB. Neglected Taxonomy and Continuing Extinctions of Tuatara (Sphenodon). Nature. 1990;347(6289):177–9.
Isaac NJB, Mallet J, Mace GM. Taxonomic inflation: its influence on macroecology and conservation. Trends Ecol Evol. 2004;19(9):464–9.
Padial JM, De la Riva I. Taxonomic inflation and the stability of species lists: The perils of ostrich's behavior. Syst Biol. 2006;55(5):859–67.
Carstens BC, Dewey TA. Species Delimitation Using a Combined Coalescent and Information-Theoretic Approach: An Example from North American Myotis Bats. Syst Biol. 2010;59(4):400–14.
Carstens BC, Pelletier TA, Reid NM, Satler JD. How to fail at species delimitation. Mol Ecol. 2013;22(17):4369–83.
Edwards DL, Knowles LL. Species detection and individual assignment in species delimitation: can integrative data increase efficacy? P Roy Soc B-Biol Sci. 2014;281(1777):20132765.
Ezard THG, Pearson PN, Purvis A. Algorithmic approaches to aid species' delimitation in multidimensional morphospace. BMC Evol Biol. 2010;10.
Guillot G, Renaud S, Ledevin R, Michaux J, Claude J. A Unifying Model for the Analysis of Phenotypic, Genetic, and Geographic Data. Syst Biol. 2012;61(6):897–911.
Knowles LL, Carstens BC. Delimiting species without monophyletic gene trees. Syst Biol. 2007;56(6):887–95.
Leaché AD, Koo MS, Spencer CL, Papenfuss TJ, Fisher RN, McGuire JA. Quantifying ecological, morphological, and genetic variation to delimit species in the coast horned lizard species complex (Phrynosoma). Proc Natl Acad Sci U S A. 2009;106(30):12418–23.
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol. 2006;55(4):595–609.
Puorto G, Salomao MD, Theakston RDG, Thorpe RS, Warrell DA, Wuster W. Combining mitochondrial DNA sequences and morphological data to infer species boundaries: phylogeography of lanceheaded pitvipers in the Brazilian Atlantic forest, and the status of Bothrops pradoi (Squamata : Serpentes : Viperidae). J Evolution Biol. 2001;14(4):527–38.
Solis-Lemus C, Knowles LL, Ane C. Bayesian species delimitation combining multiple genes and traits in a unified framework. Evolution. 2015;69(2):492–507.
Wiens JJ, Penkrot TA. Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Syst Biol. 2002;51(1):69–91.
Sneath PHA, Sokal RR. Numerical Taxonomy. Nature. 1962;193(4818):855.
Blackwelder. A Critique of Numerical Taxonomy. Syst Zool. 1967;16(1):64.
Sterner B. Well-Structured Biology – Numerical Taxonomy’s Epistemic Vision for Systematics. In: Hamilton A, editor. Patterns of Nature. California: University of California Press; 2014. p. 213–44.
Yeates DK, Seago A, Nelson L, Cameron SL, Joseph L, Trueman JWH. Integrative taxonomy, or iterative taxonomy? Syst Entomol. 2011;36(2):209–17.
Yang ZH, Rannala B. Unguided Species Delimitation Using DNA Sequence Data from Multiple Loci. Mol Biol Evol. 2014;31(12):3125–35.
De Queiroz K. Species concepts and species delimitation. Syst Biol. 2007;56(6):879–86.
Degnan JH, Rosenberg NA. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol. 2009;24(6):332–40.
Hudson RR, Coyne JA. Mathematical consequences of the genealogical species concept. Evolution. 2002;56(8):1557–65.
Maddison WP. Gene trees in species trees. Syst Biol. 1997;46(3):523–36.
Shaffer HB, Thomson RC. Delimiting species in recent radiations. Syst Biol. 2007;56(6):896–906.
Slowinski JB, Knight A, Rooney AP. Inferring species trees from gene trees: A phylogenetic analysis of the elapidae (Serpentes) based on the amino acid sequences of venom proteins. Mol Phylogenet Evol. 1997;8(3):349–62.
