
Abstract
Background
Population structure and genetic diversity of marine organisms in the Northwestern Pacific Ocean exhibited complex patterns. Saccharina japonica is a commercially and ecologically important kelp species widely distributed along the coast of Japan Sea. However, it is still poorly known about population genetics and phylogeographic patterns of wild S. japonica populations on a large geographic scale, which is an important contribution to breeding and conservation of this marine crop.
Results
We collected 612 mitochondrial COI and trnW-trnL sequences. Diversity indices suggested that S. japonica populations along the coast of Hokkaido exhibited the highest genetic diversity. Bayesian Analysis of Population Structure (BAPS) revealed four clusters in the kelp species (cluster 1: Hokkaido and South Korea; cluster 2: northwestern Hokkaido; cluster 3: Far Eastern Russia; cluster 4: China). The network inferred from concatenated data exhibited two shallow genealogies corresponding to two BAPS groups (cluster 2 and cluster 3). We did not detect gene flow between the two shallow genealogies, but populations within genealogy have asymmetric gene exchange. Bayesian skyline plots and neutrality tests suggested that S. japonica experienced postglacial expansion around 10.45 ka.
Conclusions
The coast of Hokkaido might be the origin and diversification center of S. japonica. Gene exchange among S. japonica populations could be caused by anthropogenic interference and oceanographic regimes. Postglacial expansions and gene exchange apparently led to more shared haplotypes and less differentiation that in turn led to the present shallow phylogeographical patterns in S. japonica.
Similar content being viewed by others
Background
The commercially and ecologically important seaweed Saccharina japonica (Aresch.) C.E. Lane, C. Mayes, Druehl & G.W. Saunders is widely distributed along the coast of Japan Sea. Taxonomically, this species was initially named Laminaria japonica by Areschoug in 1851 [1]. Recently, Lane et al. [2] reported a new genus Saccharina Stackhouse from the genus Laminaria Lamouroux based on multiple lines of molecular data and proposed to use S. japonica to replace L. japonica. The systematic survey in Laminariales from the Far Eastern Seas of Russia also supported to transfer L. japonica into the genus Saccharina [3]. For S. japonica along the coast of Hokkaido, integrative morphological and phylogenetic analyses have identified four varieties, S. religiosa (Miyabe) C.E. Lane, C. Mayes, Druehl & G.W. Saunders, S. ochotensis (Miyabe) C.E. Lane, C. Mayes, Druehl & G.W. Saunders and S. diabolica (Miyabe) C.E. Lane, C. Mayes, Druehl & G.W. Saunders [4, 5]. In this study, we did not distinguish these varieties and treated them as one species for phylogeographic analyses.
Global climate change and over exploitation has drastically reduced the resource of S. japonica in recent years [6]. Understanding genetic diversity and population structure of wild S. japonica populations will aid in the management, conservation and breeding of this marine macroalga. However, current genetic researches of S. japonica mainly focused on cultivated populations, and involved wild populations only on a limited geographic scale [7, 8]. The broad picture of wild S. japonica populations in the Japan Sea needs clarification, especially regarding population genetic and phylogeographic patterns on a large geographic scale.
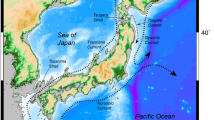
Population structure and genetic diversity of marine organisms in the Northwestern Pacific Ocean exhibited complex patterns which mainly resulted from biotic factors (e.g. reproductive strategies and intrinsic life-history characteristics) and abiotic factors (e.g. complex costal topography, dynamic currents and habitat discontinuities) [9–13]. The semi-isolated marginal Japan Sea is connected with the Sea of Okhostk, the North Pacific, and the East China Sea through four shallow straits of less than 130 m depth [14] (Fig. 1). During the last glacial maximum (LGM), shallow straits restricted or completely blocked inflow of the warm Tsushima current into the Japan Sea via the Tsushima and Tsugaru Straits, reducing sea water temperatures and salinity. After the LGM, higher sea level allowed relatively warm water to flow into the Japan Sea and increased seawater temperature and salinity. Since the mid-Holocene, the modern oceanographic regime of the Japan Sea was established [15–17] and Paleoclimatic oscillations and intricate tectonic topography may have affected the genetic diversity and genetic structure of macroalgae in the Northwestern Pacific [18, 19]. In addition, dispersal processes played an important role in facilitating gene flow in macroalgal populations and structuring the established populations [20–23]. Along with short-lived spores and gametes, macroalgae have relatively poor dispersal ability [24]. However, ocean currents facilitated dispersal over long distances for organisms such as Laminaria digitata (Hudson) J.V. Lamouroux and Macrocystis pyrifera (Linnaeus) C. Agardh [21, 22]. Incident anthropogenic introduction of S. japonica influenced dispersal and gene flow of brown seaweed, especially economic seaweeds [7, 8, 25]. In the wild environment, which kinds of factors affected the genetic structure and phylogeographic pattern of S. japonica are rarely known.

Map showing the sampling locations, and schematic oceanic currents around the Japan Sea. Pink arrows indicate warm currents and blue ones cold currents [14]
Mitochondrial COI and trn DNA markers have been intensively used to explore intra-specific phylogeographic patterns of brown seaweeds [18, 26–32], including the kelp S. japonica [5]. In particular, COI marker has been demonstrated to has more polymorphism sites than nuclear (ITS) and plastid (rbcLS), and has been verified in S. japonica [33]. In the present study, we chose mitochondrial COI and trnW-L and applied them in 26 S. japonica wild populations to explore intraspecific genetic diversity, population structure and demographic history and to find potential abiotic and/or biotic factors associated with genetic differentiation in S. japonica.
Methods
Sample collection
Between 2011 and 2013, 26 populations of S. japonica (612 individuals) were collected, ranging from Sakhalin, Russia (48°50′ N) to Shandong, China (37°09′ N) (Fig. 2; Additional file 1: Table S1). The collection covered most kelp distribution locations along the coast of the Northwestern Pacific Ocean.

Distribution of the 53 combined mtDNA haplotypes detected in the 26 Saccharina japonica populations. Pie charts are labeled with the population numbers as shown in Table 1. The size of divisions inside each pie chart is proportional to the frequency of occurrence for individual haplotypes. Colors represent the shared haplotypes and private haplotypes are shown in white
DNA extraction, PCR and sequencing
S. japonica genomic DNA was extracted using the Plant genomic DNA kit (Tiangen, Beijing) according to manufacturer’s instructions and mitochondrial markers COI and trnW-L were adopted. One COI partial region was amplified with primers SacCOIF and SacCOIR [33] and the trnW-L partial region from the 3′ end of the trnW gene to the 5′end of the trnL was amplified with primers 5′-GGTTCAAGTCCCTCTCTTTCTGT-3′ (trnW-LBF, forward) and 5′-AACCTAAACCCAGCGTGTAT-3′ (trnW-LBR, reverse) based on the mitochondrial genome sequence of S. japonica [5]. The PCR mixture (50 μL) containing 50 ng genomic DNA, 0.2 mM dNTP, 1 × Taq buffer, 0.4 μM of each primer, 0.5 unit Taq polymerase and sterile water (Transgen, Beijing) was amplified on a Takara thermocycler (Takara Bio, Japan) with initial denaturation of 95 °C for 5 min, 35 cycles of 95 °C denaturation for 30 s, 52 °C (COI)/55 °C (trnW-L) annealing for 30 s, a 72 °C extension for 2 min (COI) or 1 min (trnW-L) and final extension of 72 °C for 5 min. Amplified products were sequenced on an ABI PRISIM 3730 automatic sequencer (Applied Biosystems, USA) and sequenced data were edited and aligned with BioEdit v7.1 software [34].
Data analysis
Genetic diversity and haplotype network construction
The number of segregating sites (S), average number of nucleotide differences (K), nucleotide diversity (Pi) and haplotype diversity (Hd) were calculated using DNASP v5 [35]. NETWORK v4.5.1.6 [36] was used to generate the maximum parsimony media-joining network, which exhibited genealogical relation to the haplotypes of S. japonica. To test mutation rate, COI and trnW-L sequence data were concatenated and the combined mtDNA haplotype network was constructed.
Population structure and gene flow
With concatenated data of two mtDNA loci, Bayesian analysis of population structure was conducted using BAPS v6.0 with a spatial model to define groups [37, 38]. Calculations were performed with the upper number of K clusters varying from 2 to 26, providing the highest posterior probability for reasonable partitioning. Hierarchical molecular variance analysis (AMOVA) was performed on the four BAPS groups with 10,000 permutations using Arlequin v3.5 [39]. Estimated F ST values of pairwise populations and pairwise BAPS groups were conducted for genetic differentiation analysis using 10,000 permutations with Arlequin v3.5.
The mantel test for isolation-by-distance (IBD) established relationships between genetic and geographic distances [40] and genetic distances (F ST/ (1–F ST)) were regressed against the logarithm of geographic distances within location distances (estimated with Google Earth 6.0). To visualize IBD patterns [41], 1000 randomizations were analyzed with IBDWS (http://ibdws.sdsu.edu/~ibdws/).
To further test migration rate and divergence time between neighboring populations in Far Eastern Russia and Hokkaido, an isolation-with-migration model was applied using software IMa2 [42, 43]. Migration rate for population pairs in IMa2 had to be estimated due to computational challenges related to the high number of parameters from multiple populations. Preliminary runs were performed to optimize upper bounds on prior distributions (q = 10, t = 10, m = 10; where q = population size, t = divergence time, and m = migration rate) and to optimize heating schemes. Final analyses consisted of three runs of 10–60 geometrically heated chains with burn-in of 500,000 steps. The heating scheme used a geometric model with parameters ha = 0.96 and hb = 0.9. A total of 120,000 genealogies were saved after the three long runs and used to calculate parameter values and likelihood ratio tests of nested speciation models [43].
Molecular clock calibration
According to the study of Silberfeld et al. [44] on brown algae, the divergence time of Saccharina occurred approximately 20 Ma ago. Therefore, 626 bp COI was used to re-construct the phylogeny of Laminariales (3 genera and 5 species) and 20 Ma was used as the ancestral time of the common ancestor (t MRCA ) of Saccharina, Pelagophycus Areschoug and Nereocystis Postels & Ruprecht, which all are implemented in BEAST v1.7.4 [45] with the uncorrelated log-normal (UCLN) relaxed-clock model and the HKY model based on the Bayesian information criterion analysis in PartitionFinder v1.1.0 [46]. The maximum clade credibility tree was constructed with TreeAnnotator v1.7.5 [47].