Bossu CM, Near TJ. Gene Trees Reveal Repeated Instances of Mitochondrial DNA Introgression in Orangethroat Darters (Percidae: Etheostoma). Syst Biol. 2009;58(1):114–29.
Keck BP, Near TJ. A young clade repeating an old pattern: diversity in Nothonotus darters (Teleostei: Percidae) endemic to the Cumberland River. Mol Ecol. 2010;19(22):5030–42.
Wu CA, Campbell DR. Cytoplasmic and nuclear markers reveal contrasting patterns of spatial genetic structure in a natural Ipomopsis hybrid zone. Mol Ecol. 2005;14(3):781–92.
Edwards SV, Liu L, Pearl DK. High-resolution species trees without concatenation. Proc Natl Acad Sci U S A. 2007;104(14):5936–41.
Heled J, Drummond AJ. Bayesian Inference of Species Trees from Multilocus Data. Mol Biol Evol. 2010;27(3):570–80.
Kubatko LS, Carstens BC, Knowles LL. STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics. 2009;25(7):971–3.
O'Meara BC. New Heuristic Methods for Joint Species Delimitation and Species Tree Inference. Syst Biol. 2010;59(1):59–73.
Yang ZH, Rannala B. Bayesian species delimitation using multilocus sequence data. Proc Natl Acad Sci U S A. 2010;107(20):9264–9.
Simmons LW. Sexual selection and genital evolution. Austral Entomol. 2014;53(1):1–17.
Fraley C, Raftery AE, Brendan M, Scrucca L. mclust Version 4 for R: Normal Mixture Modeling for Model-Based Clustering, Classification, and Density Estimation. In: Technical Report No 597. University of Washington: Department of Statistics; 2012.
Fraley C, Raftery AE. Model-based clustering, discriminant analysis, and density estimation. J Am Stat Assoc. 2002;97(458):611–31.
Ahrens D. The phylogeny of Sericini and their position within the Scarabaeidae based on morphological characters (Coleoptera : Scarabaeidae). Syst Entomol. 2006;31(1):113–44.
Eberle J, Fabrizi S, Lago P, Ahrens D. A historical biogeography of megadiverse Sericini—another story “out of Africa”? Cladistics; 2016. doi:10.1111/cla.12162.
Eberhard WG. Sexual Selection and Animal Genitalia. Cambridge: Harvard University Press; 1985.
Ahrens D, Lago PK. Directional asymmetry reversal of male copulatory organs in chafer beetles (Coleoptera : Scarabaeidae): implications on left-right polarity determination in insect terminalia. J Zool Syst Evol Res. 2008;46(2):110–7.
Dalla Torre KW. Scarabaeidae: Melolonthinae I. Coleopterorum Catalogus 45. 1912.
Beckett M. The distribution patterns in Pleophylla species (Coleptera: Scarabaeidae) – indicators of ancient forest distributions. Bonn: Rheinische Friedrich-Wilhelms-Universität Bonn; 2012.
Rohlf FJ. TPSDig 2.1. http://life.bio.sunysb.edu/morph/. Accessed Mar 2009.
MacLeod N. Generalizing and extending the eigenshape method of shape space visualization and analysis. Paleobiology. 1999;25(1):107–38.
Macleod N, Rose KD. Inferring Locomotor Behavior in Paleogene Mammals Via Eigenshape Analysis. Am J Sci. 1993;293a:300–55.
Krieger JD. Measure LMs 4.0. Morpho-tools http://www.morpho-tools.net. Accessed Mar 2009.
Fraley C, Raftery AE. Bayesian regularization for normal mixture estimation and model-based clustering. J Classif. 2007;24(2):155–81.
Celeux G, Govaert G. Gaussian Parsimonious Clustering Models. Pattern Recogn. 1995;28(5):781–93.
McLachlan GJ, Basford KE. Mixture Models: Inference and Applications to Clustering. New York: Marcel Dekker; 1988.