Demographic history
The duration of transplantation and domestication to S. japonica in China and Korea is short compared with terrestrial crops, so neutrality tests and mismatch distribution analysis were used to explore the demographic history of the 22 S. japonica populations (representing all populations examined in this study except for four populations in China and Korea. Tajima’s D [48] and Fu’s F S [49] were estimated and a mismatch distribution test was administered with reference to the method of Schneider et al. [50]. A Bayesian skyline plot (BSP) was applied for the historical demographic analysis of S. japonica using BEAST v1.7.4 [51]. COI or trnW-L marker and concatenated data (COI and trnW-L) was used for data analysis, with mean substitution/site selected at 1.0 for the time estimation to the units of substitution/site. The HKY + I substitution model was selected for COI, the HKY substitution model was applied for trnW-L and the HKY + G substitution model was selected for combined sequences as identified in PartitionFinder v1.1.0 [46]. The number of grouped intervals was set at 15, and each run was initiated with random starting trees having chain lengths of 3 × 107 and 108. For each marker, multiple analyses were conducted with different random seeds to test convergence, and results from replicate runs were pooled with LogCombiner v2.1.2 and parameters were assessed using Tracer v1.4.1 [51].
Results
Haplotype patterns and network
With alignment and trimming, the remaining sequences were 1528 bp for COI and 362 bp for trnW-L. In the COI and trnW-L sequences from 612 individuals, 35 and 20 segregating sites respectively were identified, possessing 30 COI haplotypes (GenBank accession number: KT963115-KT963144) and 22 trnW-L haplotypes (GenBank accession number: KT963093-KT963114).
Calculated genetic variations are summarized in Table 1. For the COI marker, nucleotide diversity (Pi) range was 0.00000–0.00191 and haplotype diversity (Hd) range was 0.0000–0.68254. For the trnW-L marker, haplotype diversity was 0.00000–0.0042 and nucleotide diversity was 0.00000–0.76190. Data from both COI and trnW-L showed highest diversity indices in Hokkaido, Japan. For the whole dataset (COI + trnW-L sequences, 1890 bp), there were 55 segregating sites and 53 haplotypes in all the 26 populations (Table 1). Haplotype diversity (Hd) of total populations was 0.72165, with a range of 0.0000–0.76190 (Table 1), and haplotype diversity in Hokkaido (Hd = 0.72528) was higher than in other populations (Russia 0.58537; Korea 0.03333; China 0.55306) (Table 1). Highest nucleotide diversity was also detected in Hokkaido populations (0.00079) (Table 1). Generally, diversity parameters yielded from the two combined mtDNA markers indicated that pop7 and pop9 possessed the highest nucleotide and haplotype diversity and pop21 (Russia) and pop24 (China) exhibited the lowest nucleotide and haplotype diversity (Table 1). All genetic diversity parameters indicated that the S. japonica populations along the coast of Hokkaido exhibited the highest genetic diversity.
There were 10 shared haplotypes and 43 private haplotypes, which were unique to a single population based on combined sequences (Fig. 2). The most frequent and widespread haplotype H1 was found in all geographic regions and the main haplotype H33 lay in Far Eastern Russia. H20 was shared by Hokkaido pop 4, pop9, pop12 and pop26. H30 was shared by Hokkaido pop 4 and Sakhalin pop 13. H1 was in 48.69 % of individuals and 76.92 % of populations. Moreover, 30 haplotypes were definable with a single mutation difference from H1, with H2 seven mutation steps removed from H1 (Fig. 3).

Median-joining network constructed using the haplotypes of the concatenated sequences. Circle size is proportional to sample size for each population (n = 1-298 individuals). Missing haplotypes are shown as dots in the network. Each connecting line indicates one mutation step between haplotypes and each black dot represents one mutation step. Colors represent shared haplotypes and private haplotypes are shown in white
Genetic structure and gene flow
Bayesian Analysis of Population Structure (BAPS) revealed four groups (Additional file 2: Figure S1). Cluster 1 contained 14 populations not otherwise partitioned. Cluster 2 included pop1, pop2, pop7 and pop11 in Hokkaido, while cluster 3 contained 6 populations, which located in Far Eastern Russia and cluster 4 contained 2 wild populations (pop24 and pop25) in northern China. AMOVA tests indicated significant differentiation among all populations (F CT = 0.546, P < 0.001) defined by BAPS, which accounted for 54.79 % of variation (Table 2). Pairwise F ST values among the four clusters ranged from 0.22309 (cluster 1 and cluster 4) to 0.87016 (cluster 2 and cluster 3) (Additional file 3: Table S2). Pairwise F ST tests exhibited low or moderate genetic differentiation (Additional file 4: Table S3). Overall, pop1 and pop2 in Hokkaido have deep divergence with other populations (F ST: 0.384–0.9336). Pairwise F ST values indicated that pop24 and pop25 in China diverged significantly from the other 24 populations (F ST: 0.5304–1.000) (Additional file 4: Table S3).