Schwarz G. Estimating Dimension of a Model. Ann Stat. 1978;6(2):461–4.
Venables WN, Ripley BD. Modern Applied Statistics. 4th ed. New York: Springer; 2002.
Revell LJ. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 2012;3(2):217–23.
Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol Biol Evol. 2012;29(6):1695–701.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–90.
Stamatakis A, Hoover P, Rougemont J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst Biol. 2008;57(5):758–71.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–5.
Marshall DC. Cryptic Failure of Partitioned Bayesian Phylogenetic Analyses: Lost in the Land of Long Trees. Syst Biol. 2010;59(1):108–17.
Rannala B, Zhu TQ, Yang ZH. Tail Paradox, Partial Identifiability, and Influential Priors in Bayesian Branch Length Inference. Mol Biol Evol. 2012;29(1):325–35.
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. Plos Biol. 2006;4(5):699–710.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Papadopoulou A, Anastasiou I, Vogler AP. Revisiting the Insect Mitochondrial Molecular Clock: The Mid-Aegean Trench Calibration. Mol Biol Evol. 2010;27(7):1659–72.
Papadopoulou A, Jones AG, Hammond PM, Vogler AP. DNA taxonomy and phylogeography of beetles of the Falkland Islands (Islas Malvinas). Mol Phylogenet Evol. 2009;53(3):935–47.
Bouckaert R, Heled J. DensiTree 2: Seeing Trees Through the Forest. doi:10.1101/012401 2014.
Joly S, McLenachan PA, Lockhart PJ. A Statistical Approach for Distinguishing Hybridization and Incomplete Lineage Sorting. Am Nat. 2009;174(2):E54–70.
Joly S. JML: testing hybridization from species trees. Mol Ecol Resour. 2012;12(1):179–84.
Templeton AR, Crandall KA, Sing CF. A Cladistic-Analysis of Phenotypic Associations with Haplotypes Inferred from Restriction Endonuclease Mapping and DNA-Sequence Data.3. Cladogram Estimation. Genetics. 1992;132(2):619–33.
Puillandre N, Lambert A, Brouillet S, Achaz G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol. 2012;21(8):1864–77.
Ezard THG, Fujisawa T, Barraclough TG. SPLITS: Species’ Limits by Threshold Statistics R package. 2009. http://barralab.bio.ic.ac.uk. Accessed May 2012.
Fontaneto D, Herniou EA, Boschetti C, Caprioli M, Melone G, Ricci C, Barraclough TG. Independently evolving species in asexual bdelloid rotifers. Plos Biol. 2007;5(4):914–21.
Zhang JJ, Kapli P, Pavlidis P, Stamatakis A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 2013;29(22):2869–76.
Ahrens D, Fujisawa T, Krammer HJ, Eberle J, Fabrizi S, Vogler AP. Rarity and Incomplete Sampling in DNA-based Species Delimitation. Syst Biol. 2016. [Epub ahead of print].
Hart MW, Sunday J. Things fall apart: biological species form unconnected parsimony networks. Biol Letters. 2007;3(5):509–12.
Britton T, Anderson CL, Jacquet D, Lundqvist S, Bremer K. Estimating divergence times in large phylogenetic trees. Syst Biol. 2007;56(5):741–52.
Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19(2):301–2.
Rambaut A, Charleston M. TreeEdit 1.0. http://tree.bio.ed.ac.uk/software/treeedit/. Accessed May 2012.
Powell JR. Accounting for uncertainty in species delineation during the analysis of environmental DNA sequence data. Methods Ecol Evol. 2012;3(1):1–11.
Vogler AP, Monaghan MT. Recent advances in DNA taxonomy. J Zool Syst Evol Res. 2007;45(1):1–10.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Leaché AD, Fujita MK. Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). P Roy Soc B-Biol Sci. 2010;277(1697):3071–7.
Ahrens D, Monaghan MT, Vogler AP. DNA-based taxonomy for associating adults and larvae in multi-species assemblages of chafers (Coleoptera : Scarabaeidae). Mol Phylogenet Evol. 2007;44(1):436–49.