The mantel test indicated no significant correlation between genetic distance (F ST/(1–F ST)) and geographical distance (22 populations except populations in China and Korea) (Additional file 5: Figure S2). Regression coefficients (R2 < 0.01) indicated that the population genetic structure of S. japonica did not fit the IBD model.
Posterior probability distributions, peak posterior point estimates and 95 % confidence intervals of parameters are in Table 3. Two lineages (cluster 2 and cluster 3) had clear geographic distribution and were divided into 5 neighboring units: Validivostok (VL, pop21 and pop22), west coast of Sakahin (WS, pop15–18), Wakkanai (WA, pop1 and pop2), Shari (SH, pop12) and Hakodate (HA, pop7). IMa2 was ran for three pairs of populations in S. japonica (pair 1: WS versus VL; pair 2: WA versus SH; pair 3: WA, HA & SH versus VL & WS) reaching convergence with high ESS values across all parameters. Migration between cluster 2 and cluster 3 (pair 3) seemed to be negligible in both directions (HiPt: 0.02359, 95 % HPD: 0.00–7.147; in the opposite direction, HiPt: 0.007412, 95 % HPD: 0.00–2.780). Effective population sizes (HiPt: 1.3 × 105, 95 % HPD: 6.8 × 104–2.6 × 105) were approximately two times higher in cluster 2 than in cluster 3 (HiPt: 5.4 × 105, 95 % HPD: 2.2 × 104–1.1 × 105). Peak posterior distribution of migration rate estimated asymmetrical gene flow between WA and SH, where migration to SH was significant (HiPt: 44.98, 95 % HPD: 0.0–2822) and migration to WA was negligible (HiPt: 0.3907, 95 % HPD: 0.0–2.030). A similar asymmetrical gene flow was observed between VL and WS, with gene flow from VL to WS was significantly higher than in the opposite direction. Estimated split time between cluster 2 and cluster 3 was 0.0554 Ma (HiPt: 0.0554, 95 % HPD 0.0226–1.3667), with all statistics sharing broadly overlapping 95 % HPD intervals (Table 3, Fig. 4a).

Results of pairwise IMa analysis of Saccharina japonica populations based on combined mtDNA sequences. a Posterior densities for splitting time (t0); (b) Posterior densities for population sizes (q0, q1, qA); (c, d) Posterior densities for population migration (2N0m0 > 1 and 2N1m1 > 0)
Demographic history
The significantly negative Tajima’s D and Fu F S values indicated that S. japonica populations might experience demographic expansion (Additional file 6: Figure S3). Using a single COI marker or trn W-L marker for mismatch distribution, displayed L-shaped and reflected shallow haplotype phylogeny in S. japonica populations (Additional file 6: Figure S3a and b). Moreover, the mismatch distribution of combined haplotypes showed a unimodal pattern, indicating that S. japonica populations might have expanded. In addition, Bayesian skyline plots analyses showed that S. japonica population underwent population expansion (Additional file 7: Figure S4).
With a maximum clade credibility tree based on the COI data (Additional file 8: Figure S5), divergent time between the S. japonica and S. augustata (Kjellman) C.E. Lane, C. Mayes, Druehl & G.W. Saunders was assumed to be 4.84 Ma. Calculated COI region divergence between S. japonica (AP011493) and S. augustata (AP011498) was 4.633 % and estimated divergent rate was 0.9572 %/Ma (4.633 %/4.84 Ma, rounded conservatively) [52]. Since mutation rate should be half of divergence rate [53, 54], estimated mutation rate was 0.4786 %/Ma. Bayesian skyline plots and the calculated molecular clock of COI suggested that S. japonica populations started to expand at 10.45 Ka (Additional file 7: Figure S4a).
Discussion
Two shallow genealogies existed in S. japonica populations
Data sets revealed a shallow genetic structure in S. japonica, with many shared haplotypes in four genetic clusters (Fig. 2; Additional file 2: Figure S1). The combined network does not have large divergences between haplotype groups and two shallow genealogies corresponded to two BAPS groups (cluster 2 and cluster 3) (Fig. 3; Additional file 2: Figure S1). Most kelp populations in these two genealogies were isolated by the Soya Strait, a major outlet of the Tsushima Warm Current [55]. The current flow direction has hindered dispersal of kelp, producing low levels of gene flow (2 Nm < 1) in both directions between these two genealogies (Table 3; Fig. 4c and d). The Tsushima Warm Current flows through the Soya Strait and enhanced the sea water temperature and salinity [55], so the northern coast of Hokkaido has higher sea temperature and lower salinity than the west coast of Sakhalin, which might lead to different release time of spores, different discharge time of sperm and different egg fertilization time [56, 57]. Distinct marine environmental factors and non-simultaneous reproductive duration likely caused these two genealogies isolation and influenced the formation of them.