Ahrens D, Vogler AP. Towards the phylogeny of chafers (Sericini): Analysis of alignment-variable sequences and the evolution of segment numbers in the antennal club. Mol Phylogenet Evol. 2008;47(2):783–98.
Bocak L, Barton C, Crampton-Platt A, Chesters D, Ahrens D, Vogler AP. Building the Coleoptera tree-of-life for > 8000 species: composition of public DNA data and fit with Linnaean classification. Syst Entomol. 2014;39(1):97–110.
Hunt T, Bergsten J, Levkanicova Z, Papadopoulou A, John OS, Wild R, Hammond PM, Ahrens D, Balke M, Caterino MS et al. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science. 2007;318(5858):1913–6.
Heled J, Bryant D, Drummond AJ. Simulating gene trees under the multispecies coalescent and time-dependent migration. BMC Evol Biol. 2013;13.
Olave M, Sola E, Knowles LL. Upstream Analyses Create Problems with DNA-Based Species Delimitation. Syst Biol. 2014;63(2):263–71.
Zhang C, Rannala B, Yang ZH. Bayesian Species Delimitation Can Be Robust to Guide-Tree Inference Errors. Syst Biol. 2014;63(6):993–1004.
Rannala B. Are molecular taxonomists lost upstream? In: Phylogeny etc. Meditations on Phylogenetic Inference. http://phylogenyetc.tumblr.com/post/78791524128/are-molecular-taxonomists-lost-upstream. Accessed Dec 2015.
Fujisawa T, Barraclough TG. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst Biol. 2013;62(5):707–24.
Yang ZH. The BPP program for species tree estimation and species delimitation. Curr Zool. 2015;61(5):854–65.
Mallet J. Mayr's view of Darwin: was Darwin wrong about speciation? Biol J Linn Soc. 2008;95(1):3–16.
Hausdorf B, Hennig C. Species Delimitation Using Dominant and Codominant Multilocus Markers. Syst Biol. 2010;59(5):491–503.
Carstens BC, Satler JD. The carnivorous plant described as Sarracenia alata contains two cryptic species. Biol J Linn Soc. 2013;109(4):737–46.
Fujita MK, Leache AD, Burbrink FT, McGuire JA, Moritz C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol Evol. 2012;27(9):480–8.
Sites JW, Marshall JC. Operational criteria for delimiting species. Annu Rev Ecol Evol S. 2004;35:199–227.
de Queiroz K. The general lineage concept of species, species criteria, and the process of speciation. In: Howard SJ BS, editor. Endless Forms: Species and Speciation. New York: Oxford University Press; 1998.
de Queiroz K. Ernst Mayr and the modern concept of species. Proc Natl Acad Sci U S A. 2005;102:6600–7.
Camargo A, Morando M, Avila LJ, Sites Jr JW. Species delimitation with ABC and other coalescent-based methods: a test of accuracy with simulations and an empirical example with lizards of the Liolaemus darwinii complex (Squamata: Liolaemidae). Evolution. 2012;66(9):2834–49.
Ence DD, Carstens BC. SpedeSTEM: a rapid and accurate method for species delimitation. Mol Ecol Resour. 2011;11(3):473–80.
Jones G. Species delimitation and phylogeny estimation under the multispecies coalescent. bioRxiv 2015. doi:10.1101/010199.
Jones G, Aydin Z, Oxelman B. DISSECT: an assignment-free Bayesian discovery method for species delimitation under the multispecies coalescent. Bioinformatics. 2015;31(7):991–8.
O'Meara BC, Ane C, Sanderson MJ, Wainwright PC. Testing for different rates of continuous trait evolution using likelihood. Evolution. 2006;60(5):922–33.
Edwards SV. Is a New and General Theory of Molecular Systematics Emerging? Evolution. 2009;63(1):1–19.