Postglacial expansion, oceanic current and anthropogenic introduction influence the current genetic structure
Mismatch distributions, neutrality tests and BSP analyses all suggested that S. japonica expanded in the Northwestern Pacific Ocean and calculations using the COI molecular clock suggested that such population expansion occurred about 10.45 Ka. Post-LGM expansion (25–15 Ka) significantly influenced the demography of seaweeds [28, 60]. The paleoceanographic record in the Japan Sea shows that sea water temperature and salinity were lower (drop 140 m of sea level) during the LGM (25–15 Ka) and seawater exchange through straits was blocked. After the LGM, sea water temperature rose in the early Holocene (8–10 ka) due to the inflow of Tsushima Warm Current and sea temperature rose 6–7 °C. Meanwhile, the salinity of seawater in the Japan Sea increased during the interglacial period. With the cold-adapted and lower salinity tolerant characters, S. japonica could survive severe ancient conditions and colonized suitable coastal territories when the sea level increased after the LGM. Judging from the constructed haplotype network (Fig. 3) and IMa analyses (Fig. 4 and Table 3), pink and green genealogies had no contemporary gene flow since they diverged in mid-Pleistocene, showing that these two genealogies might be existed at least in two glacial refugia in the Japan Sea coast during LGM and afterwards, expanded their populations to the current distribution in the interglacial period.
Low genetic differentiation in the populations of S. japonica suggested gene exchange among detected populations. In addition, there were several shared haplotypes and no significant population differentiation corresponding to geographic distribution (Fig. 3; Additional file 2: Figure S1), indicating gene flow in kelp populations. Shared haplotype H30 and H20 indicated that some populations in Sakhalin and Hokkaido had gene exchange. Lower gene flow existed in brown algae such as Fucus ceranoides Linnaeus and Laminaria digitata (Hudson) Lamouroux [22, 58] due to factors such as short-lived spores and gametes and reproductive strategies [20, 22, 24]. Macroalgae are species with relatively poor dispersal ability, but S. japonica in the present study has high gene flow among most populations. In S. japonica, due to limited gametes and spores dispersal ability, the floating or drifting thallus by oceanic currents was regarded as the main dispersal factor to enhance the gene flow in the kelp populations [8, 57]. The warm Tsushima Current, flows northward along the western coast of Hokkaido, and enters into Sea of Okhostk through the Soya Strait, then forwards along coast of northern Hokkaido. The Tsushima current is likely the major force responsible for drifting kelp thallus from the eastern coast of Hokkaido to Kunashir Island, evidenced by asymmetric gene flow between WA and SH (Table 3).
Marine transportation also promotes kelp population genetic homogeneity. Pop 21 (Primorye costal region) and pop 15–18 in Sakhalin shared also one haplotype (H33), and pop19 (Sakhalin) and pop22 (Primorye costal region) shared one haplotype (H27), indicating gene exchange in Far Eastern Russia populations. In addition, IMa analyses indicated that an asymmetric gene flow existed between WS and VL in Far Eastern Russia (Table 3). Marine transportation seems closely related to kelp introduction from west coast of Sakhalin to Primorye costal region, because most locations are near harbors. Besides ballast water in ships likely promoted gene exchange in macroalgae such as Undaria pinnatifida (Harvey) Suringar [59].
Artificial cultivation enhances kelp distribution and impacts population genetic patterns [26, 54]. Gene flow reduces population differences and promotes shallow population genetic structure. Shared haplotypes H1 in China and H20 in Japan indicated that S. japonica in China was mainly from Hokkaido areas, confirming suggestions of Tseng et al., [25]. Pop23 in Korea shared haplotypes H27 and H29 with pop4 and pop5 in Hokkaido. Apparently, artificial cultivation facilitates the gene exchange among the populations in China, South Korea and Japan.
Genetic diversity pattern
Ancestral Laminariales occurred along the coast of Hokkaido, which is regarded as the diversity center for kelps in the Northwestern Pacific Ocean [61]. Similar to Undaria pinnatifida and Gracilaria vermiculophylla (Ohmi) Papenfuss [30, 62], S. japonica exhibited the highest genetic diversity in Hokkaido. Among the 55 haplotypes analyzed with the COI and trnW-L marker systems, H1 was the most common in almost all populations (20/26), so H1 might be the ancestral haplotype. Moreover, pop7 and pop9 presented the highest genetic diversity among all populations (Fig. 2; Table 1), suggesting that the origin and diversification center may be on the Southwest coast of Hokkaido.
The invasive populations have lower genetic diversity than the source populations as reported in other introduced seaweeds, including Gracilaria vermiculophylla (Ohmi) Papenfuss [30], Codium fragile (Suringar) Hariot [63] and Caulerpa taxifolia (M. Vahl) C. Agardh [64]. S. japonica is not native to China and this kelp was firstly found in the vicinity of Dalian harbor around 1930s [25, 65], possibly because it was accidentally introduced by rafts or ships from Japan to China. It is interesting that natural populations in China have relatively high genetic diversity based on the nucleotide diversity and haplotype diversity (Table 1). We presumed that human mediated multiple introductions for breeding and cultivation contributed to high genetic diversity in China populations. Recently, selection, breeding and cultivation have reduced the genetic diversity and narrowed the genetic base to the cultivated kelp populations in China [7, 8]. In this study, the identified wild kelp populations with high genetic diversity might make important contributions to improve and enhance domesticated genepool of cultivated S. japonica in China.
Conclusions
This study examined the phylogeographic architecture and population genetic diversity of S. japonica in its native range. The shallow phylogeographic architecture suggests a complex interaction of anthropogenic interference (marine transportation and aquaculture) and post-LGM population expansion. The coast of Hokkaido might be the origin center for S. japonica in the northwestern Pacific. Extensive selection and multi-generation inbreeding have already reduced genetic diversity of S. japonica and caused genetic degeneration of cultivars in China. The generated knowledge about the levels of diversity of wild S. japonica populations is an important contribution for efficient breeding and rejuvenation of cultivated Saccharina by improving and enhancing domesticated genepool of S. japonica in China.
Availability of supporting data
The newly obtained COI and trnW-L haplotype sequences: GenBank accessions number: KT963115-KT963144 for COI; KT963093-KT963114 for trnW-L.
References
AlgaeBase. World-wide electronic publication, National University of Ireland, Galway. 2015. http://www.algaebase.org. Accessed 19 Oct 2015.
Lane CE, Mayes C, Druehl LD, Saunders GW. A multi-gene molecular investigation of the kelp (Laminariales, Phaeophyceae) supports substantial taxonomic re-organization1. J Phycol. 2006;42:493–512.
Selivanova ON, Zhigadlova GG, Hansen GI. Revision of the systematics of algae in the order Laminariales (Phaeophyta) from the Far-Eastern Seas of Russia on the basis of molecular-phylogenetic data. Russ J Mar Biol. 2007;33:278–89.
Yotsukura N, Kawashima S, Kawai T, Abe T, Druehl LD. Asystematic re-examination of four Laminariaspecies: L. japonica, L. religiosa, L. ochotensis and L. diabolica. J Jpn Bot. 2008;83:165–76.
Yotsukura N, Shimizu T, Katayama T, Druehl LD. Mitochondrial DNA sequence variation of four Saccharina species (Laminariales, Phaeophyceae) growing in Japan. J Appl Phycol. 2009;22:243–51.
Yotsukura N, Nagai K, Kimura H, Morimoto K. Seasonal changes in proteomic profiles of Japanese kelp: Saccharina japonica (Laminariales, Phaeophyceae). J Appl Phycol. 2010;22:443–51.
Liu FL, Yao JT, Wang XL, Repnikova A, Galanin DA, Duan DL. Genetic diversity and structure within and between wild and cultivated Saccharina japonica (Laminariales, Phaeophyta) revealed by SSR markers. Aquaculture. 2012;358–359:139–45.
Shan TF, Pang SJ, Zhang YR, Yakovleva IM, Skriptsova AV. An AFLP-based survey of genetic diversity and relationships of major farmed cultivars and geographically isolated wild populations of Saccharina japonica (Phaeophyta) along the northwest coasts of the Pacific. J Appl Phycol. 2010;23:35–45.
Liu JX, Gao TX, Wu SF, Zhang YP. Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol Ecol. 2007;16:275–88.
Shen KN, Jamandre BW, Hsu CC, Tzeng WN, Durand JD. Plio-Pleistocene sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus. BMC Evol Biol. 2011;11:83.
Hu ZM, Zhang J, Lopez-Bautista J, Duan DL. Asymmetric genetic exchange in the brown seaweed Sargassum fusiforme (Phaeophyceae) driven by oceanic currents. Mar Biol. 2013;160:1407–14.
Cheang CC, Tsang LM, Ng WC, Williams GA, Chu KH, Chan BKK. Phylogeography of the cold-water barnacle Chthamalus challengeri in the north-western Pacific: effect of past population expansion and contemporary gene flow. J Biogeography. 2012;39:1819–35.
Xu JW, Chan TY, Tsang LM, Chu KH. Phylogeography of the mitten crab Eriocheir sensu stricto in East Asia: Pleistocene isolation, population expansion and secondary contact. Mol Phylogenet Evol. 2009;52:45–56.
Oba T, Kato M, Kitazato H, Koizumi I, Omura A, Sakai T, et al. Paleoenvironmental changes in the Japan Sea during the last 85,000 years. Paleoceanography. 1991;6:499–518.
Ishiwatari R, Uzaki M, Yamada K. Carbon-isotope composition of individual N-alkanes in recent sediments. Org Geochem. 1994;21:801–8.
Tada R, Irino T, Koizumi I. Land-ocean linkages over orbital and millennial timescales recorded in late Quaternary sediments of the Japan Sea. Paleoceanography. 1999;14:236–47.
Kitamura A, Takano O, Takata H, Omote H. Late Pliocene-early Pleistocene paleoceanographic evolution of the Sea of Japan. Palaeogeogr Palaeocl. 2001;172:81–98.
Cheang CC, Chu KH, Ang Jr PO. Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Mol Ecol. 2010;19:2933–48.
Hu ZM, Uwai S, Yu SH, Komatsu T, Ajisaka T, Duan DL. Phylogeographic heterogeneity of the brown macroalga Sargassum horneri (Fucaceae) in the northwestern Pacific in relation to late Pleistocene glaciation and tectonic configurations. Mol Ecol. 2011;20:3894–909.