Kubatko LS, Gibbs HL, Bloomquist EW. Inferring Species-Level Phylogenies and Taxonomic Distinctiveness Using Multilocus Data in Sistrurus Rattlesnakes. Syst Biol. 2011;60(4):393–409.
Niemiller ML, Near TJ, Fitzpatrick BM. Delimiting Species Using Multilocus Data: Diagnosing Cryptic Diversity in the Southern Cavefish, Typhlichthys Subterraneus (Teleostei: Amblyopsidae). Evolution. 2012;66(3):846–66.
Lim GS, Balke M, Meier R. Determining Species Boundaries in a World Full of Rarity: Singletons, Species Delimitation Methods. Syst Biol. 2012;61(1):165–9.
Janzen DH, Hajibabaei M, Burns JM, Hallwachs W, Remigio E, Hebert PDN. Wedding biodiversity inventory of a large and complex Lepidoptera fauna with DNA barcoding. Philos T Roy Soc B. 2005;360(1462):1835–45.
Collins RA, Cruickshank RH. Known knowns, known unknowns, unknown unknowns and unknown knowns in DNA barcoding: a comment on Dowton et al. Syst Biol. 2014;63(6):1005–9.
Dowton M, Meiklejohn K, Cameron SL, Wallman J. A Preliminary Framework for DNA Barcoding, Incorporating the Multispecies Coalescent. Syst Biol. 2014;63(4):639–44.
Dupuis JR, Roe AD, Sperling FA. Multi-locus species delimitation in closely related animals and fungi: one marker is not enough. Mol Ecol. 2012;21(18):4422–36.
Eberle J, Warnock RCM, Ahrens D (2016) Data from: Bayesian species delimitation in Pleophylla chafers (Coleoptera) – the importance of prior choice and morphology. Zenodo. doi:10.1186/s12862-016-0659-3.
Acknowledgements
We would like to thank Simon Joly for advice using the JML software, Norman MacLeod for advice with the eigenshape analysis, the referees for their helpful comments on an earlier draft of the manuscript, Silvia Fabrizi for assisting with the lab work, and Pia Addison and Cate Bazelet for help with the collection permit for the Cape Province. This project was supported by a studentship from the Natural Environment Research Council to R.C.M.W (NE/E522891/1and NE/I528250/1), and grants from the German Science Association to D.A. (DFG/AH175/1 and AH175/3). For providing D.A. with research and collection permits, we thank the various governmental institutions and departments in Eastern Cape (Permit No.: WRO 122/07WR and WRO123/07WR), Gauteng (Permit No.: CPF6 1281), Limpopo (Permit No.: CPM-006-00001), Mpumalangma (Permit No.: MPN-2009-11-20-1232), Cape Province (Permit No: AAA0007-00097-0056), and Kwazulu-Natal (Permit Nos.: OP3752/2009, 1272/2007, 3620/2006). This work was partially supported by the computational facilities of the Advanced Computing Research Centre, University of Bristol and of the Zoological Research Museum A. Koenig, Bonn.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RCMW: molecular lab work, sequence assembly and alignments, phylogenetic inference, DNA-based species delimitation; JE: clustering analyses, DNA-based species delimitation, integrative species delimitation; DA: conducted fieldwork collections, conceived the study. RCMW, JE, DA: morphometric analyses and drafted the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Supplementary text, figures S1-S8, tables S1-S8, S10-S12. (PDF 1909 kb)
Additional file 2:
3d morphospace. (GIF 6172 kb)
Additional file 3:
Supplementary table S9. (XLSX 64 kb)
Additional file 4:
Supplementary table S13. (XLS 185 kb)
Additional file 5:
Supplementary table S14. (XLS 40 kb)
Additional file 6:
Supplementary table S15. (XLS 38 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Eberle, J., Warnock, R.C.M. & Ahrens, D. Bayesian species delimitation in Pleophylla chafers (Coleoptera) – the importance of prior choice and morphology. BMC Evol Biol 16, 94 (2016). https://doi.org/10.1186/s12862-016-0659-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-016-0659-3