Muhlin JF, Engel CR, Stessel R, Weatherbee RA, Brawley SH. The influence of coastal topography, circulation patterns, and rafting in structuring populations of an intertidal alga. Mol Ecol. 2008;17:1198–210.
Alberto F, Raimondi PT, Reed DC, Watson JR, Siegel DA, Mitarai S, et al. Isolation by oceanographic distance explains genetic structure for Macrocystis pyrifera in the Santa Barbara Channel. Mol Ecol. 2011;20:2543–54.
Brennan G, Kregting L, Beatty GE, Cole C, Elsasser B, Savidge G, et al. Understanding macroalgal dispersal in a complex hydrodynamic environment: a combined population genetic and physical modelling approach. J R Soc Interface. 2014;11:20140197.
White C, Selkoe KA, Watson J, Siegel DA, Zacherl DC, Toonen RJ. Ocean currents help explain population genetic structure. Proc R Soc B. 2010;277:1685–94.
Billot C, Engel CR, Rousvoal S, Kloareg B, Valero M. Current patterns, habitat discontinuities and population genetic structure: the case of the kelp Laminaria digitata in the English Channel. Mar EcolProg Ser. 2003;253:21.
Tseng CK. Algal biotechnology industries and research activities in China. J Appl Phycol. 2001;13:375–80.
Voisin M, Engel CR, Viard F. Differential shuffling of native genetic diversity across introduced regions in a brown alga: aquaculture vs. maritime traffic effects. Proc Natl Acad Sci USA. 2005;102:5432–7.
McDevit DC, Saunders GW. A DNA barcode examination of the Laminariaceae (Phaeophyceae) in Canada reveals novel biogeographical and evolutionary insights. Phycologia. 2010;3:235–48.
Olsen JL, Zechman FW, Hoarau G, Coyer JA, Stam WT, Valero M, et al. The phylogeographic architecture of the fucoid seaweed Ascophyllum nodosum: an intertidal ‘marine tree’ and survivor of more than one glacial-interglacial cycle. J Biogeography. 2010;37:842–56.
Coyer JA, Hoarau G, Van Schaik J, Luijckx P, Olsen JL. Trans-Pacific and trans-Arctic pathways of the intertidal macroalga Fucus distichus L. reveal multiple glacial refugia and colonizations from the North Pacific to the North Atlantic. J Biogeography. 2011;4:756–71.
Kim SY, Weinberger F, Boo SM. Genetic data hint at a common donor region for invasive Atlantic and Pacific populations of Gracilaria vermiculophylla (Gracilariales, Rhodophyta). J Phycol. 2010;46:1346–9.
Yow YY, Lim PE, Phang SM. Genetic diversity of Gracilaria changii (Gracilariaceae, Rhodophyta) from west coast, Peninsular Malaysia based on mitochondrial cox1 gene analysis. J Appl Phycol. 2010;23:219–26.
Hu ZM, Li JJ, Sun ZM, Oak JH, Zhang J, Fresia P, et al. Phylogeographic structure and deep lineage diversification of the red alga Chondrus ocellatus Holmes in the Northwest Pacific. Mol Ecol. 2015;19:5020–33.
Balakirev ES, Krupnova TN, Ayala FJ. DNA variation in the phenotypically-diverse brown alga Saccharina japonica. BMC Plant Biol. 2012;12:108.
Hall AT. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Corander J, Waldmann P, Marttinen P, Sillanpaa MJ. BAPS 2: enhanced possibilities for the analysis of genetic population structure. Bioinformatics. 2004;20:2363–9.
Corander J, Marttinen P, Siren J, Tang J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics. 2008;9:539.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Slatkin M. Isolation by distance in equilibrium and nonequilibrium populations. Evolution. 1993;47:264–79.
Rousset F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997;145:1219–28.
Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–60.
Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA. 2007;104:2785–90.
Silberfeld T, Leigh JW, Verbruggen H, Cruaud C, de Reviers B, Rousseau F. A multi-locus time-calibrated phylogeny of the brown algae (Heterokonta, Ochrophyta, Phaeophyceae): Investigating the evolutionary nature of the “brown algal crown radiation”. Mol Phylogenet Evol. 2010;56:659–74.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;6:1695–701.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Tajima F. Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Schneider S, Excoffier L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates very among sites: Application to human mitochondrial DNA. Genetics. 1999;152:1079–89.
Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22:1185–92.
Hoarau G, Coyer JA, Veldsink JH, Stam WT, Olsen JL. Glacial refugia and recolonization pathways in the brown seaweed Fucus serratus. Mol Ecol. 2007;16:3606–16.
Chan SW, Cheang CC, Chirapart A, Gerung G, Tharith C, Ang P. Homogeneous population of the brown alga Sargassum polycystum in Southeast Asia: possible role of recent expansion and asexual propagation. PLoS ONE. 2013; doi: 10.1371/journal.pone.0077662.
Ni G, Li QI, Kong L, Yu H. Comparative phylogeography in marginal seas of the northwestern Pacific. Mol Ecol. 2014;23:534–48.
Bae SW, Lee KE, Park Y, Kimoto K, Ikehara K, Harada N. Sea surface temperature and salinity changes near the Soya Strait during the last 19 ka. Quatern Int. 2014;344:200–10.
Reed DC, Schroeter SC, Raimondi PT. Spore supply and habitat availability as sources of recruitment limitation in the giant kelp Macrocystis pyrifera (Phaeophyceae). J Phycol. 2004;40:275–84.
Li J, Pang SJ, Liu F, Shan TF, Gao SQ. Spermatozoid life-span of two brown seaweeds, Saccharina japonica and Undaria pinnatifida, as measured by fertilization efficiency. Chin J Oceanol Limn. 2013;31:774–81.
Neiva J, Pearson GA, Valero M, Serrão EA. Drifting fronds and drifting alleles: range dynamics, local dispersal and habitat isolation shape the population structure of the estuarine seaweed Fucus ceranoides. J Biogeography. 2012;39:1167–78.
Voisin M, Engel C, Viard F. Unravelling the mechanisms behind introduction through genetic approaches: Undaria Pinnatifida (Alariaceae) as a model species. Phycologia. 2005;44:106–7.
Marko PB, Hoffman JM, Emme SA, Mcgovern TM, Keever C, Cox LN. The “Expansion-Contraction” model of pleistocene biogeography: rocky shores suffer a sea change? Integr Comp Biol. 2010;50:108.
Bolton JJ. The biogeography of kelps (Laminariales, Phaeophyceae): a global analysis with new insights from recent advances in molecular phylogenetics. Helgoland Mar Res. 2010;64:263–79.
Uwai S, Nelson W, Neill K, Wang WD, Aguilar-Rosas LE, Boo SM, et al. Genetic diversity in Undaria pinnatifida (Laminariales, Phaeophyceae) deduced from mitochondria genes – origins and succession of introduced populations. Phycologia. 2006;45:687–95.
Provan J, Booth D, Todd NP, Beatty GE, Maggs CA. Tracking biological invasions in space and time: elucidating the invasive history of the green alga Codium fragile using old DNA. Divers Distrib. 2008;14:343–54.
Jousson O, Pawlowski J, Zaninetti L, Zechman FW, Dini F, Di Guiseppe G, et al. Invasive alga reaches California - the alga has been identified that threatens to smother Californian coastal ecosystems. Nature. 2000;408:157–8.
Tseng CK. The past, present and future of phycology in China. Hydrobiologia. 2004;512:11–20.
Acknowledgement
We acknowledge anonymous reviewers for the critical comments and suggestions for the manuscript, thanks are due to Prof. S.M. Kim for kelp samples from Korea and Mr. Donald Sturge for English revision. This research was supported by NSFC (31302188) to J.T. Yao, and National Key Technology Research and Development Program (2013BAB01B01), and the Ocean Public Welfare Scientific Research Project (201405040) to D.L. Duan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
There are no financial or non-financial competing interests.
Authors’ contributions
JZ carried out the molecular genetic studies, performed the statistical analysis and drafted the manuscript. ZMS, GF, DAG, NC and MT participated in the collection of samples and biological data. ZMH helped to analyze and interpret the data and revised the manuscript. JTY and DLD participated in the design of the study and obtained funding. DLD conceived the study, participated in its design and coordination, and helped to draft the manuscript. All authors have read and approved the final version of the manuscript.
Additional files
Additional file 1: Table S1.
Collection details of the 26 populations of Saccharina japonica studied. (PDF 48 kb)
Additional file 2: Figure S1.
Tessellation illustration of Bayesian analysis of population structure. Each cell of the tessellation corresponds to the physical neighborhood of an observed data point. Various colors indicate genetic groupings as retrieved by phylogenetic analyses. (PDF 238 kb)
Additional file 3: Table S2.
Genetic differentiation (F ST) between the pairs of four genetic clusters. **,P< 0.001; *,P< 0.05 (PDF 48 kb)
Additional file 4: Table S3.
Pairwise F ST estimates among Saccharina japonica populations. *P< 0.05; **P< 0.001; ns not significant. (PDF 64 kb)
Additional file 5: Figure S2.
Isolation-by distance (IBD) analyses within 22 populations (except for four populations in China and Korea). Regression of genetic differentiation (estimated by F ST/(1- F ST)) against logarithm of geographical distances (km). (PDF 36 kb)
Additional file 6: Figure S3.
Pairwise mismatch distributions for Saccharina japonica inferred from mtDNA sequences. The abscissa indicates the number of pairwise differences between compared sequences. The ordinate is frequency for each value. Bar represent the observed distribution of pairwise frequencies, while the solid line shows the expected distribution. (PDF 340 kb)
Additional file 7: Figure S4.
Bayesian skyline plots showing effective population size as a function of time. (a) inferred from single sequence COI; (b) inferred from single sequence trnW-L;(c) inferred from combined mtDNA sequences (COI + trnW-L). The upper and lower limits of light blue trend represent the 95 % confidence intervals of HPD analysis. (PDF 150 kb)
Additional file 8: Figure S5.
Maximum clade credibility coalescent tree, based on COI. (PDF 57 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, J., Yao, JT., Sun, ZM. et al. Phylogeographic data revealed shallow genetic structure in the kelp Saccharina japonica (Laminariales, Phaeophyta). BMC Evol Biol 15, 237 (2015). https://doi.org/10.1186/s12862-015-0517-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-015-0517-8